
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroalkynylative cyclization of 1,6-enynes with terminal alkynes†
Qi
Teng
a,
Nuligonda
Thirupathi
a,
Chen-Ho
Tung
 a and
Zhenghu
Xu
*ab
a and
Zhenghu
Xu
*ab
aKey Lab for Colloid and Interface Chemistry of Education Ministry, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, People's Republic of China. E-mail: xuzh@sdu.edu.cn
bState Key Laboratory of Organometallic Chemistry Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, PR China
First published on 13th June 2019
Abstract
A rhodium-catalyzed highly regio- and enantioselective hydroalkynylation, generating cis-hydrobenzofuranone-tethered enynes has been developed. The reaction proceeds with a selective head-to-head insertion and symmetry breaking Michael addition cascade. One product was produced from tens of possible isomers through precise control of chemo-, regio-, and stereoselectivities using a single rhodium catalyst. Notable features of this method include 100% atom-economy, mild reaction conditions and a very broad substrate scope.
Introduction
Conjugated enynes are a type of privileged structure distributed in a number of natural products1 and luminescent materials.2 They are also very important building blocks widely used in organic synthesis.3 The most attractive and atom-economic pathway to conjugated enynes is the transition-metal catalyzed direct addition of a terminal alkyne C–H bond to a second alkyne moiety.4,5 Generally the acceptor alkynes are electron-deficient internal alkynes.4 Hydroalkynylation6 of a second terminal alkyne, also termed cross-dimerization of two different terminal alkynes is more challenging. This reaction could in principal produce six possible cross-dimerization products and six homodimerization side products (Scheme 1a). Ideally, the metal catalyst should selectively produce one isomer from 12 similar isomers, and consequently very few examples have been reported.5 Taking advantage of the steric and electronic differences of two alkynes, so controlling chemo- and regioselectivity is a possible solution. For instance, silyl alkynes were usually chosen to couple with aryl or aliphatic alkynes to give different products depending on the choice of catalyst.5d,e However, in all these reactions, undesired isomers were always produced together with the major product.5 Consequently, the development of a new synthetically useful strategy to realize selective cross-hydroalkynylation reactions to access conjugated enynes7 is important.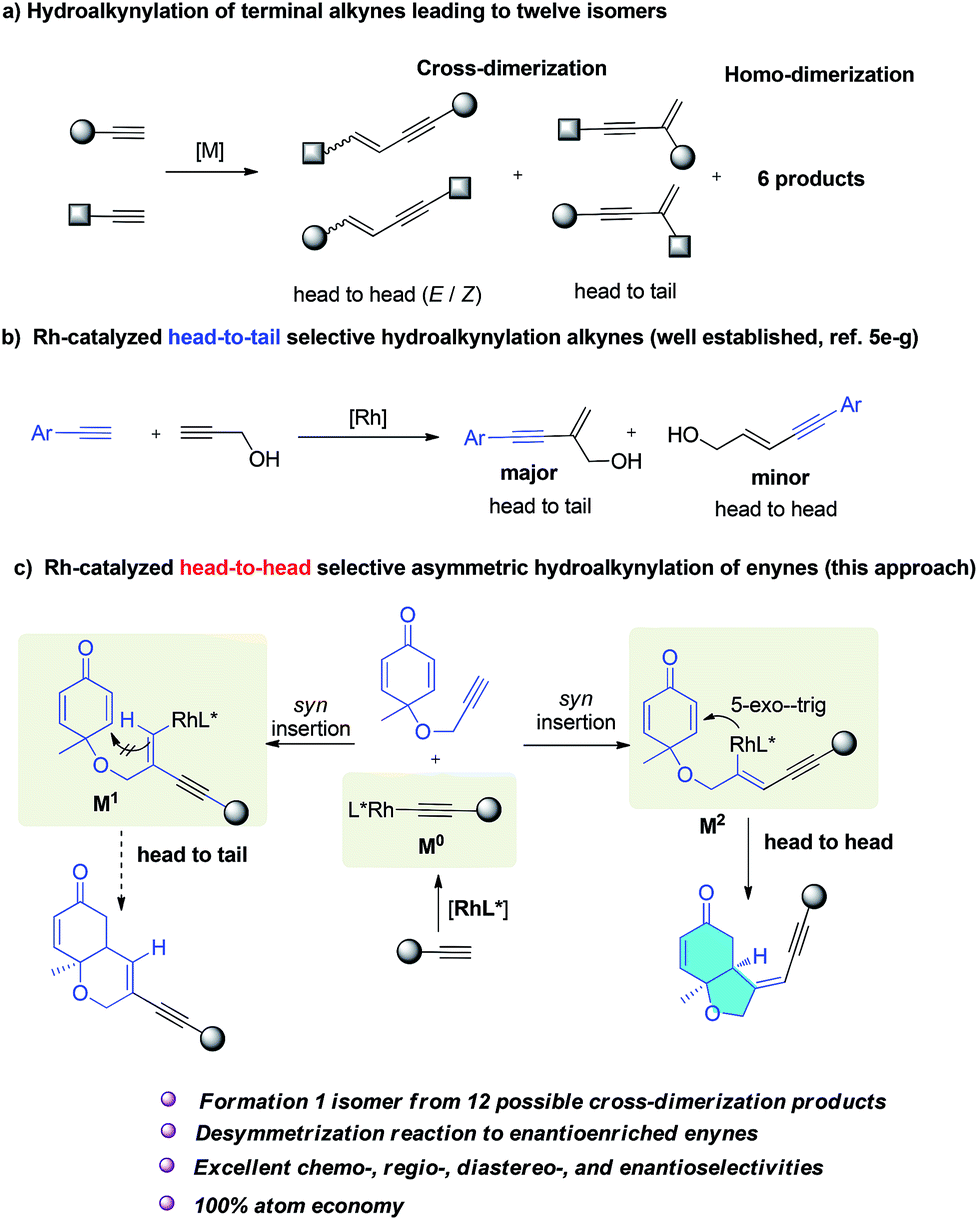 | ||
| Scheme 1 Rh-catalyzed cross-hydroalkynylation of terminal alkynes. | ||
Generally, a metal acetylide complex was selectively formed, and subsequent syn insertion into the other triple bond usually led to two different alkenyl metal intermediates because both head-to-head and head-to-tail insertion are possible.
Completely selective formation of one intermediate over another similar intermediate is generally unlikely and most reactions reported always led to one major product together with other isomers as minor products.5 When aryl alkynes were reacted with propargyl alcohol, head-to-head dimers were obtained as a major product in the presence of a palladium catalyst,5c,j but when rhodium catalyst was employed, head-to-tail dimers were obtained as a major product, together with some head-to-head isomers as byproducts (Scheme 1b).5e–g Herein, we report the first Rh-catalyzed asymmetric cross-hydroalkynylation of 1,6-enynes via a selective head-to-head insertion and symmetry breaking Michael addition cascade. As illustrated in Scheme 1c, rhodium acetylide (M0) was selectively formed from a terminal alkyne and its subsequent syn addition to the alkyne moiety of enynes could generate two vinyl rhodium intermediates (M1, M2). M2 can undergo a fast 5-exo-trig cyclization to form the final bicyclic products. However, M1 bearing a trans C–Rh bond cannot undergo the Michael addition to form stabilized ring products. A possible E/Z isomerization8 of M1 might generate a cis C–Rh bond which could undergo Michael addition, but this will involve a much higher activation energy. Consequently, the formation of M2 would be preferred, delivering the head-to-head products selectively. More importantly, when a chiral phosphine ligand was applied, enantioenriched enynes bearing an important chiral cis-hydrobenzofuran moiety were obtained. This is a unique scaffold present in many bioactive natural products including incarviditone,9a isoambrox9b haterumaimide I,9c and milling-tonine A.9d To date, no examples of synthesis of chiral enynes by asymmetric cross-hydroalkynylation process have been reported.
Results and discussion
A cyclohexadienone tethered terminal alkyne (1a) was selected as one partner in this reaction, because this prochiral electron-deficient diene is an excellent Michael acceptor with which to build chiral cis-hydrobenzofuranones by organo- or transition-metal-catalyzed desymmetrization reactions.10,11 The aromatic alkyne (2a), bearing an alkyne proton which is more acidic than that in aliphatic alkynes,12 was chosen as the second partner to react with the enyne (1a) in the presence of a rhodium catalyst. The rhodium catalyst might react with 2a to form phenyl-ethynyl rhodium, and upon subsequent hydroalkynylation of 1a, deliver the cyclization products. To test this hypothesis, the reaction was performed in dichloroethane (DCE) at rt overnight in the presence of 5 mol% of [Rh(COD)Cl]2 and 10 mol% of S-BINAP. The desired head-to-head bicyclo[4.3.0] product (3a) was obtained in 72% isolated yield as a single diastereomer, and no head-to-tail bicyclo[4.4.0] product was observed in the reaction (Table 1, entry 1). These results are contrary to previous rhodium-catalyzed reactions which give head-to-tail structures as major products5e–g (Scheme 1b) and indicate the viability of the proposed reaction shown in Scheme 1c. In addition, HPLC analysis shows that the enantioselectivity of this reaction is as high as 94% ee.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Solvent | Yieldb/% | eec/% |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), catalyst (5 mol%), and ligand (10 mol%), solvent (2 mL), rt. b Isolated yield. c Determined by HPLC analysis on a chiral stationary phase. d 1a (0.2 mmol), 2a (0.4 mmol), [Rh(cod)Cl]2 (2.5 mol%), and L1 (5 mol%). | |||||
| 1 | [Rh(cod)Cl]2 | L1 | DCE | 72 | 94 |
| 2 | [Rh(cod)Cl]2 | L2 | DCE | 94 | 79 |
| 3 | [Rh(cod)Cl]2 | L3 | DCE | 50 | 83 |
| 4 | [Rh(cod)Cl]2 | L4 | DCE | 98 | 77 |
| 5 | [Rh(cod)Cl]2 | L5 | DCE | 99 | 88 |
| 6 | [Rh(cod)Cl]2 | L6 | DCE | 48 | 98 |
| 7 | [Rh(cod)Cl]2 | L1 | MeOH | 62 | 99 |
| 8 | [Rh(cod)Cl]2 | L1 | THF | 0 | — |
| 9 | [Rh(cod)Cl]2 | L1 | DMF | Trace | — |
| 10 | [Rh(cod)Cl]2 | L1 | Toluene | 59 | 92 |
| 11 | [Rh(cod)Cl]2 | L1 | DCE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH = 3:1 :MeOH = 3:1 |
81 | 98 |
| 12 | [Rh(cod)Cl]2 | L1 | DCE:MeOH = 1:1 |
66 | 97 |
| 13 | [Rh(cod)Cl] 2 | L1 |
DCE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :MeOH = 3:1 :MeOH = 3:1
|
92 | 99 |
| 14 | [Rh(cp*)Cl2]2 | L1 | DCE | 0 | — |
| 15 | NiCl2·6H2O | L1 | DCE | 0 | — |
| 16 | Ni(acac)2 | L1 | DCE | 0 | — |
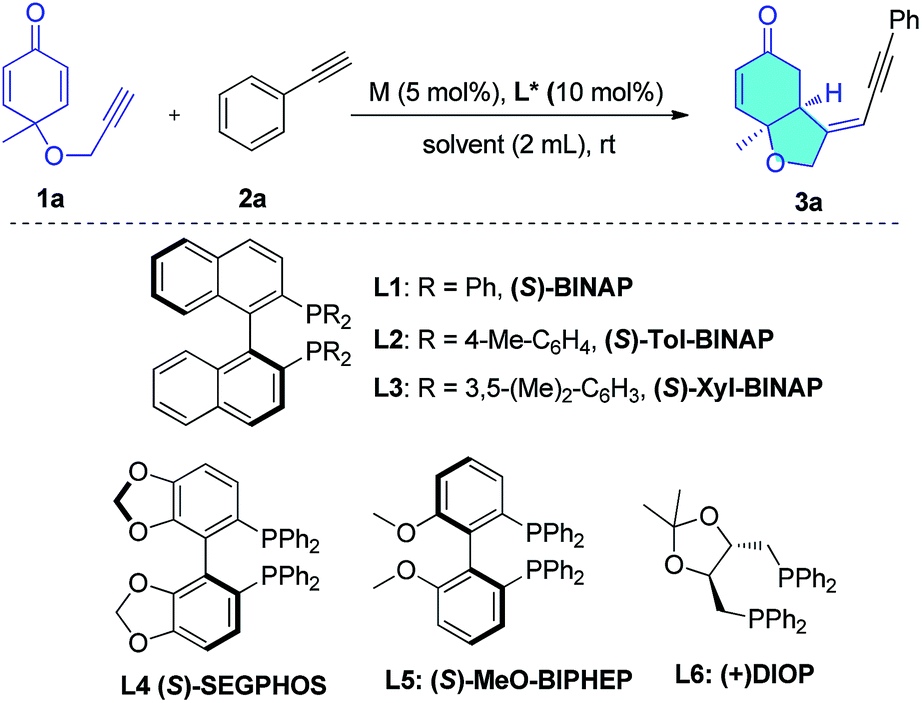
Next, we sought to optimize the reaction conditions, obtaining the results shown in Table 1. We first examined various diphosphine ligands (entries 1–6). Increasing the steric hindrance of the phosphine substituents led to lower enantioselectivities (entries 2 and 3). Biphenyl ligands such as SEGPHOS (L4) and MeO-BIPHEP (L5) gave almost quantitative yields but lower enantioselectivity (entries 4 and 5). When DIOP (L6) was employed, the highest 98% ee was obtained but with much lower yield (entry 6). The solvent effect was then investigated (entries 7–12). The reaction in MeOH was much more rapid, leading to 99% ee but lower isolated yield (62%, entry 7). We observed that when DCE was added to the MeOH solvent, a higher reaction yield was obtained without any obvious decrease in enantioselectivity (entries 11 and 12). Increasing the concentration of substrates and lowering the catalyst loading to 5 mol% in the mixed solvent (DCE:MeOH = 3:1) led to 92% isolated yield and 99% ee, and represents the optimal conditions (entry 13). When the Rh(I) catalyst was replaced by a Rh(III) catalyst (entry 14) or nickel catalysts (entries 15 and 16), no product was formed. Under the optimized conditions, this atom-economic reaction produces chiral hydrobenzofuranone-tethered enynes in high yield with extremely high enantioselectivity. Notably, the rhodium catalyst produced a single product from tens of possible products counting two new chiral centers by precise control of the chemo-, regio-, diastereo-, and enantioselectivities. Such reactions are very rare in organic synthesis.
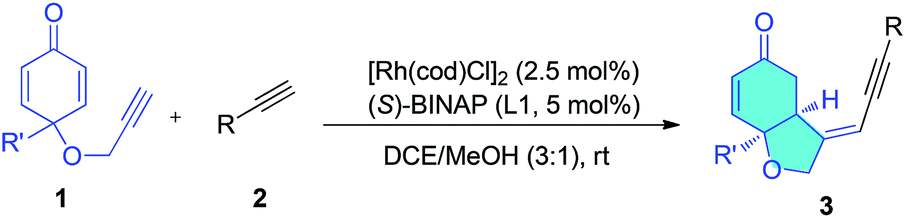
Having established the optimal conditions for this asymmetric enyne hydroalkynylation reaction, the scope of terminal alkynes was investigated. As shown in Table 2, various aromatic terminal alkynes reacted with enyne 1a under the optimized conditions, and the corresponding head-to-head hydroalkynylation products (3a–3q) were produced in good yields (50–97%) and excellent enantioselectivity (92% to >99% ee). Various electron-withdrawing or electron-donating functional groups at the para, ortho, or meta- positions do not affect this reaction (entries 1–11). Electron-donating groups generally favor the reaction and electron-withdrawing groups lead to slightly lower yields. Functional groups such as alkoxyl (3c), halogens (3d–3f), CF3 (3c), and even an unprotected hydroxyl group (3i, 3j) are all tolerated. In the case of 2j, the 2-hydroxymethyl group led to decreased enantioselectivity (92% ee), but when the hydroxyl group was protected, 99% ee was obtained (entries 10 and 11). Terminal alkynes containing heterocycles, including indole and thiophene also reacted smoothly under the standard conditions, delivering corresponding enantioenriched enynes in good yields and with 96–99% ee (entries 14–16). Remarkably, the ferrocene-derived alkyne (2q) was also suitable for this reaction and gave the cyclization product (3q) in 80% yield and with over 99% ee (entry 17). Two single crystals of products 3m and 3p were grown to confirm their structures and stereochemistry. The absolute configuration of these two products were established as S and S by X-ray crystallography (Scheme 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 (R′) | 2 (R) | 3 | Yieldb/% | eec/% |
| a Reaction conditions: 1a (0.2 mmol), 2 (0.4 mmol), [Rh(cod)Cl]2 (2.5 mol%), and L1 (5 mol%), rt. b Isolated yield. c Determined by HPLC analysis on a chiral stationary phase. | |||||
| 1 | 1a (Me) | 2a (C6H5) | 3a | 92 | 99 |
| 2 | 1a | 2b (4-MeC6H4) | 3b | 83 | 98 |
| 3 | 1a | 2c (4-MeOC6H4) | 3c | 90 | 98 |
| 4 | 1a | 2d (4-FC6H4) | 3d | 92 | >99 |
| 5 | 1a | 2e (4-ClC6H4) | 3e | 67 | >99 |
| 6 | 1a | 2f (4-BrC6H4) | 3f | 66 | >99 |
| 7 | 1a | 2g (4-CF3C6H4) | 3g | 73 | 99 |
| 8 | 1a | 2h (3-MeC6H4) | 3h | 80 | 99 |
| 9 | 1a | 2i (3-OHC6H4) | 3i | 64 | 99 |
| 10 | 1a | 2j (2-CH2OHC6H4) | 3j | 50 | 92 |
| 11 | 1a | 2k (2-CH2OTBSC6H4) | 3k | 58 | 99 |
| 12 | 1a | 2l (1-naphthyl) | 3l | 81 | 99 |
| 13 | 1a | 2m (2-naphthyl) | 3m | 78 | 98 |
| 14 | 1a | 2n (3-indolyl) | 3n | 68 | 96 |
| 15 | 1a | 2o (3-thienyl) | 3o | 96 | >99 |
| 16 | 1a | 2p (2-thienyl) | 3p | 97 | 99 |
| 17 | 1a | 2q (ferrocenyl) | 3q | 80 | >99 |
| 18 | 1a | 2r (cyclopropyl) | 3r | 89 | >99 |
| 19 | 1a | 2s (C6H5CH2CH2CH2–) | 3s | 55 | >99 |
| 20 | 1a | 2t (C6H5OCH2–) | 3t | 46 | 98 |
| 21 | 1a |
|
3u | 96 | >99 |
| 22 | 1b (Et-) | 2a (C6H5) | 3v | 93 | >99 |
| 23 | 1b (Et-) | 2d (4-FC6H4) | 3w | 81 | >99 |
| 24 | 1c (i-propyl) | 2a | 3x | 86 | 97 |
| 25 | 1d (C6H5) | 2a | 3y | 89 | 99 |
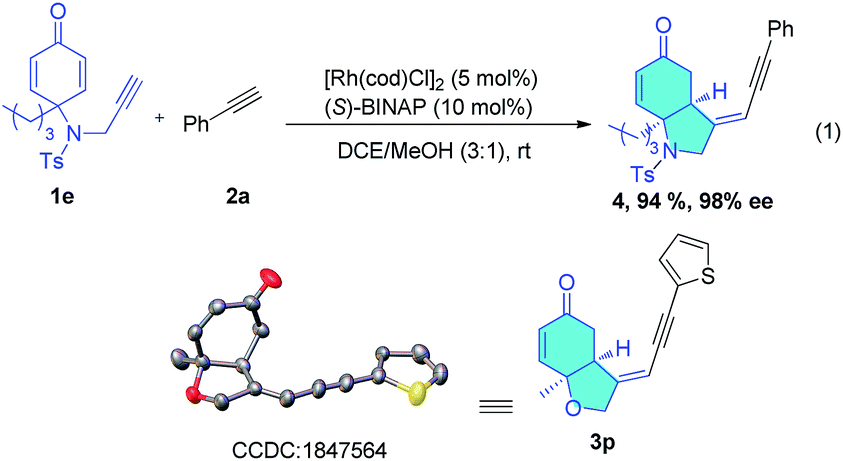 | ||
| Scheme 2 X-ray crystallographic structure of 3p. | ||
Selective cross-dimerization of two aliphatic alkynes is more challenging and is unprecedented. The cyclopropyl alkyne (2r) and cyclohexenyl alkyne (2u) react equally well under these conditions and give the corresponding bicyclic products (3r, 3u) in excellent yields (89% and 96%) and enantioselectivity (>99% ee), (entries 18 and 21). Simple linear aliphatic alkynes (2s, 2t) were converted to the corresponding bicyclic enynes in acceptable yields with excellent enantioselectivities (99% and 98% ee), (entries 19 and 20). The scope of the enynes was also studied. Enynes containing aliphatic groups such as ethyl or the bulkier isopropyl, and a phenyl group at the prochiral quaternary carbon center all reacted smoothly to give the corresponding desymmetrization products in excellent yields and enantioselectivities (entries 22–25). The nitrogen bridged enyne (1e) was also amenable to this transformation, giving the nitrogen heterocycle (4) in 94% yield and 98% ee (eqn (1)).
In a large scale reaction, 1.14 g of 3a was isolated in 86% yield and 99% ee with low catalyst loading, demonstrating the remarkable practicality of this method (eqn (2)). Further synthetic transformations of the bicyclic enynes were explored (Scheme 3). Selective reduction of the ketone group by NaBH4 afforded an allyl alcohol (5) in 93% yield with 98% ee. Pd/C-catalyzed hydrogenation of 3a with a balloon of H2 led to complete hydrogenation of the enyne moiety in the side chain and enone affording ketone (6) in 95% yield. Conjugated borylation of 3a with B2Pin2 in the presence of CuI gave the borate (7) in 72% yield and 99% ee. The silyl enol ether (8) was prepared to undergo a gold-catalyzed intramolecular Conia-ene reaction, the addition of enol ether to alkyne moiety to yield cyclized products, but the desired tricyclic product was not formed, and instead, a phenol tethered enyne (9) was produced in 85% yield through an aromatized C–O bond cleavage. It should be noted that formally, this is a meta-functionalization reaction of phenol, which is very difficult to achieve by known methods.
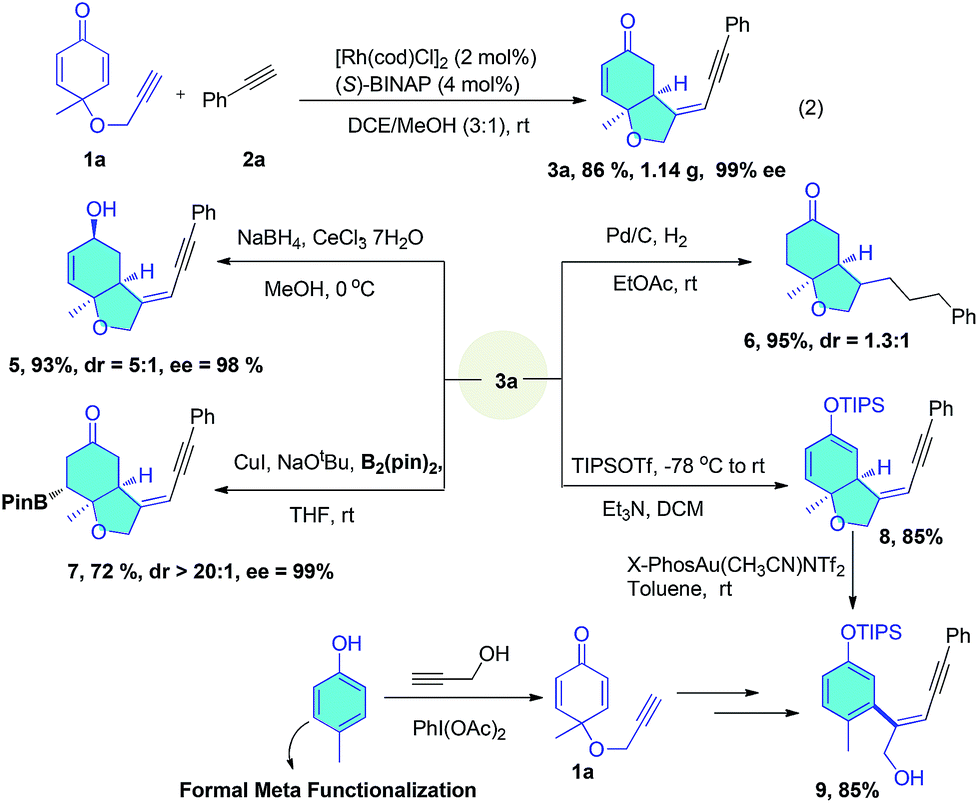 | ||
| Scheme 3 Scale-up experiment and further synthetic applications of bicyclic enynes. | ||
To gain insight into the hydroalkynylation process, we conducted deuterium labelling experiments (Scheme 4). The standard reaction in MeOD led to 3a-D with 50% deuterium content at the methylene position adjacent to the ketone (eqn (3)). When the deuterated alkyne 2c-D was subjected to the reaction in MeOH, the CH2 was not deuterated at all, but when the reaction was performed in DCE solution, the proton was 22% deuterated (eqn (4)). Product 3a was not deuterated under standard conditions in MeOD (eqn (5)). These results indicate that the origin of this hydrogen could be the alkyne proton and solvent. Based on these results and previous reports, a plausible mechanism is proposed and shown in Scheme 4. Rh(I) catalyst reacted with 2a forming Rh(I) acetylide M0 and released catalytic amount of HCl, which might exchange with solvent methanol and played a very important role in the final protonation step. Rh(I) acetylide M0 went through head-to-head insertion afforded the vinyl rhodium intermediate (M2). Asymmetric Michael addition to this prochiral diene gave an Rh(I) enolate (M3), which was protonated by HCl/MeOH to form the product (3a) and Rh(I)Cl. This final unselective protonation of Rh(I) enolate (M3) with HCl/MeOD could result in both protons of the methylene group being deuterated, which would be consistent with eqn (3). If the first formation of Rh(I) acetylide M0 was oxidative addition, then the final step should be reductive elimination and the proton of the methylene group should come from the terminal alkyne, which fails to agree with eqn (3). If [Rh(COD)OMe]2 catalyst was used instead of chloride catalyst, much slower reaction and lower yield was observed, because this catalyst reacted with terminal alkyne only produce neutral methanol, but not acid.
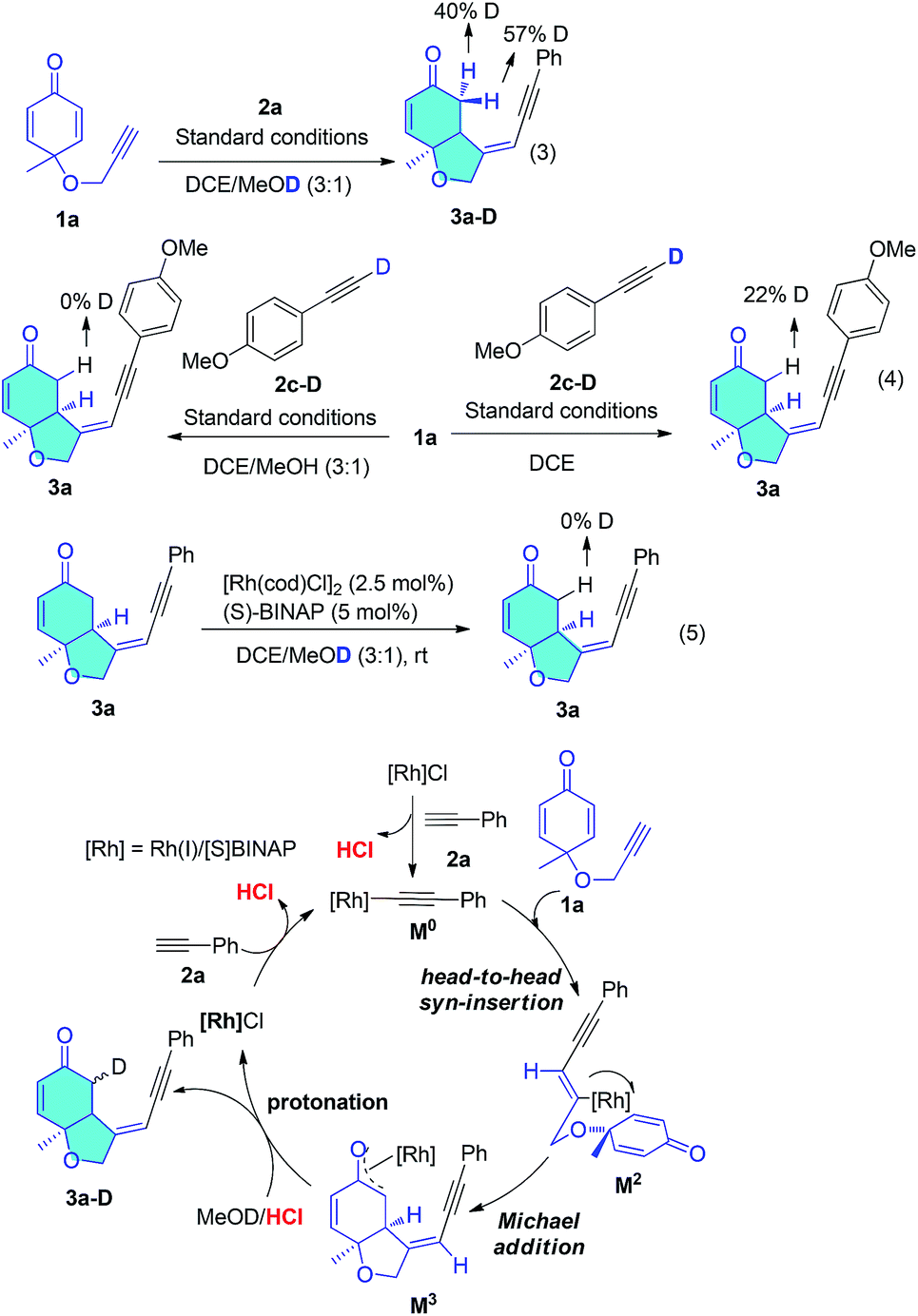 | ||
| Scheme 4 Control experiments and proposed mechanism. | ||
Conclusions
In summary, we have developed a highly enantioselective Rh-catalyzed hydroalkynylation reaction that can be used for the synthesis of enantioenriched cis-hydrobenzofuranone-tethered enynes. Although many isomers are possible, a single head-to-head regioisomer was obtained in this reaction. Notable features of this approach include 100% atom-economy, mild reaction conditions, a very broad substrate scope, and precise control of chemo-, regio-, diastereo- and enantioselectivity by a single catalyst. Further application of this strategy is in progress in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Natural Science Foundation of China (no. 21572118), and Tang scholar award and The Fundamental Research Funds of Shandong University. We thank Prof. Dr Di Sun who carried out the X-ray crystallographic analysis.Notes and references
- (a) P. Lan, L. E. White, E. S. Taher, P. E. Guest, M. G. Banwell and A. C. Willis, J. Nat. Prod., 2015, 78, 1963 CrossRef CAS; (b) P. Nussbaumer, I. Leitner, K. Mraz and A. Stütz, J. Med. Chem., 1995, 38, 1831 CrossRef CAS PubMed.
- Y. Liu, M. Nishiura, Y. Wang and Z. Hou, J. Am. Chem. Soc., 2006, 128, 5592 CrossRef CAS PubMed.
- (a) P. McGee, G. Bétournay, F. Barabé and L. Barriault, Angew. Chem., Int. Ed., 2017, 56, 6280 CrossRef CAS PubMed; (b) J. Cheng and T.-P. Loh, J. Am. Chem. Soc., 2015, 137, 42 CrossRef CAS PubMed; (c) Y. Wang, P. Zhang, D. Qian and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14849 CrossRef CAS PubMed; (d) W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, J. Am. Chem. Soc., 2010, 132, 3664 CrossRef CAS PubMed.
- (a) B. M. Trost, M. T. Sorum, C. Chan, A. E. Harms and G. Rühter, J. Am. Chem. Soc., 1997, 119, 698 CrossRef CAS; (b) B. M. Trost, B. R. Taft, J. T. Masters and J.-P. Lumb, J. Am. Chem. Soc., 2011, 133, 8502 CrossRef CAS PubMed.
- For a review, see: (a) B. M. Trost and J. T. maters, Chem. Soc. Rev., 2016, 45, 2212 RSC; For recent examples, see: (b) Q. Liang, K. M. Osten and D. Song, Angew. Chem., Int. Ed., 2017, 56, 6317 CrossRef CAS PubMed; (c) M. G. Lauer, B. R. Headford, O. M. Gobble, M. B. Weyhaupt, D. L. Gerlach, M. Zeller and K. H. Shaughnessy, ACS Catal., 2016, 6, 5834 CrossRef CAS; (d) O. Rivada-Wheelaghan, S. Chakraborty, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 6942 CrossRef CAS PubMed; (e) R. Azpíroz, L. Rubio-Pérez, R. Castarlenas, J. J. Pérez-Torrente and L. A. Oro, ChemCatChem, 2014, 6, 2587 CrossRef; (f) H.-D. Xu, R. Zhang, X. Li, S. Huang, W. Tang and W.-H. Hu, Org. Lett., 2013, 15, 840 CrossRef CAS PubMed; (g) H. M. Peng, J. Zhao and X. Li, Adv. Synth. Catal., 2009, 351, 1371 CrossRef CAS; (h) R. H. Platel and L. L. Schafer, Chem. Commun., 2012, 48, 10609 RSC; (i) C.-C. Lee, Y.-C. Lin, Y.-H. Liu and Y. Wang, Organometallics, 2005, 24, 136 CrossRef CAS; (j) C. Jahier, O. V. Zatolochnaya, N. V. Zvyagintsev, V. P. Ananikov and V. Gevorgyan, Org. Lett., 2012, 14, 2846 CrossRef CAS PubMed; (k) C.-H. Jun, Z. Lu and R. H. Crabtree, Tetrahedron Lett., 1992, 33, 7119 CrossRef CAS.
- Rh-catalyzed asymmetric hydroalkynylation of alkene moiety has been well developed, see: (a) X.-Y. Bai, W.-W. Zhang, Q. Li and B.-J. Li, J. Am. Chem. Soc., 2018, 140, 506 CrossRef CAS PubMed; (b) K.-L. Choo and M. Lautens, Org. Lett., 2018, 20, 1380 CrossRef CAS PubMed; (c) Y. Zhi, J. Huang, N. Liu, T. Lu and X. Dou, Org. Lett., 2017, 19, 2378 CrossRef CAS PubMed; (d) X.-Y. Bai, Z.-X. Wang and B.-J. Li, Angew. Chem., Int. Ed., 2016, 55, 9007 CrossRef CAS PubMed; (e) X. Dou, Y. Huang and T. Hayashi, Angew. Chem., Int. Ed., 2016, 55, 1133 CrossRef CAS PubMed; (f) Q. Yang, P. Y. Choy, B. Fan and F. Y. Kwong, Adv. Synth. Catal., 2015, 357, 2345 CrossRef CAS; (g) B.-M. Fan, Q.-J. Yang, J. Hu, C.-L. Fan, S.-F. Li, L. Yu, C. Huang, W. W. Tsang and F. Y. Kwong, Angew. Chem., Int. Ed., 2012, 51, 7821 CrossRef CAS PubMed; (h) T. Nishimura, T. Sawano and T. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 8057 CrossRef CAS PubMed; (i) T. Nishimura, X.-X. Guo, N. Uchiyama, T. Katoh and T. Hayashi, J. Am. Chem. Soc., 2008, 130, 1576 CrossRef CAS PubMed; (j) D. F. Cauble, J. D. Gipson and M. J. Krische, J. Am. Chem. Soc., 2003, 125, 1110 CrossRef CAS PubMed.
- Z. Yan, X.-A. Yuan, Y. Zhao, C. Zhu and J. Xie, Angew. Chem., Int. Ed., 2018, 57, 12906 CrossRef CAS PubMed.
- C. Clarke, C. A. Incerti-Pradillos and H. W. Lam, J. Am. Chem. Soc., 2016, 138, 8068 CrossRef CAS PubMed.
- (a) Y.-Q. Chen, Y.-H. Shen, Y.-Q. Su, L.-Y. Kong and W.-D. Zhang, Chem. Biodiversity, 2009, 6, 779 CrossRef CAS PubMed; (b) K. Zhao, G.-J. Cheng, H. Yang, H. Shang, X. Zhang, Y.-D. Wu and Y. Tang, Org. Lett., 2012, 14, 4878 CrossRef CAS PubMed; (c) J. M. Castro, S. Salido, J. Altarejos, M. Nogueras and A. Sánchez, Tetrahedron, 2002, 58, 5941 CrossRef CAS; (d) M. J. Uddin, S. Kokubo, K. Ueda, K. Suenaga and D. Uemura, J. Nat. Prod., 2001, 64, 1169 CrossRef CAS PubMed; (e) T. Hase, K. Ohtani, R. Kasai, K. Yamasaki and C. Picheansoonthon, Phytochemistry, 1996, 41, 317 CrossRef CAS.
- For a review on desymmetrization reactions: (a) X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330 CrossRef CAS PubMed; For organo-catalyzed desymmetrization of cyclohexadienone, see: (b) X. Su, W. Zhu, Y. Li and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 6874 CrossRef CAS PubMed; (c) S. Takizawa, T. M.-N. Nguyen, A. Grossmann, D. Enders and H. Sasai, Angew. Chem., Int. Ed., 2012, 51, 5423 CrossRef CAS PubMed; (d) Q. Gu and S.-L. You, Chem. Sci., 2011, 2, 1519 RSC; (e) Q. Gu, Z.-Q. Rong, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2010, 132, 4056 CrossRef CAS PubMed; (f) N. T. Vo, R. D. M. Pace, F. O'Hara and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 404 CrossRef CAS PubMed; (g) Q. Liu and T. Rovis, J. Am. Chem. Soc., 2006, 128, 2552 CrossRef CAS PubMed; (h) W. Yao, X. Dou, S. Wen, J. Wu, J. J. Vittal and Y. Lu, Nat. Commun., 2016, 7, 13024 CrossRef PubMed.
- For metal-catalyzed desymmetrization of cyclohexadienone, see: (a) T. Shu, L. Zhao, S. Li, X.-Y. Chen, C. von Essen, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 10985 CrossRef CAS PubMed; (b) X. Zhou, Y. Pan and X. Li, Angew. Chem., Int. Ed., 2017, 56, 8163 CrossRef CAS PubMed; (c) J. Chen, X. Han and X. Lu, Angew. Chem., Int. Ed., 2017, 56, 14698 CrossRef CAS PubMed; (d) R. Kumar, Y. Hoshimoto, E. Tamai, M. Ohashi and S. Ogoshi, Nat. Commun., 2017, 8, 32 CrossRef PubMed; (e) Z.-T. He, X.-Q. Tang, L.-B. Xie, M. Cheng, P. Tian and G.-Q. Lin, Angew. Chem., Int. Ed., 2015, 54, 14815 CrossRef CAS PubMed; (f) P. Liu, Y. Fukui, P. Tian, Z.-T. He, C.-Y. Sun, N.-Y. Wu and G.-Q. Lin, J. Am. Chem. Soc., 2013, 135, 11700 CrossRef CAS PubMed; (g) Z.-T. He, B. Tian, Y. Fukui, X. Tong, P. Tian and G.-Q. Lin, Angew. Chem., Int. Ed., 2013, 52, 5314 CrossRef CAS PubMed; (h) J. Keilitz, S. G. Newman and M. Lautens, Org. Lett., 2013, 15, 1148 CrossRef CAS PubMed; (i) T. L. N. Nguyen, C. A. Incerti-Pradillos, W. Lewis and H. W. Lam, Chem. Commun., 2018, 54, 5622 RSC; (j) K. K. Gollapelli, S. Donikela, N. Manjula and R. Chegondi, ACS Catal., 2018, 8, 1440 CrossRef CAS.
- A. J. Kresge and P. Pruszynski, J. Org. Chem., 1991, 56, 4808 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1845186 for 3m and 1847564 for 3p. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02341k |
| This journal is © The Royal Society of Chemistry 2019 |