DOI:
10.1039/C9SC01631G
(Edge Article)
Chem. Sci., 2019,
10, 6007-6012
Stable radical versus reversible σ-bond formation of (porphyrinyl)dicyanomethyl radicals†
Received
3rd April 2019
, Accepted 13th May 2019
First published on 13th May 2019
Abstract
(Porphyrinyl)dicyanomethyl radicals were produced by oxidation of dicyanomethyl-substituted porphyrins with PbO2. These radicals constitute a rare example displaying stable radical versus dynamic covalent chemistry (DCC) depending upon the substitution position of the dicyanomethyl radical. meso-Dicyanomethyl-substituted radicals exist as stable monomeric species and do not undergo any dimerization processes either in the solid state or in solution. In contrast, β-dicyanomethyl-substituted radicals are isolated as σ-dimers that are stable in the solid-state but display reversible σ-dimerization behavior in solution; monomeric radical species exist predominantly at high temperatures, while σ-dimerization is favoured at low temperatures. This dynamic behaviour has been confirmed by variable-temperature 1H NMR, UV-vis and EPR measurements. The structures of the stable radical and σ-dimer have been revealed by single-crystal X-ray diffraction analysis. The observed different reactivities of the two (porphyrinyl)dicyanomethyl radicals have been rationalized in terms of their spin delocalization behaviours.
Introduction
Radical-based dynamic covalent chemistry (DCC) has continued to attract considerable attention in recent years due to its intriguing nature to form stable molecular self-assemblies via a clean radical–radical coupling process.1–3 In particular, the stimuli-responsive nature of radicals to form σ-bonds in a reversible manner has been used as an attractive tool for various applications.2 In recent studies, a number of dicyanomethyl-substituted aromatic compounds were reported to show such reversible self-dimerization-dissociation processes.3 Among them, the study by Seki and co-workers demonstrated that p-diphenylamino-substituted dicyanomethyl radical 1 underwent clean DCC to form its σ-dimer (1)2 in a reversible manner.3a In addition to this, they have also reported reversible formation of the π-dimer of the more stable aryl-dicyanomethyl radical 2 possessing a fused nitrogen atom (Chart 1).3d Recently, Chi and co-workers have reported three-dimensional DCC of aryl-dicyanomethyl radicals, which resulted in the construction of a large cyclic hexamer, whose structure was elucidated by single-crystal X-ray analysis.3e Further, Winter and co-workers described the effect of the structure–activity relationship on aryl-dicyanomethyl radicals.3g
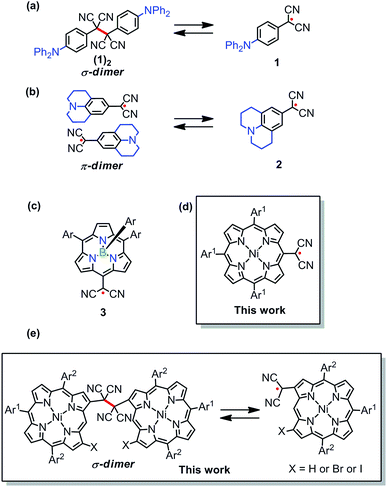 |
| | Chart 1 (a–c) Reported dicyanomethyl based radicals. (d and e) Porphyrin based stable radical vs. reversible σ-dimer (DCC). Ar = 4-methylphenyl, Ar1 = 3,5-di-tert-butylphenyl and Ar2 = mesityl. | |
Following these results, we have reported that (meso-subporphyrinyl)dicyanomethyl radical 3 is quite stable and does not undergo any dimerization.4 As an extension, we examined the chemistry of (porphyrinyl)dicyanomethyl radicals, since porphyrins have been demonstrated to be a nice platform to stabilize neighboring radicals owing to effective spin-delocalization.5,6 While various types of porphyrin-based stable radicals have been explored, those undergoing reversible σ-dimerization are still quite rare,7 except the important examples.8 In the present work, (meso- and β-porphyrinyl)dicyanomethyl radicals were examined and displayed contrasting behaviors in solutions. (Porphyrinyl)dicyanomethyl radicals constitute a rare example displaying stable radical versus dynamic covalent chemistry (DCC) depending upon the substitution position of the dicyanomethyl radical. In particular, (meso-porphyrinyl)dicyanomethyl radicals do not form any σ- or π-dimers and exist predominantly as a monomer radical in solution, while (β-porphyrinyl)dicyanomethyl radicals show DCC by forming σ-dimers in a reversible manner. These results contrast sharply with the previous reports that oxidation of 5,10- and 5,15-dicyanomethyl-substituted porphyrins gave closed shell quinonoid compounds.9
Results and discussion
(meso-Porphyrinyl)dicyanomethyl radicals
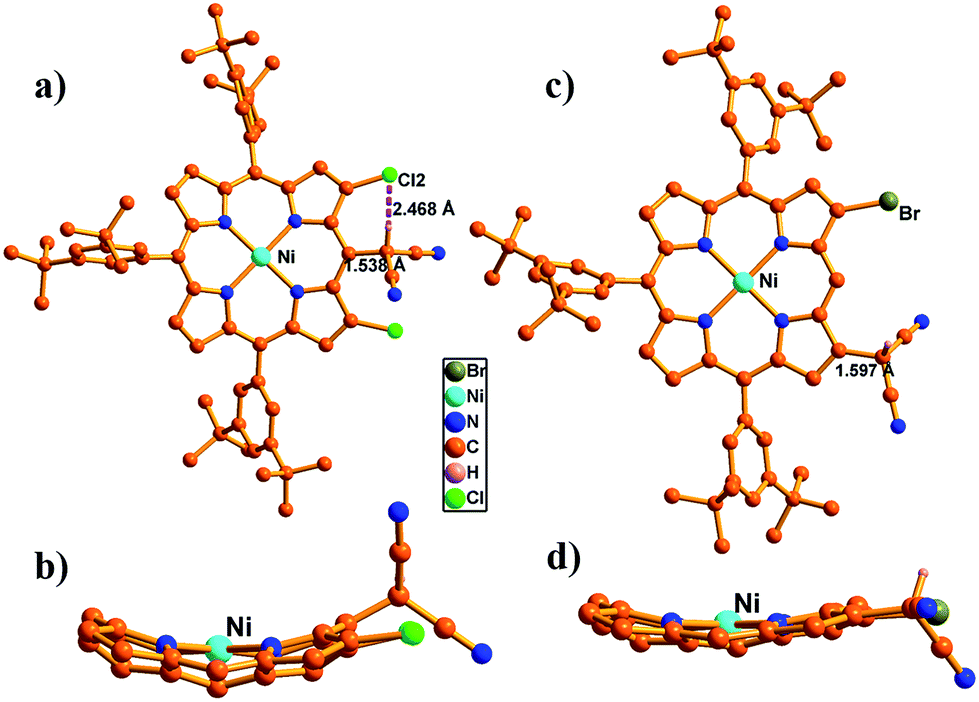
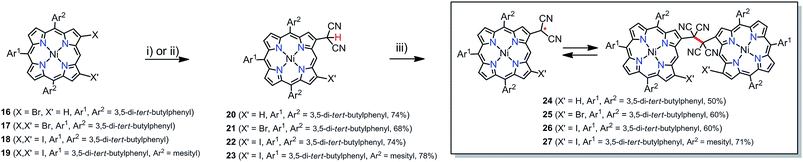
In the synthesis of meso-dicyanomethylporphyrins, we employed a simple nucleophilic aromatic substitution (SNAr) of meso-halogenated porphyrins44–7 with malononitrile in the presence of NaH in 1,3-dimethyl-2-imidazolidinone (DMI) at 60 °C, which produced the desired products 8–11 in 65–70% yields as stable compounds (Scheme 1). Notably, these SNAr reactions proceeded nicely even for sterically hindered substrates 5, 6 and 7. The 1H NMR spectrum of 8 shows a singlet at 7.01 ppm due to the dicyanomethyl group (Fig. S17†), while peaks of 9–11 are observed in the range of 7.35–8.00 ppm (Fig. S19, S21 and S23†). These trends are comparable with the previously reported subporphyrin results,4 and have been ascribed to effective hydrogen bonding interaction between the methine proton in the dicyanomethyl group and the β-halogens. The structure of 9 was confirmed by X-ray crystallographic analysis (Fig. 1a), which revealed the distance between the methine hydrogen and β-chlorine atom to be 2.468(1) Å, suggesting intramolecular hydrogen bonding interaction. The meso-dicyanomethyl group is largely tilted by 52.17(9)° from the mean plane of the Ni(II) porphyrin.
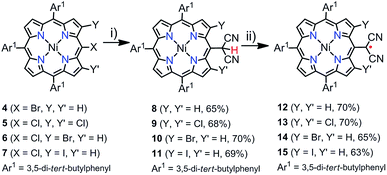 |
| | Scheme 1 Synthesis of 8–15. (i) 10 equiv. malononitrile, 10 equiv. NaH, DMI, 60 °C, 1 h. (ii) 200 equiv. PbO2, CH2Cl2, rt, 2 h. | |
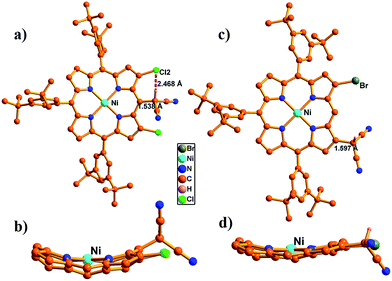 |
| | Fig. 1 Single crystal X-ray structure of 9 and 21. (a and c) Top view and (b and d) side view. The meso-aryl groups in (b and d) and hydrogen atoms in (a–d) are omitted for clarity. | |
Oxidation of meso-dicyanomethyl porphyrins 8–11 with an excess amount of PbO2 in CH2Cl2 gave the corresponding dicyanomethyl radicals 12–15 in 63–70% yields (Scheme 1). All these radicals were stable and tolerant towards moisture and air and separable by silica-gel column chromatography. These radicals were NMR silent even at low temperatures and EPR active (Fig. S45†). The structure of radical 12 was revealed by single crystal X-ray analysis (Fig. 2). In the unit cell, three radicals of slightly different structures formed an offset stacking aggregate with interporphyrin separations of ca. 3.5 and 3.7 Å. Therefore, the structure of 12 was studied based on averaged values. The dicyanomethyl radical group was tilted towards the porphyrin plane with a small dihedral angle of 38.08° and a C–C bond distance of 1.395 Å, which is shorter as compared to the regular C–C single bond (1.45 Å), indicating a double bond character and effective spin delocalization over the porphyrin ring. These structural features are comparable with those of the previously reported subporphyrins and other dicyanomethyl derivatives.3d,g,4
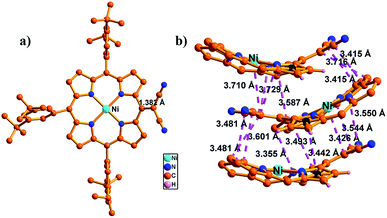 |
| | Fig. 2 Single crystal X-ray structure of 12. (a) Top view and (b) close interaction between radical pairs. The meso-aryl groups in (b) and the peripheral hydrogen atoms in (a and b) have been omitted for clarity. | |
The absorption spectra of 8–11 in CH2Cl2 showed Soret-like bands around 420 nm and Q-like bands between 550 and 600 nm as typical absorption features of porphyrin (Fig. S47–S50†). The absorption spectra of radicals 12–15 in CH2Cl2 showed red shifted Soret-like bands at around 470 nm and Q-like bands between 600 and 900 nm along with a radical-characteristic broad absorption band extending to 1600 nm (Fig. S47–S50†). Importantly, there were no significant changes of the absorption spectra of 12–15 upon temperature change in line with the discussion above (Fig. S51–S55†).
Further, the magnetic properties of radicals 12–15 in the solid state were examined by SQUID magnetometry (Fig. S46†). The observed χT–T curves clearly indicated intermolecular weak anti-ferromagnetic interactions, which were reproduced with a modified Bleaney–Bowers model. The best simulation plots of 12–15 were obtained with parameters of (f1, f2, J1/kB) = (0.713, 0.105, −4.85 K), (0.225, 0.313, −33.7 K), (0.170, 0.374, −29.1 K) and (0.141, 0.397, −24.7 K), respectively.10 Collectively, the dicyanomethyl radicals 12–15 are concluded to exist as a monomeric stable species in solution and in the solid-state.
(β-Porphyrinyl)dicyanomethyl radicals
Our initial attempt to produce simple β-dicyanomethyl-substituted porphyrin 20 by nucleophilic aromatic substitution of β-bromoporphyrin 16 did not proceed well, while the same reaction proceeded nicely for β,β-dihalogenated porphyrins 17–19 with malononitrile in the presence of NaH in DMI at 100 °C to afford 21–23 in moderate yields (Scheme 2). The simple β-dicyanomethyl porphyrin 20 was prepared in 74% yield by a Pd-catalyzed coupling reaction of 16 with malononitrile in the presence of NaH in 1,2-dimethoxyethane (DME) at 80 °C (Scheme 2). The structure of 21 was confirmed by single crystal X-ray analysis (Fig. 1c), which revealed the bond length between the dicyanomethyl unit and porphyrin to be 1.597(5) Å and the tilt angle of the dicyanomethyl unit from the mean plane of the porphyrin to be 49.83(2)°.
 |
| | Scheme 2 Synthesis of 20–27. For synthesis of 20, (i) 5 equiv. malononitrile, 8 equiv. NaH, DME, Pd(PPh3)4 (20 mol%), 80 °C, 2 h. For synthesis of 21–23 (ii) 10 equiv. malononitrile, 10 equiv. NaH, DMI, 100 °C, 6 h. (iii) 200 equiv. PbO2, CH2Cl2, rt, 2 h. | |
Oxidation of 20–23 proceeded smoothly with an excess amount of PbO2 in CH2Cl2 to give dimers (24)2–(27)2 as stable compounds. These dimers were separated by silica gel column chromatography in 50–71% yields. The molecular ion peaks of (24)2–(27)2 were not detected but the half values were observed at m/z = 993.5096, 1073.4174, 1119.4040 and 979.2468 (M+), respectively, matching with the expected radical compositions (Fig. S13–S16†).
Dimers (24)2–(27)2 showed common temperature-dependent 1H NMR spectral features. As a typical example, the 1H NMR spectra of (27)2 at various temperatures are shown in Fig. 3a. The spectrum was rather broad at 298 K but became increasingly sharper upon decreasing the temperature, which, judging from the previous related studies,3 has been interpreted in terms of DCC between the monomeric radical 27 and its σ-dimer (27)2. At high temperatures a certain amount of monomeric radical exists but the σ-dimer dominates at 193 K. We succeeded in the X-ray structural determination of (27)2. Fig. 3b and c show the structure of (27)2 which is a covalently linked syn-dimer, in which the formed C–C bond length is very long, 1.635(1) Å, which is marginally longer than that of the reported examples undergoing DCC.3a,e
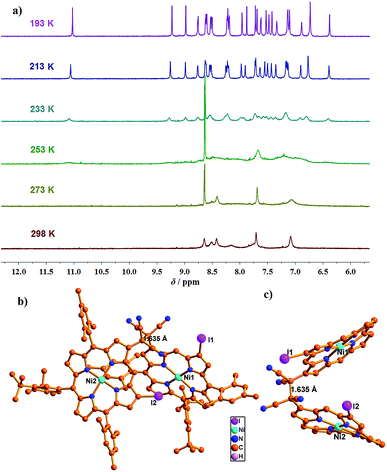 |
| | Fig. 3 (a) 1H-NMR spectrum of (27)2 in CD2Cl2 at various temperatures; (b and c) X-ray crystal structure of (27)2. meso-Aryl groups in (c) and hydrogen atoms in (b and c) have been omitted for clarity. | |
In line with DCC, (24)2–(27)2 showed reversible temperature dependent absorption spectra. As the representative example, the variable temperature absorption spectra of (27)2 in toluene are shown in Fig. 4. Upon increasing the temperature, the typical absorption bands of normal porphyrins, i.e. the Soret band at 414 nm and Q-bands at 535 and 571 nm, decreased and broad bands around 490 and 672 nm as well as a very broad tail in the NIR range of 1000–1400 nm emerged. These spectral changes have been interpreted to indicate that (27)2 exists predominantly at room temperature and undergoes homolysis to give persistent dicyanomethyl radical 27 at high temperatures. This interpretation was strongly reinforced by variable temperature EPR measurement, which actually showed increased EPR signal intensity at higher temperatures (Fig. 4, inset). Similar variable temperature absorption spectra and EPR spectra were observed for (24)2 (Fig. S60†), (25)2 (Fig. S61†) and (26)2 (Fig. S62†). On the basis of these experiments, the thermodynamic parameters of DCC were determined (Table 1). Overall, the VT-NMR, VT-UV, and VT-EPR studies clearly supported the conclusion that the products exist in an equilibrium state between the σ-dimers and radicals.
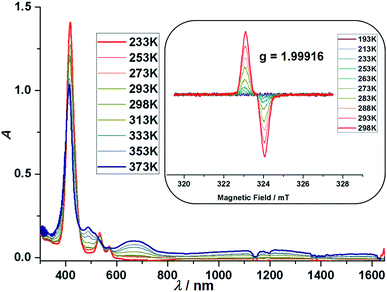 |
| | Fig. 4 Absorption spectra of (27)2 in toluene at various temperatures. Inset: EPR spectra of (27)2 in toluene at various temperatures. | |
Table 1 Thermodynamic parameters of (24)2–(27)2
|
|
ΔHdim/kJ mol−1 |
ΔSdim/J K−1 mol−1 |
ΔGdim,298 K/kJ mol−1 |
|
24
|
−55.7 |
−77.8 |
−32.5 |
|
25
|
−60.6 |
−89.5 |
−33.9 |
|
26
|
−66.3 |
−112.2 |
−32.9 |
|
27
|
−60.6 |
−91.7 |
−33.3 |
Cyclic voltammograms and DFT calculations
To gain further insight into the electronic properties, electrochemical analysis was accomplished by cyclic voltammetry in CH2Cl2 solution, and the redox potentials for 12–15 are summarized in Table 2. The stable neutral radicals 12–15 exhibit two reversible oxidation waves and two reversible reduction waves. The energy gap between the first oxidation and reduction potentials of compounds 12–15 was very small, characteristic of radical species. We also examined the electrochemical properties of the σ-dimers (24)2–(27)2. Cyclic voltammetry showed two similar reversible oxidation waves and two reversible reduction waves and the electrochemical redox gaps were determined to be 0.83, 0.86, 0.85, and 0.85 eV for (24)2–(27)2, respectively. Because the redox gaps of (24)2–(27)2 (<1 eV) were too small considering their optical HOMO–LUMO gaps (∼2 eV), the observed electrochemically active species were determined to be monomeric radicals 24–27 that were in equilibrium with the corresponding dimers.
Table 2 Electrochemical potentials of 12–15 and (24)2–(27)2
|
|
E
OX1
1/2/V |
E
OX2
1/2/V |
E
RED1
1/2/V |
E
RED2
1/2/V |
ΔEHL/eV |
|
12
|
0.26 |
1.21 |
−0.33 |
−2.05 |
0.59 |
|
13
|
0.31 |
1.21 |
−0.29 |
−1.95 |
0.60 |
|
14
|
0.29 |
1.22 |
−0.31 |
−2.02 |
0.60 |
|
15
|
0.28 |
1.20 |
−0.30 |
−1.95 |
0.58 |
| (24)2 |
0.50 |
1.29 |
−0.33 |
−1.99 |
0.83 |
| (25)2 |
0.55 |
1.33 |
−0.31 |
−1.88 |
0.86 |
| (26)2 |
0.55 |
1.37 |
−0.30 |
−1.88 |
0.85 |
| (27)2 |
0.54 |
1.39 |
−0.31 |
−1.96 |
0.85 |
To investigate the spin density distribution, spin-unrestricted density functional theory (DFT) calculations were conducted at the UB3LYP/6-311g(d) level (Fig. S75–S78†).11 As representative examples, the spin density maps of 12 and 24 are shown in Fig. 5. For meso-dicyanomethyl radicals 12–15, the spin density maps clearly show that the spin is fully delocalized over the whole π-network of porphyrin backbones (Fig. S73†). On the other hand, the spin density in β-dicyanomethyl radicals 24–27 is less delocalized and large spin densities are found at the dicyanomethyl unit and neighbouring β-carbon. Spin densities at the dicyanomethyl center clearly indicate a difference depending on the substitution position: 0.19–0.29 for 12–15 and 0.47–0.49 for 24–27. For 12–15, the spin density at the dicyanomethyl center largely depends on the neighboring β-substituent(s): 0.289 (12) > 0.242 (14) ∼ 0.234 (15) > 0.191 (13). This trend is related to the average tilt angles of the dicyanomethyl group toward the porphyrin plane; 12 (32.3°) < 14 (39.1°) ∼ 15 (39.7°) < 13 (44.7°) (Fig. S85†). We consider that the less delocalized spin nature makes 24–27 suitable for DCC.
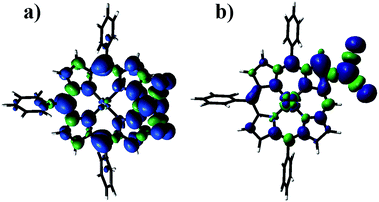 |
| | Fig. 5 Spin density distribution plots of (a) 12 and (b) 24 calculated at the UB3LYP/6-311G* level (isovalue: 0.001). Consistent with the electrochemical data, the energy gaps between the SOMO(α) and SOMO(β) of 12–15 (1.14–1.17 eV) are distinctly smaller than those of 24–27 (1.35–1.48 eV). Further TD-DFT calculations were carried out to interpret the absorption features of 12–15 (Fig. S86–S89†). The calculated absorption transitions were consistent with the observed data, which were similar to the previously reported subporphyrin radicals. | |
Conclusions
In summary, we synthesized (meso-porphyrinyl)dicyanomethyl radicals and (β-porphyrinyl)dicyanomethyl radicals by a SNAr reaction and Pd-catalyzed coupling reaction of the corresponding porphyrin halides with malononitrile and subsequent oxidation with PbO2. (meso-Porphyrinyl)dicyanomethyl radicals are very stable, exist as monomeric species and do not undergo any recombination in solution or in the solid state. On the other hand, (β-porphyrinyl)dicyanomethyl radicals are isolated as covalent σ-dimers, which are stable in the solid state but display DCC behavior in solution; namely, they undergo homolysis to form a persistent dicyanomethyl radical at high temperatures. This marked difference can be ascribed to better spin delocalization in (meso-porphyrinyl)dicyanomethyl radicals as compared with (β-porphyrinyl)dicyanomethyl radicals. Further studies on (porphyrinyl)dicyanomethyl radicals are actively ongoing in our laboratories.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers 25220802, 18H03910, and 18K19074. B. A. thanks the JSPS for a Postdoctoral Fellowship. D. S. acknowledges a JSPS Fellowship for Young Scientists. We acknowledge the Research Center for Molecular-Scale Nanoscience, Institute for Molecular Science (IMS), Okazaki, Japan. The authors are also grateful to Prof. Dr Takahisa Ikeue (Shimane University) for EPR studies and Prof. Dr Hideki Yorimitsu (Kyoto University) for HRMS measurements.
Notes and references
-
(a) H. Yokoi, S. Hiroto and H. Shinokubo, J. Am. Chem. Soc., 2018, 140, 4649 CrossRef CAS PubMed
 ;
(b) S. N. Spisak, A. V. Zabula, M. Alkan, A. S. Filatov, A. Y. Rogachev and M. A. Petrukhina, Angew. Chem., Int. Ed., 2018, 57, 6171 CrossRef CAS PubMed ;
(c) R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2010, 10, 14 CrossRef PubMed ;
(d) Y. Wang, X. Tan, Y.-M. Zhang, S. Zhu, I. Zhang, B. Yu, K. Wang, B. Yang, M. Li, B. Zou and S. X.-A. Zhang, J. Am. Chem. Soc., 2015, 137, 931 CrossRef CAS PubMed .
;
(b) S. N. Spisak, A. V. Zabula, M. Alkan, A. S. Filatov, A. Y. Rogachev and M. A. Petrukhina, Angew. Chem., Int. Ed., 2018, 57, 6171 CrossRef CAS PubMed ;
(c) R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2010, 10, 14 CrossRef PubMed ;
(d) Y. Wang, X. Tan, Y.-M. Zhang, S. Zhu, I. Zhang, B. Yu, K. Wang, B. Yang, M. Li, B. Zou and S. X.-A. Zhang, J. Am. Chem. Soc., 2015, 137, 931 CrossRef CAS PubMed .
-
(a) Y. Kishimoto and J. Abe, J. Am. Chem. Soc., 2009, 131, 4227 CrossRef CAS PubMed ;
(b) K. Imato, J. C. Natterodt, J. Sapkota, R. Goseki, C. Weder, A. Takahara and H. Otsuka, Polym. Chem., 2017, 8, 2115 RSC ;
(c) K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2015, 54, 6168 CrossRef CAS PubMed ;
(d) K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138 CrossRef CAS PubMed .
-
(a) H. D. Hartzler, J. Org. Chem., 1966, 31, 2654 CrossRef CAS ;
(b) T. Kobashi, D. Sakamaki and S. Seki, Angew. Chem., Int. Ed., 2016, 55, 8634 CrossRef CAS PubMed ;
(c) J. P. Peterson, M. R. Geraskina, R. Zhang and A. H. Winter, J. Org. Chem., 2017, 82, 6497 CrossRef CAS PubMed ;
(d) K. Okino, S. Hira, Y. Inoue, D. Sakamaki and S. Seki, Angew. Chem., Int. Ed., 2017, 56, 16597 CrossRef CAS PubMed ;
(e) L. Yuan, Y. Han, T. Tao, H. Phan and C. Chi, Angew. Chem., Int. Ed., 2018, 57, 9023 CrossRef CAS PubMed ;
(f) D. Wang, C. C. Ferron, J. Li, S. Gamez-Valenzuela, R. P. Ortiz, J. T. L. Navarrete, V. H. Jolin, X. Yang, M. P. Alvarez, V. G. Baonza, F. Hartl, M. C. R. Delgado and H. Li, Chem.–Eur. J., 2017, 23, 13776 CrossRef CAS PubMed ;
(g) R. Zhang, J. P. Peterson, L. J. Fischer, A. Ellern and A. H. Winter, J. Am. Chem. Soc., 2018, 140, 14308 CrossRef CAS PubMed .
- B. Adinarayana, D. Shimizu and A. Osuka, Chem.–Eur. J., 2019, 25, 1706 CrossRef CAS PubMed .
- D. Shimizu and A. Osuka, Chem. Sci., 2018, 9, 1408 RSC .
-
(a) T. Koide, G. Kashiwazaki, M. Suzuki, K. Furukawa, M.-C. Yoon, S. Cho, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 9661 CrossRef CAS PubMed ;
(b) D. Shimizu, J. Oh, K. Furukawa, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 6613 CrossRef CAS PubMed ;
(c) D. Shimizu, J. Oh, K. Furukawa, D. Kim and A. Osuka, J. Am. Chem. Soc., 2015, 137, 15584 CrossRef CAS PubMed ;
(d) K. Kato, W. Cha, J. Oh, K. Furukawa, H. Yorimitsu, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 8711 CrossRef CAS PubMed ;
(e) D. Shimizu, K. Furukawa and A. Osuka, Angew. Chem., Int. Ed., 2017, 56, 7435 CrossRef CAS PubMed ;
(f) K. Kato, K. Furukawa, T. Mori and A. Osuka, Chem.–Eur. J., 2018, 24, 572 CrossRef CAS PubMed ;
(g) D. Shimizu and A. Osuka, Angew. Chem., Int. Ed., 2018, 57, 3733 CrossRef CAS PubMed ;
(h) K. Kato, K. Furukawa and A. Osuka, Angew. Chem., Int. Ed., 2018, 57, 9491 CrossRef CAS PubMed .
- Only one report which related to norcorrole system. B. Liu, T. Yoshida, X. Li, M. Stępień, H. Shinokubo and P. J. Chmielewski, Angew. Chem., Int. Ed., 2016, 55, 13142 CrossRef CAS PubMed .
-
(a) J. H. Fuhrhop, S. Besecke, J. Subramanian, C. Mengersen and D. Riesner, J. Am. Chem. Soc., 1975, 97, 7141 CrossRef CAS ;
(b) A. L. Balch, B. C. Noll, S. M. Reid and E. P. Zovinka, J. Am. Chem. Soc., 1993, 115, 2531 CrossRef CAS .
-
(a) I. M. Blake, H. L. Anderson, D. Beljonne, J.-L. Brédas and W. Clegg, J. Am. Chem. Soc., 1998, 120, 10764 CrossRef CAS ;
(b) I. M. Blake, L. H. Rees, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed., 2000, 39, 1818 CrossRef CAS ;
(c) M. J. Smith, W. Clegg, K. A. Nguyen, J. E. Rogers, R. Pachter, P. A. Fleitz and H. L. Anderson, Chem. Commun., 2005, 2433 RSC ;
(d) Q. Chen, Y.-Z. Zhu, Q.-J. Fan, S.-C. Zhang and J.-Y. Zheng, Org. Lett., 2014, 16, 1590 CrossRef CAS PubMed ;
(e) K.-i. Yamashita, S. Sakamoto, A. Suzuki and K.-i. Sugiura, Chem.–Asian J., 2016, 11, 1004 CrossRef CAS PubMed .
- The dimeric structure was not fixed by strong interaction such as covalent bond, we assumed that some of radicals were not involved in the dimeric packing. Thus, we used f1 and f2 to as weights for the dimeric diradical and isolated radicals, respectively.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed .
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ko
Furukawa
b and
Atsuhiro
Osuka
a,
Ko
Furukawa
b and
Atsuhiro
Osuka