DOI:
10.1039/C9SC01686D
(Edge Article)
Chem. Sci., 2019,
10, 8129-8134
A sterically hindered asymmetric D–A–D′ thermally activated delayed fluorescence emitter for highly efficient non-doped organic light-emitting diodes†
Received
8th April 2019
, Accepted 3rd July 2019
First published on 3rd July 2019
Abstract
Thermally activated delayed fluorescence (TADF) materials have opened a new chapter for high-efficiency and low-cost organic light-emitting diodes (OLEDs). Herein, we describe a novel and effective design strategy for TADF emitters which includes introducing a carbazole donor unit at the ortho-position, at which the donor and acceptor groups are spatially in close proximity to guarantee the existence of intramolecular electrostatic attraction and through-space charge transfer, leading to reduced structural vibrations, suppressed non-radiative decay and rapid radiative decay to avoid excited state energy loss. As a result, a green TADF emitter (2Cz-DPS) showing high solid-state photoluminescence quantum efficiency (91.9%) and excellent OLED performance was produced. Theoretical simulations reveal that the non-adiabatic coupling accelerates the reverse intersystem crossing of 2Cz-DPS, resulting in a state-of-the-art non-doped OLED with an extremely high external quantum efficiency of 28.7%.
Introduction
Organic-based emissive materials that exhibit thermally activated delayed fluorescence (TADF) properties can harvest both singlet and triplet excitons for light emission via reversible intersystem crossing (RISC) from the lowest triplet (T1) to singlet (S1) excited states, leading to a maximum internal quantum efficiency (IQE) of electroluminescence of 100%. Accordingly, fully utilizing the TADF mechanism in “third-generation” organic light-emitting diodes (OLEDs) has attracted considerable attention.1 TADF-based emitters are composed of donor (D) and acceptor (A) groups in various structural configurations which help to achieve a small overlap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), reducing the spin exchange energy (J) which ultimately leads to a small energy gap between the S1 and T1 (ΔEST), which facilitates the RISC process. However, linear D–A linkages tend to decrease the radiative transition oscillator strength (f) and induce intramolecular D–A rotation and other vibrational modes of the excited-state molecules that dissipate energy. These internal motions can be reduced by introducing large steric hindrance between the D and A units, which can not only restrict non-radiative pathways,2 but also enable rapid radiative decay from the excited-state molecules,3 leading to high-efficiency TADF emitters. In general, high-performance TADF-OLEDs are fabricated by dispersing the TADF emitters into host materials to alleviate exciton annihilation,4 leading to issues such as complicated fabrication processes and possible host–guest phase separation. Therefore, the non-doping technique, which enables ease of fabrication along with more reliable and reproducible output manufacturing of the fabricated devices, is more attractive. Accordingly, aggregation-induced emission (AIE) materials are ideal for utilization as non-doped emissive layers (EMLs) due to their unusually strong fluorescence in the “aggregate-state” (film state).5 In this regard, in 2014 we proposed a structural design strategy that involved the merging of the two concepts, AIE and TADF, together in one molecule.
Inserting an electron accepting sulfonyldibenzene core in between carbazole and phenothiazine electron donating units afforded an asymmetric TADF material with outstanding near-quantitative solid-state photoluminescence quantum yield (PLQY) (97.3%).6 Thereafter, this strategy has been validated many times towards the exploration of efficient TADF emitters and fabrication of non-doped OLEDs7 and can achieve very good OLED performance, such as an external quantum efficiency (EQE) ∼20%.7,8 Despite these advances, there are still no reports regarding TADF emitters that provide non-doped OLEDs with EQEs close to 30%, which is the upper limit of common bottom-emitting OLEDs.
In this work, we present a novel and effective structural design strategy for TADF emitters with strong solid-state emission properties, wherein D and A groups are linked at the ortho-position to afford a spatially close D–A interaction, leading to reduced vibrations and suppressed non-radiative pathways. In addition, 2Cz-DPS possesses dual-charge transfer pathways (through-bond charge transfer and through-space charge transfer), resulting in rapid prompt and delayed decay. Significantly, the designed asymmetric TADF emitter 2Cz-DPS with a highly twisted conformation exhibits a high PLQY as a neat film (AIE property) and excellent solid-state thermal stability. Most importantly, 2Cz-DPS affords a record-high EQE of 28.7% amongst other reported non-doped OLEDs.
Results and discussion
Molecular design strategy and theoretical simulations
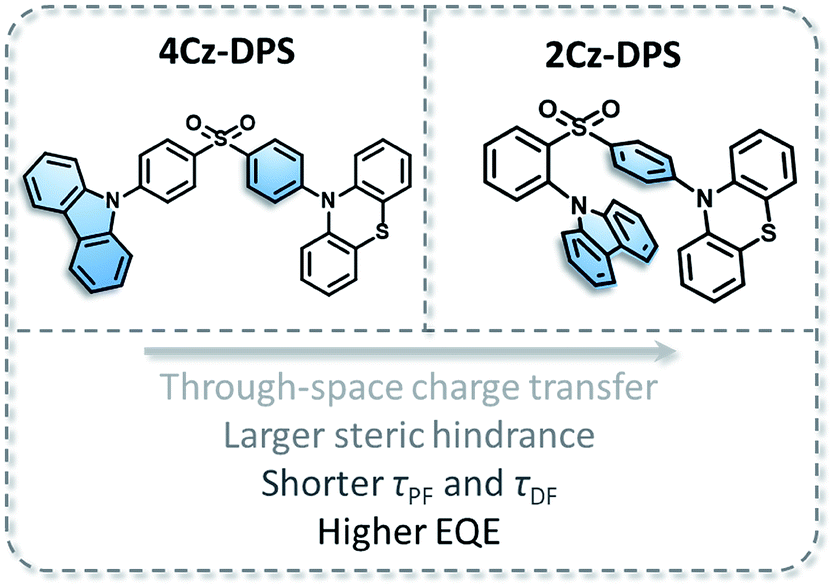
2Cz-DPS is derived from our previously reported compound 4Cz-DPS, in which the carbazole group has been relocated to the ortho-position as opposed to the original para-position. The molecular structures of 4Cz-DPS and 2Cz-DPS based on the carbazole and phenothiazine donor groups and the diphenyl-sulfone acceptor group are illustrated in Fig. 1, in which 4Cz-DPS was synthesized according to our previous report6 and 2Cz-DPS was synthesized in three steps (Scheme S1 in the ESI†) using cheap and common reagents. Structural characterization was conducted by 1H NMR, 13C NMR, EI-MS and HRMS (Fig. S1–S9, ESI†) and single crystal analysis. Similar to AIE-active 4Cz-DPS, 2Cz-DPS with a highly twisted conformation also possesses AIE properties (more details are shown in Fig. S10, ESI†). The THF/water solutions of 2Cz-DPS with low water fractions show very weak emission, while highly intense green emission is observed when a large amount of water (fw = 90%) is added (Fig. S10a†), and nanoaggregates with effective diameters of 186 nm are evidenced by dynamic light scattering study (Fig. S10b†). Moreover, 2Cz-DPS has a PLQY of only 0.9% even in dilute non-polar hexane, which excludes the polarity quenching of the charge transfer (CT) emitters in the solution state,9 while the PLQY is greatly enhanced to 91.9% in the solid state. These results indeed demonstrate that 2Cz-DPS is AIE active.
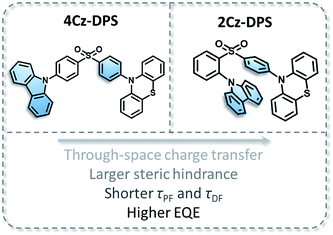 |
| | Fig. 1 Molecular structures of 4Cz-DPS and 2Cz-DPS. | |
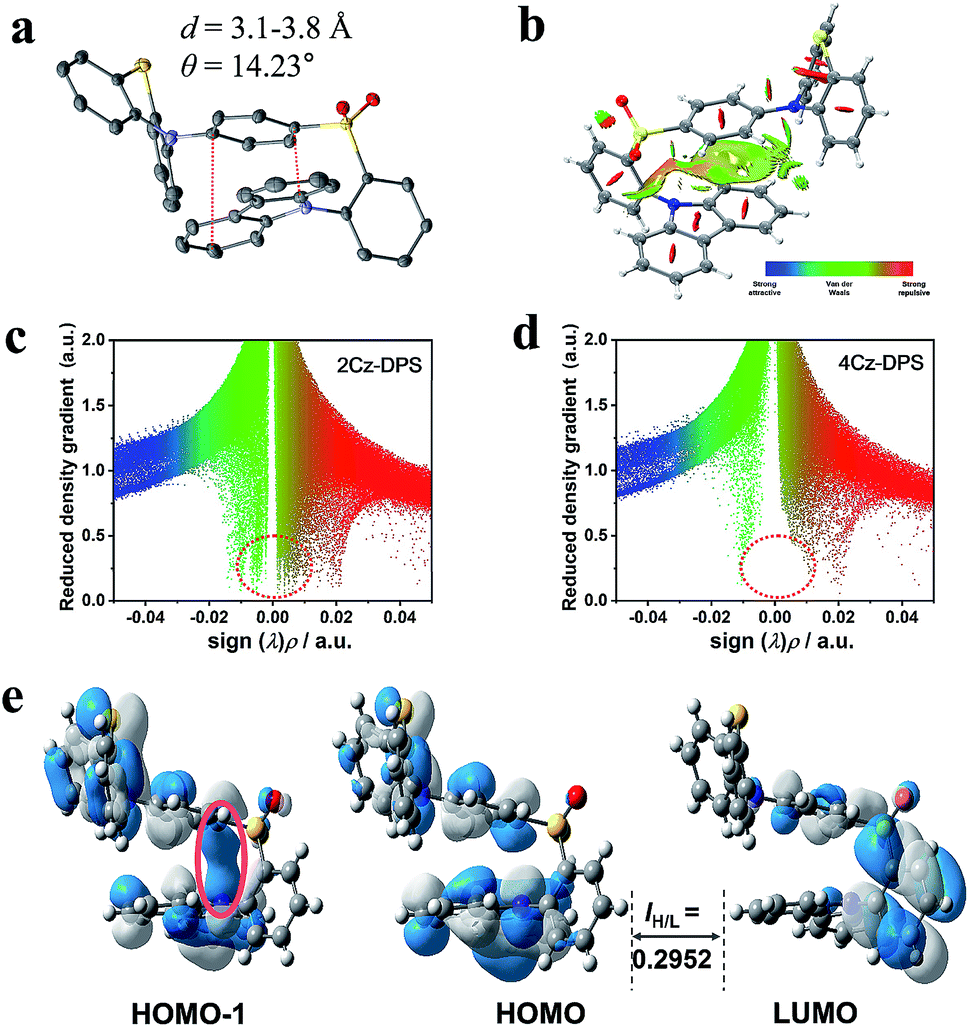
As demonstrated from the conformation in the single crystal of 2Cz-DPS (Fig. 2a), the carbazole donor group is nearly parallel to the phenyl ring attached to the diphenyl-sulfone acceptor group with a co-facial alignment at a distance of 3.1–3.8 Å and a tilt angle (θ) of 14.23°, suggesting strong intramolecular interactions between D and A. On the other hand, the single crystal structure of 4Cz-DPS (Fig. S11a, ESI†) demonstrates orthotropic attachment between the carbazole group and the diphenyl-sulfone group. To reveal the intramolecular non-covalent interactions in 2Cz-DPS, the functions of reduced density gradient (RDG) and Sign (λ2)ρ were calculated using Multiwfn software.10 RDG analysis (Fig. 2b and c) confirms the presence of obvious intramolecular attractive interactions (green region) and larger steric hindrance (brown region) between D and A segments in 2Cz-DPS, which could effectively restrict the intramolecular vibrations and prevent energy loss of the excited molecules. In contrast, the linearly linked conformation of 4Cz-DPS exhibits weak intramolecular interactions (Fig. 2d), which is a common phenomenon found in general TADF molecules. According to time-dependent density functional theory (TD-DFT) calculations based on the conformations extracted from single crystal analysis, the HOMOs are mainly distributed on the phenyl-phenothiazine or carbazole units (D), while the LUMO is mostly located on the diphenyl-sulfone core (A) of the 4Cz-DPS and 2Cz-DPS compounds (Fig. 2e and S11b†). Interestingly, as observed from the HOMO-1 of 2Cz-DPS, there is an obvious orbital overlap between the parallel carbazole and phenyl planes, confirming the through-space charge transfer characteristics between D and A.11 Due to the close proximity between D and A in 2Cz-DPS, intramolecular charge transfer occurs not only through the molecular conjugated backbone, but also by through-space conjugation mediated by intramolecular D–A interactions.11b,12 The dual charge transfer pathways greatly enhance the oscillator strength (f = 0.0207) for faster radiative decay, in comparison to the isomer 4Cz-DPS (f = 0.0074). As a result, the strong intramolecular interactions generated in 2Cz-DPS help to increase the molecular rigidity, decrease the relaxation of the excited states, and enhance the radiative transition rate,3 which can also be evidenced from shortened transient and delayed lifetimes (Fig. S12, ESI†), thus facilitating the occurrence of efficient TADF. The charge transfer (CT) character can also be verified from the large Stokes shift with increasing solvent polarity (Fig. S13 and S14, ESI†).13 When the solvent polarity is increased from n-hexane to DMF, the PL spectral peaks are red-shifted from 475 nm to 565 nm for 4Cz-DPS and from 471 nm to 567 nm for 2Cz-DPS (Fig. S13†), while their absorption spectra are relatively insensitive to the solvent polarity (Fig. S14†). In contrast to the solutions showing dual emission, the 4Cz-DPS and 2Cz-DPS films give structure-less PL spectra (Fig. S15, ESI†), due to the highly strengthened and dominant intermolecular CT process in the solid-sate films.14
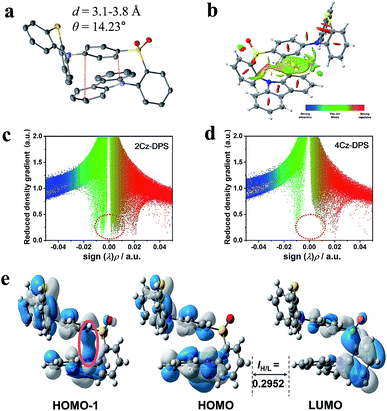 |
| | Fig. 2 (a) Single crystal and (b) reduced density gradient (RDG) isosurface map with an isovalue of 0.5 for 2Cz-DPS. The functions of RDG and Sign (λ2)ρ for (c) 2Cz-DPS and (d) 4Cz-DPS. (e) HOMO and LUMO distribution of 2Cz-DPS, showing the overlap integral extents IH/L. | |
TADF property characterization
Temperature dependent steady-state spectra of the 2Cz-DPS film from 17 to 298 K (Fig. S16a, ESI†) show that the emission intensity gradually increases as the temperature rises. Furthermore, transient PL spectra of the 2Cz-DPS film were measured (Fig. S16b, ESI†) and display two-component decays, including the prompt and the delayed components. With increasing temperature from 17 to 298 K, the delayed component progressively increases, leading to prompt and delayed lifetimes of 4.4 ns and 19.1 μs for 2Cz-DPS at 298 K, respectively, and 6.3 ns and 62.2 μs for 4Cz-DPS, respectively (Fig. S17, ESI†). The photo-physical properties of the TADF materials are summarized in Table 1. Therefore, when the substitution position changes from para- to ortho-, 2Cz-DPS shows both faster prompt decay and delayed decay at room temperature, which is quite essential for high-performance TADF-based OLEDs.15 Therefore, the steady-state spectra and transient PL spectra results confirm the TADF nature of 2Cz-DPS.
Table 1 Summary of photo-physical and thermal properties of 2Cz-DPS and 4Cz-DPS neat films
| TADF |
λ
A
(nm) |
λ
PL
(nm) |
T
d
(°C) |
T
g
(°C) |
ϕ
PF
(%) |
ϕ
DF
(%) |
τ
PF
(ns) |
τ
DF
(μs) |
k
ISC
(s−1) |
k
RISC
(s−1) |
ΔESTk (eV) |
ΔESTl (eV) |
PLQYm (%) |
E
g
(eV) |
|
Absorption peak.
PL emission peak.
Temperature of thermal decomposition at 5% weight loss.
Glass-transition temperature.
Quantum efficiency of prompt emission.
Quantum efficiency of delayed emission.
Prompt fluorescence lifetime component.
Delayed fluorescence lifetime component.
Rate constant of ISC for triplet excited states.
Rate constant of RISC for triplet excited states.
S1 and T1 energy difference determined from experiments.
S1 and T1 energy difference determined from theoretical simulations.
PL quantum yield measured under vacuum.
Energy gap calculated from the onset of absorption spectra.
|
|
4Cz-DPS
|
326 |
530 |
394 |
121 |
17.5 |
79.8 |
6.3 |
62.2 |
2.28 × 107 |
0.89 × 105 |
0.25 |
0.35 |
97.3 |
2.2 |
|
2Cz-DPS
|
341 |
520 |
389 |
111 |
26.7 |
65.2 |
4.4 |
19.1 |
4.31 × 107 |
1.74 × 105 |
0.32 |
0.41 |
91.9 |
2.5 |
The TADF feature is further demonstrated by oxygen-sensitive PL spectra (Fig. S18, ESI†). In contrast to those under air conditions, the lifetimes of the delayed components in a vacuum are increased, confirming that the T1 excitons of the TADF compounds can be deactivated by oxygen.16 It is noteworthy that the PLQYs of 4Cz-DPS and 2Cz-DPS films in air were measured to be 74.7% and 65.3%, respectively, while their PLQYs in a vacuum reach up to 97.3% and 91.9%, respectively, resulting from the reduction of oxygen quenching of the delayed component. The ΔEST of the 4Cz-DPS and 2Cz-DPS films was calculated from their fluorescence and phosphorescence spectra (Fig. S19, ESI†),17 and estimated to be 0.25 and 0.32 eV, respectively, which are relatively large values amongst other recently reported TADF emitters.1 As seen from the crystal of 2Cz-DPS and 4Cz-DPS, the axial conformation of phenothiazine accounts for the large ΔEST of the compounds.18 Compared to 4Cz-DPS, 2Cz-DPS with through-space charge transfer features possessed a slightly enlarged ΔEST which can be ascribed to the larger overlap integral of the HOMO and LUMO (IH/L = 0.2952, Fig. 1e).8a This is in contrast with previously reported through-space charge transfer TADF emitters in which through-space D–A interaction minimizes the electron-exchange energy and induces a narrow ΔEST.19 However, the larger overlap integral of frontier orbitals increases the transition dipole moment and thus results in a faster prompt decay.
Non-adiabatic coupling effect
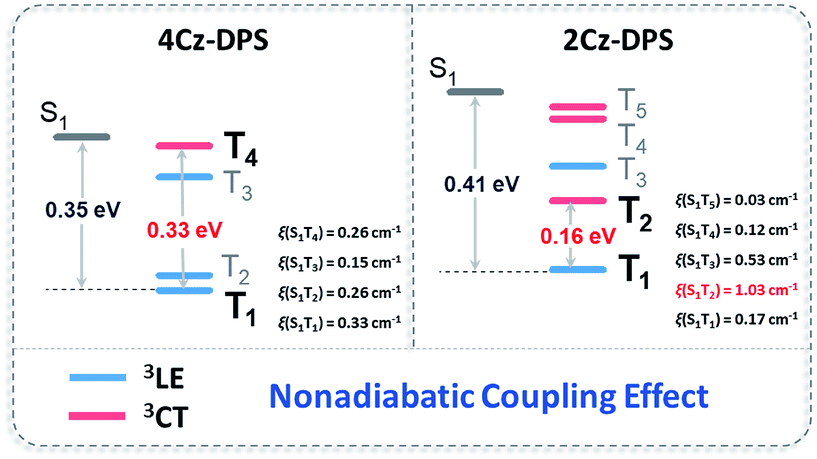
In order to evaluate the origin of the faster delayed decay in 2Cz-DPS, natural transition orbital (NTO) simulations on both 2Cz-DPS and 4-CzDPS were performed to provide insight into the transition features of the excited states. A clear CT character of the lowest S1 is expected and a strong locally excited (LE) nature of the lowest T1 is observed (Fig. S20 and S21, ESI†), which can also be demonstrated by the vibronic phosphorescence emission at 77 K. In general, the RISC process is driven by first-order spin–orbit coupling (SOC) in this type of TADF emitter (3LE as the lowest triplet state).20 In our asymmetric D–A–D′ systems, especially 2Cz-DPS, the ΔEST and SOC matrix element value of S1 and T1 are 0.41 eV and 0.17 cm−1 (Fig. 3), respectively, too large for the occurrence of efficient RISC according to the Fermi golden rule.21 However, 2Cz-DPS with a larger ΔEST and smaller SOC constant ξ(S1, T1) exhibits a much shorter delayed lifetime (τDF, 19.1 μs) than para-substituted 4Cz-DPS (62.2 μs), indicating a faster RISC. This abnormal phenomenon implies that another factor causes the acceleration process. Further analysis of the NTO pairs reveals that several triplet states exist below the S1 energy level. In 2Cz-DPS, these upper triplet states exhibit different electronic configurations. T1 and T3 show significant LE characteristics, while T2, T4 and T5 show weak CT characteristics. These dark excited triplet states have been regarded to play an important role and can result in a second order coupling (non-adiabatic coupling) of the excited states which leads to a complete understanding of the rapid RISC process, especially in TADF emitters with a large ΔEST.22 The gap between T2 (CT) and T1 (LE) is 0.16 eV in 2Cz-DPS, and it is 0.33 eV in 4Cz-DPS between T4 (CT) and T1 (LE). As reported before, the reversible energy transfer between the lowest 3LE and upper 3CT is considered as a fast intramolecular-electron exchange within a 0.2 eV gap.23 Therefore, the narrower gap between the 3CT and 3LE contributes to a faster T1–T2 equilibrium (or reverse internal conversion) by non-adiabatic coupling, resulting in a strong mixing of triplet states,24 which then gives rise to a higher RISC rate (kRISC) in 2Cz-DPS of 1.74 × 105 s−1, which is more than twice higher than that in 4Cz-DPS (0.89 × 105 s−1). Additionally, the SOC constant (ξ) of 2Cz-DPS between S1 and T2 calculated by TD-DFT is 1.03 cm−1, suggesting an effective RISC, which is consistent with the shorter τDF. Therefore, not only does ΔEST control the RISC process, but the non-adiabatic coupling between the triplet CT and LE also plays a key role in rapid RISC in 2Cz-DPS, leading to the shortened τDF.25 Compounds with shortened τDF have vast potential for highly efficient OLEDs.26
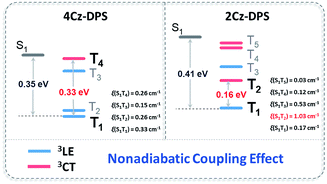 |
| | Fig. 3 Energy level diagrams, spin–orbital coupling constants (ξ) and non-adiabatic coupling effects of 4Cz-DPS and 2Cz-DPS. | |
Electrochemical and thermal properties
The electrochemical behavior of the compounds was examined by cyclic voltammetry (Fig. S22a†). The HOMO levels of 4Cz-DPS and 2Cz-DPS were determined to be 5.2 and 5.3 eV, respectively, and the bandgaps (Eg) of 4Cz-DPS and 2Cz-DPS were calculated to be 2.7 eV, from the onset of their absorption spectra in DCM solution with a concentration of 5 × 10−4 mol L−1 (Fig. S22b†). Then, the LUMO levels of 4Cz-DPS and 2Cz-DPS were estimated to be 2.6 and 2.5 eV, respectively. As demonstrated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (Fig. S23, ESI†), 4Cz-DPS and 2Cz-DPS exhibit high thermal stability with decomposition temperatures (Td: corresponding to 5% weight loss) of 394 and 389 °C and glass transition temperatures (Tg) of 121 and 111 °C, respectively, which are beneficial for maintaining stable film morphology during device operation.
Non-doped OLED characterization
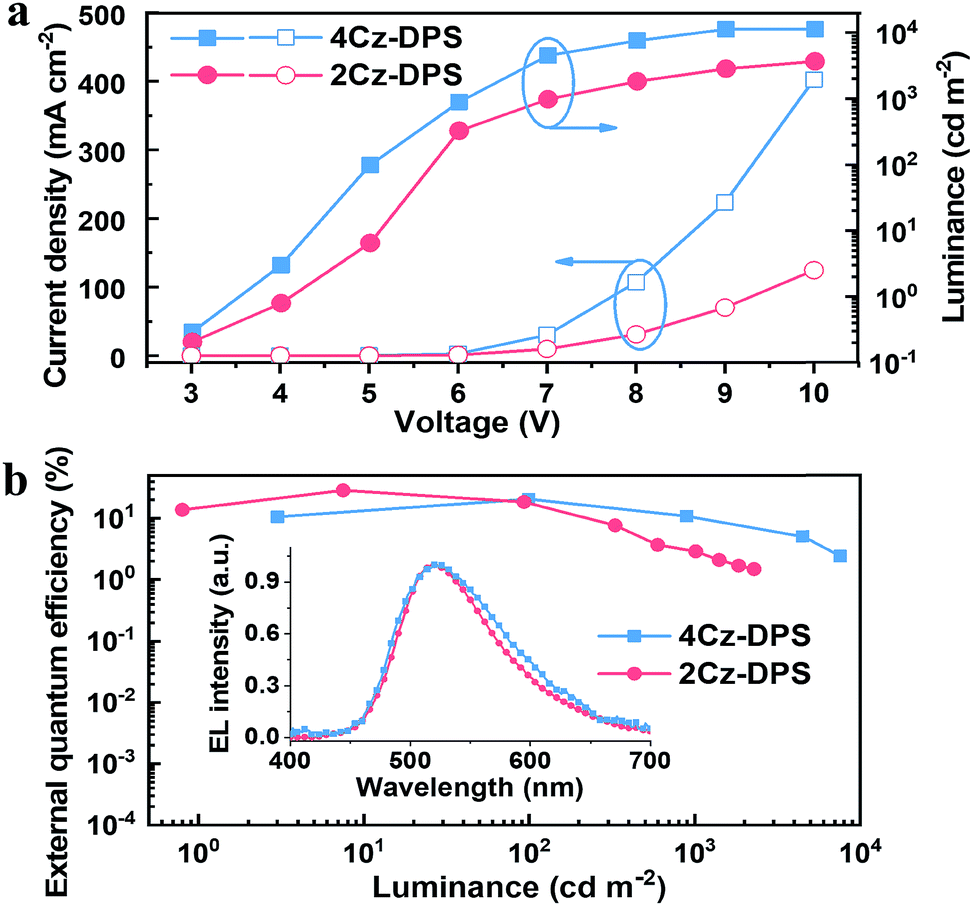
Electroluminescence (EL) properties of 4Cz-DPS and 2Cz-DPS were studied using neat EML films, and non-doped OLEDs were constructed with the device configuration ITO/PEDOT:PSS (30 nm)/m-bis(N-carbazolyl)benzene (MCP) (20 nm)/EML (30 nm)/1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBI) (40 nm)/LiF(1 nm)/Al (100 nm), wherein EML = 4Cz-DPS or 2Cz-DPS. The EL performance is shown in Fig. 4 and summarized in Table 2. With regard to the non-doped 4Cz-DPS device, the maximum EQE, current efficiency (CE) and power efficiency (PE) are obtained to be 20.7%, 61.2 cd A−1 and 38.4 lm W−1, respectively, at a luminance of 100 cd m−2, which to the best of our knowledge are amongst the best performances of non-doped TADF-OLEDs.27 Surprisingly, the non-doped 2Cz-DPS device achieves a maximum EQE, CE and PE of 28.7%, 82.3 cd A−1 and 51.7 lm W−1, respectively, at a luminance of 10 cd m−2, without the use of any light out-coupling methods. The EL spectra (inset of Fig. 4b) with peaks at 524 nm and 516 nm correspond to the emission of 4Cz-DPS and 2Cz-DPS, respectively. Furthermore, five more 2Cz-DPS devices were implemented and showed maximum EQEs ranging from 24.8% to 30.4% (Fig. S24, ESI†), thus confirming good efficiency reproducibility of the 2Cz-DPS devices. The high EQE value of the 2Cz-DPS containing device confirms the state-of-the-art performance of non-doped TADF-OLEDs,7,8 and is even comparable to that of the best performing doped OLEDs based on TADF or phosphorescence-based emitters.28 As shown by the results of single carrier devices (Fig. S25, ESI†), 2Cz-DPS shows a reduced electron mobility compared to 4Cz-DPS, due to the presence of a bulky substituent in 2Cz-DPS preventing charge transport and suppressing intermolecular interactions by larger steric hindrance.29 Importantly, more balanced hole- and electron-mobilities are observed for 2Cz-DPS than for 4Cz-DPS, which benefits the higher charge-carrier balance in the 2Cz-DPS device, and hence higher efficiencies. Despite this, the 2Cz-DPS device shows a relatively high efficiency roll-off compared to the 4Cz-DPS device, as high charge-carrier balance features alone cannot guarantee a low efficiency roll-off. It can also be observed from single carrier devices that the electron mobility of 4Cz-DPS is much higher than its hole mobility; thus the recombination zone (RZ) in the 4Cz-DPS device is confined in the EML but toward the HTL/EML interface. On the other hand, the hole mobility of 2Cz-DPS is slightly higher than its electron mobility, suggesting movement of the RZ toward the EML/ETL interface in the 2Cz-DPS device, which can induce efficiency roll-off.30 In addition, the reduced self-quenching of the 4Cz-DPS emitter, as evidenced by the higher PLQY of the 4Cz-DPS film than of the 2Cz-DPS film, could also be responsible for the lower efficiency roll-off in the 4Cz-DPS device.4a We further investigate the efficiency roll-off behaviours by taking both triplet–triplet annihilation (TTA) and singlet-polaron annihilation (SPA) into account, which are assumed to be dominant in TADF-based OLEDs.31 As seen in Fig. S26,† the main origin for efficiency roll-off in the 2Cz-DPS device is TTA (especially at high current density), while it is SPA in the 4Cz-DPS device, which is in good agreement with the results from 4Cz-DPS based single carrier devices showing abundant and imbalanced charge carriers (Fig. S25b†).
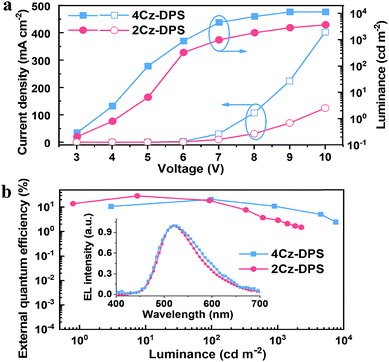 |
| | Fig. 4 (a) Current density–voltage–luminance and (b) external quantum efficiency–luminance curves of 4Cz-DPS and 2Cz-DPS devices. Inset: normalized EL spectra. | |
Table 2 EL performance of the non-doped OLEDs based on 4Cz-DPS and 2Cz-DPS
| Device |
V
on
[V] |
Luminanceb [cd m−2] |
EQE/CE/PEc [%/cd A−1/lm W−1] |
EQE/CE/PEd [%/cd A−1/lm W−1] |
λ
EL
[nm] |
FWHMf [nm] |
|
Turn-on voltage at a luminance of 1 cd m−2.
Maximum luminance.
Maximum efficiency.
Efficiency achieved at 300 cd m−2.
EL peak wavelength.
Full-width-at-half-maximum.
|
|
4Cz-DPS
|
3.5 |
11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 310 310 |
20.7/61.2/38.4 |
14.6/53.1/32.5 |
524 |
108 |
|
2Cz-DPS
|
4.1 |
4220 |
28.7/82.3/51.8 |
8.4/28.8/15.6 |
518 |
96 |
Conclusions
In summary, an ortho-substituent design strategy promoting steric hindrance has afforded excellent AIE-TADF emitters, and high-efficiency TADF-OLEDs have been subsequently demonstrated. The intramolecular interactions between the spatially close donor and acceptor moieties suppress structural vibrations, prevent the excited state energy loss and generate space conjugation. The dual intramolecular charge transfer pathways including through-bond charge transfer and through-space charge transfer enhance the radiative transition oscillator strength and accelerate the prompt decay. Although the ΔEST of 2Cz-DPS is increased by ortho-substitution, a faster delayed decay is obtained with the assistance of non-adiabatic coupling. The designed compound 2Cz-DPS exhibits high solid-state PLQY and good thermal stability in the thin film state. Most significantly, 2Cz-DPS endows the TADF-OLEDs with outstanding performance such as a record-high EQE of 28.7% by adopting a non-doped EML system. These results confirm that the presented design strategy opens up new possibilities to develop highly efficient TADF emitters and devices.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC: 51733010, 61605253, 21672267 and 51603232), Science and Technology Planning Project of Guangdong (2015B090913003 and 2015B090915003), Guangdong Natural Science Funds for Distinguished Young Scholars (2017B030306012), China Postdoctoral Science Foundation (2017M620395), Fundamental Research Funds for the Central Universities, and Shenzhen China Star Optoelectronics Semiconductor Display Technology Co., Ltd. (CSOT). We thank Chung-Chih Wu (National Taiwan University) for performing UV-vis absorption and phosphorescence spectra measurements for the TADF materials.
References
- Z. Y. Yang, Z. Mao, Z. L. Xie, Y. Zhang, S. W. Liu, J. Zhao, J. R. Xu, Z. G. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- D. Zhang, M. Cai, Y. Zhang, D. Zhang and L. Duan, Mater. Horiz., 2016, 3, 145–151 RSC.
-
(a) J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612–2615 RSC;
(b) H.-J. Park, S. H. Han and J. Y. Lee, J. Mater. Chem. C, 2017, 5, 12143–12150 RSC.
-
(a) T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Nat. Photonics, 2018, 12, 235–240 CrossRef;
(b) T.-A. Lin, T. Chatterjee, W.-L. Tsai, W.-K. Lee, M.-J. Wu, M. Jiao, K.-C. Pan, C.-L. Yi, C.-L. Chung, K.-T. Wong and C.-C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- S. D. Xu, T. T. Liu, Y. X. Mu, Y. F. Wang, Z. G. Chi, C. C. Lo, S. W. Liu, Y. Zhang, A. Lien and J. R. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS.
-
(a) J. Huang, H. Nie, J. J. Zeng, Z. Y. Zhuang, S. F. Gan, Y. J. Cai, J. J. Guo, S.-J. Su, Z. J. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2017, 56, 12971–12976 CrossRef CAS;
(b) J. J. Guo, X.-L. Li, H. Nie, W. Luo, R. Hu, A. Qin, Z. Zhao, S.-J. Su and B. Z. Tang, Chem. Mater., 2017, 29, 3623–3631 CrossRef CAS.
-
(a) R. Furue, T. Nishimoto, I. S. Park, J. Lee and Y. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171–7175 CrossRef CAS PubMed;
(b) L. Yu, Z. B. Wu, G. H. Xie, W. X. Zeng, D. G. Ma and C. L. Yang, Chem. Sci., 2018, 9, 1385–1391 RSC.
- M. Aydemir, S. Xu, C. Chen, M. R. Bryce, Z. Chi and A. P. Monkman, J. Phys. Chem. C, 2017, 121, 17764–17772 CrossRef CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
-
(a) B. He, W. Luo, S. Hu, B. Chen, S. Zhen, H. Nie, Z. J. Zhao and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 12553–12560 RSC;
(b) T. Han, H. Deng, Z. Qiu, Z. Zhao, H. Zhang, H. Zou, N. L. C. Leung, G. Shan, M. R. J. Elsegood, J. W. Y. Lam and B. Z. Tang, J. Am.
Chem. Soc., 2018, 140, 5588–5598 CrossRef CAS PubMed;
(c) L. Chen, Y. -H. Wang, B. He, H. Nie, R. Hu, F. Huang, A. J. Qin, X. -S. Zhou, Z. J. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2015, 54, 4231–4235 CrossRef CAS;
(d) H. Tsujimoto, D.-G. Ha, G. Markopoulos, H. S. Chae, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2017, 139, 4894–4900 CrossRef CAS.
- X.-L. Chen, J.-H. Jia, R. Yu, J.-Z. Liao, M.-X. Yang and C.-Z. Lu, Angew. Chem., Int. Ed., 2017, 56, 15006–15009 CrossRef CAS.
- A. Wakamiya, K. Mori and S. Yamaguchi, Angew. Chem., Int. Ed., 2007, 46, 4273–4276 CrossRef CAS.
-
(a) Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. R. Xu and Z. G. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef CAS;
(b) Z. L. Xie, Q. Y. Huang, T. Yu, L. Y. Wang, Z. Mao, W. L. Li, Z. Yang, Y. Zhang, S. W. Liu, J. R. Xu, Z. G. Chi and M. P. Aldred, Adv. Funct. Mater., 2017, 27, 1703918 CrossRef.
- Y. H. Lee, S. Park, J. Oh, J. W. Shin, J. Jung, S. Yoo and M. H. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 24035–24042 CrossRef CAS.
- H. Wang, L. Xie, Q. Peng, L. Meng, Y. Wang, Y. Yi and P. Wang, Adv. Mater., 2014, 26, 5198–5204 CrossRef CAS PubMed.
- P. Rajamalli, N. Senthilkumar, P. Gandeepan, P.-Y. Huang, M.-J. Huang, C.-Z. R. Wu, C.-Y. Yang, M.-J. Chiu, L.-K. Chu, H.-W. Lin and C.-H. Cheng, J. Am. Chem. Soc., 2016, 138, 628–634 CrossRef CAS.
-
(a) J. S. Ward, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, J. Santos, F. B. Dias and M. R. Bryce, J. Org. Chem., 2018, 83, 14431–14442 CrossRef CAS;
(b) J. S. Ward, R. S. Nobuyasu, M. A. Fox, J. A. Aguilar, D. Hall, A. S. Batsanov, Z. Ren, F. B. Dias and M. R. Bryce, J. Org. Chem., 2019, 84, 3801–3816 CrossRef CAS.
- G. A. Sommer, L. N. Mataranga-Popa, R. Czerwieniec, T. Hofbeck, H. H. Homeier, T. J. Müller and H. Yersin, J. Phys. Chem. Lett., 2018, 9, 3692–3697 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS.
- D.-H. Kim, A. D'Aléo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Brédas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS.
- X.-K. Chen, S.-F. Zhang, J.-X. Fan and A.-M. Ren, J. Phys. Chem. C, 2015, 119, 9728–9733 CrossRef CAS.
-
(a) Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed;
(b) Q. Zhang, T. Komino, S. Huang and C. Adachi, Adv. Funct. Mater., 2012, 22, 2327–2336 CrossRef CAS.
-
(a) J. Gibson, A. Monkman and T. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS;
(b) J. Gibson and T. Penfold, Phys. Chem. Chem. Phys., 2017, 19, 8428–8434 RSC.
- H. Noda, H. Nakanotani and C. Adachi, Sci. Adv., 2018, 4, 6910 CrossRef PubMed.
- H. F. Higginbotham, C.-L. Yi, A. P. Monkman and K.-T. Wong, J. Phys. Chem. C, 2018, 122, 7627–7634 CrossRef CAS.
-
(a) W.-L. Tsai, M.-H. Huang, W.-K. Lee, Y.-J. Hsu, K.-C. Pan, Y.-H. Huang, H.-C. Ting, M. Sarma, Y.-Y. Ho, H.-C. Hu, C.-C. Chen, M.-T. Lee, K.-T. Wong and C.-C. Wu, Chem. Commun., 2015, 51, 13662–13665 RSC;
(b) T. Peng, G. Li, K. Ye, C. Wang, S. Zhao, Y. Liu, Z. Hou and Y. Wang, J. Mater. Chem. C, 2013, 1, 2920–2926 RSC;
(c) S. Wu, S. Li, Q. Sun, C. Huang and M.-K. Fung, Sci. Rep., 2016, 6, 25821 CrossRef CAS.
-
(a) J. W. Sun, J. H. Lee, C. K. Moon, K. H. Kim, H. Shin and J. J. Kim, Adv. Mater., 2014, 26, 5684–5688 CrossRef CAS;
(b) K. H. Kim, E. S. Ahn, J. S. Huh, Y. H. Kim and J. J. Kim, Chem. Mater., 2016, 28, 7505–7510 CrossRef CAS;
(c) S.-Y. Kim, W.-I. Jeong, C. Mayr, Y.-S. Park, K.-H. Kim, J.-H. Lee, C.-K. Moon, W. Brütting and J.-J. Kim, Adv. Funct. Mater., 2013, 23, 3896–3900 CrossRef CAS.
- Q. Q. Liang, C. M. Han, C. B. Duan and H. Xu, Adv. Opt. Mater., 2018, 6, 1800020 CrossRef.
- W. H. Lee, D. H. Kim, P. J. Jesuraj, H. Hafeez, J. C. Lee, D. K. Choi, T.-S. Bae, S. M. Yu, M. Song, C. S. Kim and S. Y. Ryu, Mater. Res. Express, 2018, 5, 076201 CrossRef.
-
(a) S. Ying, D. Yang, X. Qiao, Y. Dai, Q. Sun, J. Chen, T. Ahamad, S. M. Alshehri and D. Ma, J. Mater. Chem. C, 2018, 6, 10793–10803 RSC;
(b) C. Li, L. Duan, D. Zhang and Y. Qiu, ACS Appl. Mater. Interfaces, 2015, 7, 15154–15159 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1891672. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01686d |
| ‡ Dr Z. Yang and Dr Z. Mao contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Chao
Xu
b,
Xiaojie
Chen
a,
Juan
Zhao
a,
Chao
Xu
b,
Xiaojie
Chen
a,
Juan
Zhao