
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVinylphosphonites for Staudinger-induced chemoselective peptide cyclization and functionalization†‡
Marc-André
Kasper§
ab,
Maria
Glanz§
ab,
Andreas
Oder
a,
Peter
Schmieder
a,
Jens P.
von Kries
a and
Christian P. R.
Hackenberger
 *ab
*ab
aLeibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Chemical Biology Department, Robert-Rössle-Strasse 10, 13125 Berlin, Germany. E-mail: hackenbe@fmp-berlin.de
bHumboldt Universität zu Berlin, Department of Chemistry, Brook-Taylor-Str. 2, 12489 Berlin, Germany
First published on 16th May 2019
Abstract
In this paper, we introduce vinylphosphonites for chemoselective Staudinger-phosphonite reactions (SPhR) with azides to form vinylphosphonamidates for the subsequent modification of cysteine residues in peptides and proteins. An electron-rich alkene is turned into an electron-deficient vinylphosphonamidate, thereby inducing electrophilic reactivity for a following thiol addition. We show that by varying the phosphonamidate ester substituent we can fine-tune the reactivity of the thiol addition and even control the functional properties of the final conjugate. Furthermore, we observed a drastic increase in thiol addition efficiency when the SPhR is carried out in the presence of a thiol substrate in a one-pot reaction. Hence, we utilize vinylphosphonites for the chemoselective intramolecular cyclization of peptides carrying an azide-containing amino acid and a cysteine in high yields. Our concept was demonstrated for the stapling of a cell-permeable peptidic inhibitor for protein–protein interaction (PPI) between BCL9 and beta-catenin, which is known to create a transcription factor complex playing a role in embryonic development and cancer origin, and for macrocyclization of cell-penetrating peptides (CPPs) to enhance the cellular uptake of proteins.
Introduction
Selective chemical modification of the sulfhydryl group of cysteine is an important tool in the life sciences.1,2 Cysteine-selective reactions have been employed to covalently modify proteins in a residue specific manner for various applications including drug delivery,3 incorporation of post-translational modifications4 and activity based protein profiling.5A plethora of different techniques have been developed in the past few decades that allow for selective modification of cysteine residues on peptides and proteins based on various reaction mechanisms. Michael additions to α,β-unsaturated carbonyl compounds in maleimides,6 sulfone reagents7 and recently developed carbonylacrylic reagents8 are the most prominent examples. Other methods rely on SN2-reactions of haloalkyl bonds such as iodoacetamides,9 on alkynylation10 and fluoroalkylation11 with hypervalent iodine reagents, on nucleophilic aromatic substitution of perfluoroaromatic compounds12 or on Pd-mediated cross-coupling.13 The level of reactivity for cysteine in these methods is predetermined by the chemical nature of the reactive group itself. Alteration of this reactivity is often difficult and requires tedious chemical manipulations. However, certain applications such as activity based protein profiling14 or covalent enzyme inhibition15 often rely on reduced reactivity that allows for modification of the active site cysteine while sparing all other free nucleophiles in the proteome. Such decreased reactivity has for instance been achieved by substituting the highly reactive iodide in α-haloacetamides with a less reactive chloride.16 Therefore, a cysteine selective modification method that utilizes easily tunable electrophilic warheads would be highly attractive for addressing challenges in peptide and protein chemistry.
Aside from protein modification, cysteine selective reactions have also been applied to peptide synthesis. One or two covalent linkages between cysteine residues in peptides enable the construction of macro or bicyclized peptides with enhanced biophysical and pharmacological properties.17 Upon macrocyclization, peptidic structures can become more stable against proteases,18 they can be locked in their structural conformation by covalently linking side chains and therefore restricting their conformational flexibility19,20 or increasing their function by rigid constraint as shown for various cyclic bioactive21 or cell penetrating peptides (cCPPs).22 The groups of Pentelute and Derda introduced perfluoroaromatic linkages for the cyclization of peptides containing two cysteines.23,24 Tricyclic peptides have also been synthesized by alkylation with tris-(bromomethyl)benzene25 and very recently by the incorporation of two chemical bridges with a set of various dibromoalkyl reagents.26 Such chemoselective modification strategies are advantageous over common cyclization techniques27 such as lactamization,28 since they can be applied after cleavage from the resin and preclude therefore tedious orthogonal protecting group strategies during SPPS. In addition, even cysteine containing proteins can be cyclized by applying chemoselective techniques, as recently demonstrated with the aid of a trimeric chloroacetamide.29
Based on the Staudinger-phosphonite reaction (SPhR), developed in our laboratory,30 we recently introduced ethynylphosphonamidates as cysteine-selective electrophiles that can be chemoselectively incorporated starting from ethynylphosphonites.31 In the current paper, we report vinylphosphonites as a new compound class for the transformation of azides into electron deficient alkenes that selectively modify cysteine residues in peptides and proteins. We observed that the SPhR with vinylphosphonites facilitates straightforward access to vinylphosphonamidates carrying different O-substituents. We discovered that the rate of the subsequent thiol addition can be drastically influenced by the electronic properties of these phosphonamidate ester substituents. In addition, we engineered an intramolecular variant of the chemically induced thiol addition for a chemoselective cyclization of peptides. We were able to demonstrate that azido modified cysteine peptides can be cyclized upon SPhR with vinylphosphonites in high yields (Scheme 1). In contrast to other chemoselective peptide cyclizations, which have been mainly used for head-to-tail cyclizations, including traceless Staudinger ligation,32 α-ketoacid–hydroxylamine amide-ligation33 or serine ligation,34 the method presented herein can be straightforwardly applied to side-chain ring closure.
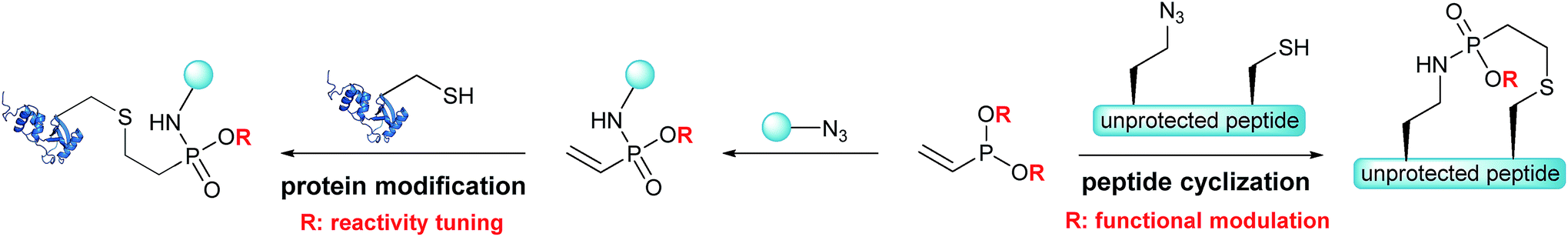 | ||
| Scheme 1 Vinylphosphonites for intramolecular peptide cyclization and cysteine selective protein modification. | ||
Results and discussion
Previously, Gao et al. have observed that methyl-substituted vinylphosphonamidates are unreactive in base mediated thiol additions,35 probably due to decreased electrophilicity, compared to their ethynyl substituted counterparts.31 We anticipated that the electrophilic reactivity of the double bond towards thiol-addition can be increased by attaching different electron-withdrawing substituents to the phosphonamidate ester moiety. Since this residue is preserved during SPhR with azides, we started our investigations by synthesizing different substituted vinylphosphonites. Here, we followed previously published procedures starting from either phosphorus trichloride (route I) or bis(diisopropylamino)-chlorophosphine (route II).36 Several attempts to isolate unprotected vinylphosphonites failed due to product decomposition, hence we decided to isolate the phosphonites in their borane-protected form and use them directly as starting materials for the subsequent SPhR. Vinylphosphonite borane adducts proved to be stable during column chromatography and we were able to obtain four vinylphosphonites with ethyl, 1a, phenyl, 1b and electron withdrawing 2-nitrobenzyl, 1c, and trifluoroethyl, 1d, substituents (Fig. 1a).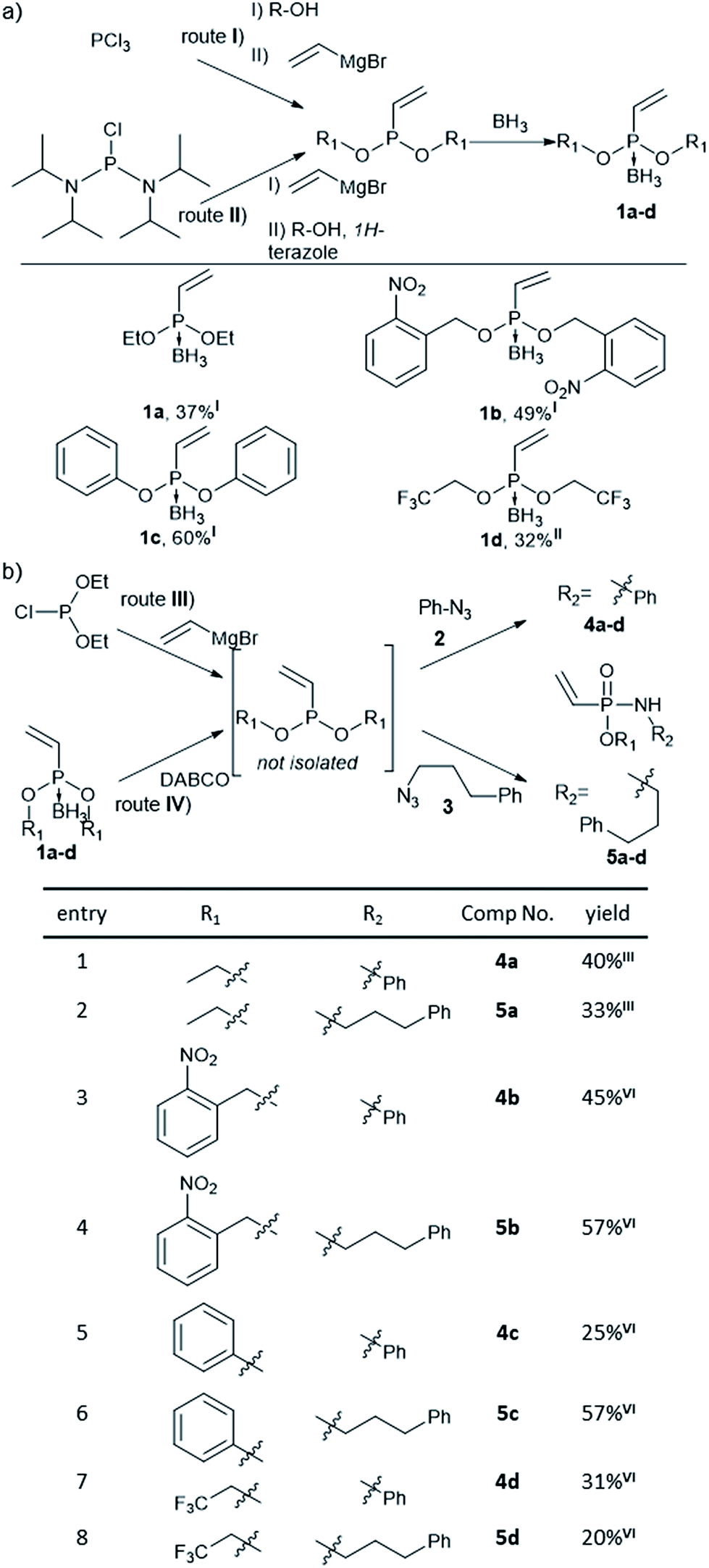 | ||
| Fig. 1 (a) Synthesis of substituted vinylphosphonites from phosphorus trichloride (route I) or bis(diisopropylamino)chlorophosphine (route II). Stated are isolated yields. (b) One-pot SPhR with vinylphosphonites and azides. Synthesis from borane protected vinylphosphonites (route III) or diethyl chlorophosphite (route IV). Stated are isolated yields. | ||
With vinylphosphonites 1a–d in hand, we continued our studies to perform the SPhR with two different model azides 2 and 3 to synthesize both N-phenyl 4a–d and N-alkyl substituted vinylphosphonamidates 5a–d. Here, we applied previously published procedures, either diethyl chlorophosphite alkylation with vinylmagnesium bromide31 (route III) or borane deprotection with DABCO in toluene37 (route IV), directly followed by azide addition and hydrolysis without isolation of the phosphonite intermediate. The SPhR was always carried out in dry solvents to prevent hydrolysis of the phosphonites. All of the tested phosphonites reacted smoothly under the tested conditions and the desired vinylphosphonamidates could be isolated by column chromatography. Electron withdrawing substituents were well tolerated in the reaction with the aryl azide 2 and with the alkyl azide 3 (Fig. 1b).
Next, we probed the reactivity of the synthesized vinylphosphonamidates towards thiol addition in aqueous solution under slightly basic conditions as previously performed for ethynylphosphonamidates31 (Fig. 2a). For this, a time course for the addition of glutathione (GSH) to vinylphosphonamidates 4a–d and 5a–d in ammonium bicarbonate buffer was recorded.
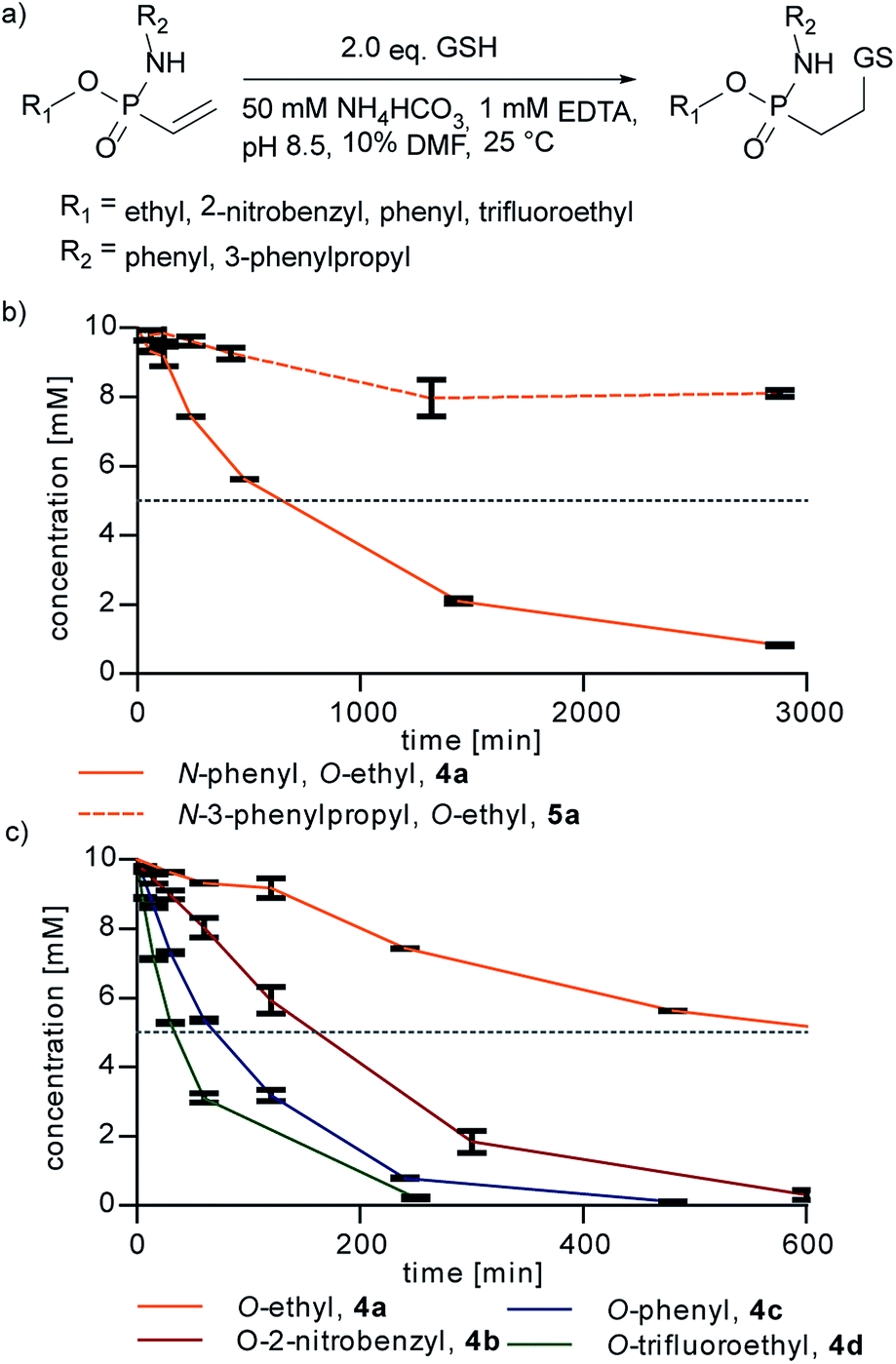 | ||
| Fig. 2 (a) Reaction of glutathione (GSH) with differently substituted vinylphosphonamidates. 10 mM vinylphosphonamidates were reacted with 20 mM GSH at pH 8.5 (NH4HCO3 buffer). Concentration of the starting materials over time, mean and error from three independent measurements, monitored by UPLC-UV. (b) N-Phenyl vs. N-alkyl comparison. (c) Comparison of different O-substituents. Dotted line depicts 50% conversion. | ||
Under the tested conditions (10 mM phosphonamidate and two equivalents of GSH) the N-phenyl vinylphosphonamidate 4a showed a good conversion of 80% to the desired thiol adduct after 24 h. In contrast to 4a, the comparable N-alkyl derivative 5a was only converted 20% under the same conditions (Fig. 2b), which is in accordance with the observations of Gao et al. on the unreactivity of N-alkyl-O-methyl-vinylphosphonamidate towards thiols; however, the previously mentioned dealkylation of the phosphonamidate ester was not observed under conditions applied here. Since we assumed that such slow kinetics would limit the applicability to protein modification, for example by undesired disulfide formation of the peptide or protein substrate, we also tested compounds with electron-deficient phosphonamidate ester substituents in the addition of glutathione. A conversion of 50% is reached after two and a half hours for 2-nitrobenzyl compound 4b and after only 30 minutes for trifluoroethyl substituted 4d. The O-phenyl compound 4c shows an accelerating effect over alkoxy-substituents as well, reaching 50% conversion in only one hour (Fig. 2c). These results clearly show that the reactivity of vinylphosphonamidates can be tuned by varying the phosphonamidate ester residue. The corresponding N-alkyl substituted compounds 5a–d also undergo rate enhancement with more electron withdrawing O-substituents, with a slower reaction compared to the N-phenyl substituted compounds 4a–d in general (ESI Fig. S1‡). Notably, the glutathione adduct was the only detectable product by UPLC/MS and no other side product was observed for any of the tested phosphonamidates (ESI Fig. S2‡). The identity of the thiol-glutathione adducts of 4a (ESI Fig. S3‡) and 4d was confirmed by NMR characterization after isolation by preparative HPLC in 60% and 89% yield, respectively.
Further kinetic investigations of the thiol addition to 4d, being the fastest compound tested in this study (ESI Fig. S1‡), revealed a second-order rate constant of 0.021 M−1 s−1 (ESI Fig. S4‡). Even though this is considerably slower than that of maleimides (734 M−1 s−1),38 ethynylphosphonamidates (0.62 M−1 s−1)31 and recently introduced dinitroimidazoles,39 the rate is still higher or in the range of other protein modification reactions such as oxime, Staudinger or Pictet–Spengler ligations.38 Next to reaction rates, an important aspect for many applications of cysteine selective protein modification reagents is susceptibility of the conjugate towards thiol exchange.1 Maleimides are in particular known to undergo retro Michael addition, followed by transfer of the modification to other cysteine containing molecules in solution.40 To prove the stability of the vinylphosphonamidate adducts in the presence of other thiols, we incubated the glutathione adducts of 4a and 4d with an excess of 100 equivalents of 2-mercaptoethanesulfonate. Even after one week of incubation, no thiol exchange could be observed for both of the constructs by UPLC/MS (ESI Fig. S5‡).
Next, we wanted to probe the reactivity of electron-poor vinylphosphonamidates towards cysteine modification on antibodies. For this, we synthesized a fluorescent EDANS-modified vinylphosphonamidate 6, analogously to compound 4d, with the N-phenyl-O-trifluoroethyl substitution pattern that has been identified to be superior in thiol addition kinetics. Based on a previously established protocol31 we reduced the four interchain disulfide bonds of cetuximab to generate 8 free cysteine residues and probed the reactivity towards 6 (Fig. 3a). Successful labelling after reduction and incubation with 20 equivalents of reagent per antibody was confirmed by in-gel fluorescence (Fig. 3b). Intact protein MS revealed a decent degree of modification of 3.8 fluorophore molecules per antibody (ESI Fig. S6‡). To probe the cysteine selectivity, a control experiment without prior antibody reduction was performed and no modification could be detected after incubation with 20 equivalents of 6 by in-gel fluorescence and MS. Only upon exposure of the unreduced antibody to high reagent excesses of more than 50 equivalents per antibody, low degrees of modification were observed (Fig. 3b). The observation of unspecific labelling at higher reagent concentrations was further investigated by mass spectrometry (LC-MS/MS) after trypsin digestion of the reduced antibody modified with 100 eq. of 4d (ESI Fig. S7‡). In addition to the inter-chain disulfide forming cysteines, a few lysine residues were also found to be modified. This cross-reactivity with lysine is also known for other cysteine labelling reagents such as iodoacetamides41 and maleimides.42 The findings are nevertheless contrary to our previously reported ethynylphosphonamidate reagents that exhibit excellent cysteine selectivity even with large reagent excesses.31 To further broaden the scope of our method, we also synthesized a biotin-modified 2-nitrobenzyl-phosphonamidate with a structure that has the same substitution pattern as compound 4b. Successful antibody labelling was confirmed by anti-biotin Western blot analysis. Analogously to the trifluoroethyl compound, only prolonged reaction times lead to minor labelling degrees without prior reduction of the antibody disulfide bonds (ESI Fig. S8‡). Taken together, we were able to show that vinylphosphonamidates can lead to efficient and selective modification of cysteine residues on proteins using only slight reagent excesses.
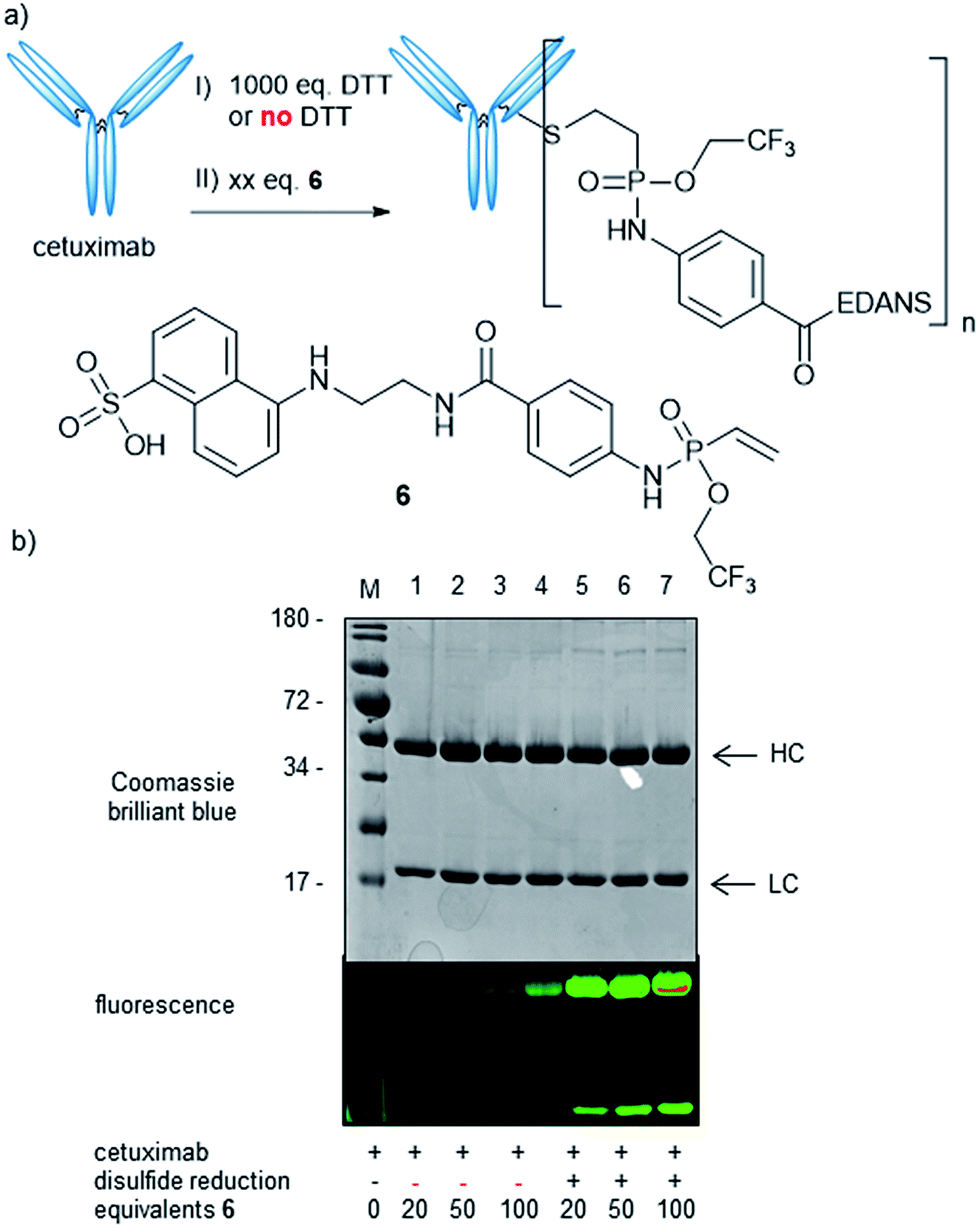 | ||
| Fig. 3 Cetuximab modification with 6. (a) Reduction and alkylation principles and structure of 6. (b) Gel analysis: reactions were carried out with varying equivalents of 6: Lane 1: untreated antibody. Lane 2–4: control reactions without prior DTT treatment. Lane 5–7: prior DTT treatment. HC: antibodies' heavy chain, LC: light chain. | ||
We further anticipated that the SPhR with subsequent thiol addition could also be applied in an intramolecular reaction. Therefore we synthesized an azidohomoalanine- and cysteine-containing peptide, aiming to initiate a covalent cyclization. As the first proof of principle study, we chose to macrocyclize the BCL9 derived peptide 8a, which has been reported to act as a potent PPI-inhibitor when stapled by ring closing metathesis (RCM).43 We decided to replace the two olefinic amino acids in the i,i+4 position with azidohomoalanine and cysteine in order to keep the linker length (10 atoms) similar to that of the literature-known peptide 7 (Scheme 2). In our initial experiments we aimed to keep a double bond in the linker structure for more rigidity and hence used an ethyl-substituted ethynylphosphonite in the reaction with peptide 8a in dry DMSO based on our previously published conditions.31 However, we found that in order to promote the initial SPhR with the alkylazide of azidohomoalanine the reaction temperature had to be raised to 50 °C, upon which the macrocyclized product formed only in low amounts. A major side product observed by UPLC-UV/MS was a peptide with an additional 28 Da, which we attribute to the alkylation of the nitrogen of the phosphonamidate. Similar N-alkylated phosphoramidates were reported previously by us after the rearrangement of phosphorimidates under elevated temperatures.44 We hypothesized that raising the reactivity of the phosphonite by using a more nucleophilic vinyl- as opposed to the ethynyl-phosphonite in the SPhR enables the use of lower reaction temperature and would thereby prevent the rearrangement to the alkylated byproduct. Therefore purified peptide 8a was reacted with crude diethyl vinylphosphonite (route III) after SPPS in an over-night reaction in DMSO. To our delight, we isolated the desired cyclized peptide 9a after preparative HPLC as the main product in a very good yield of 45%. This result is particularly remarkable in light of the slow intermolecular thiol addition to O-ethyl-N-alkyl-vinylphosphonamidates such as 5a as shown before (Fig. 2b). UPLC/MS showed a clean conversion towards the desired product without significant amounts of the side product (ESI Fig. S9‡).
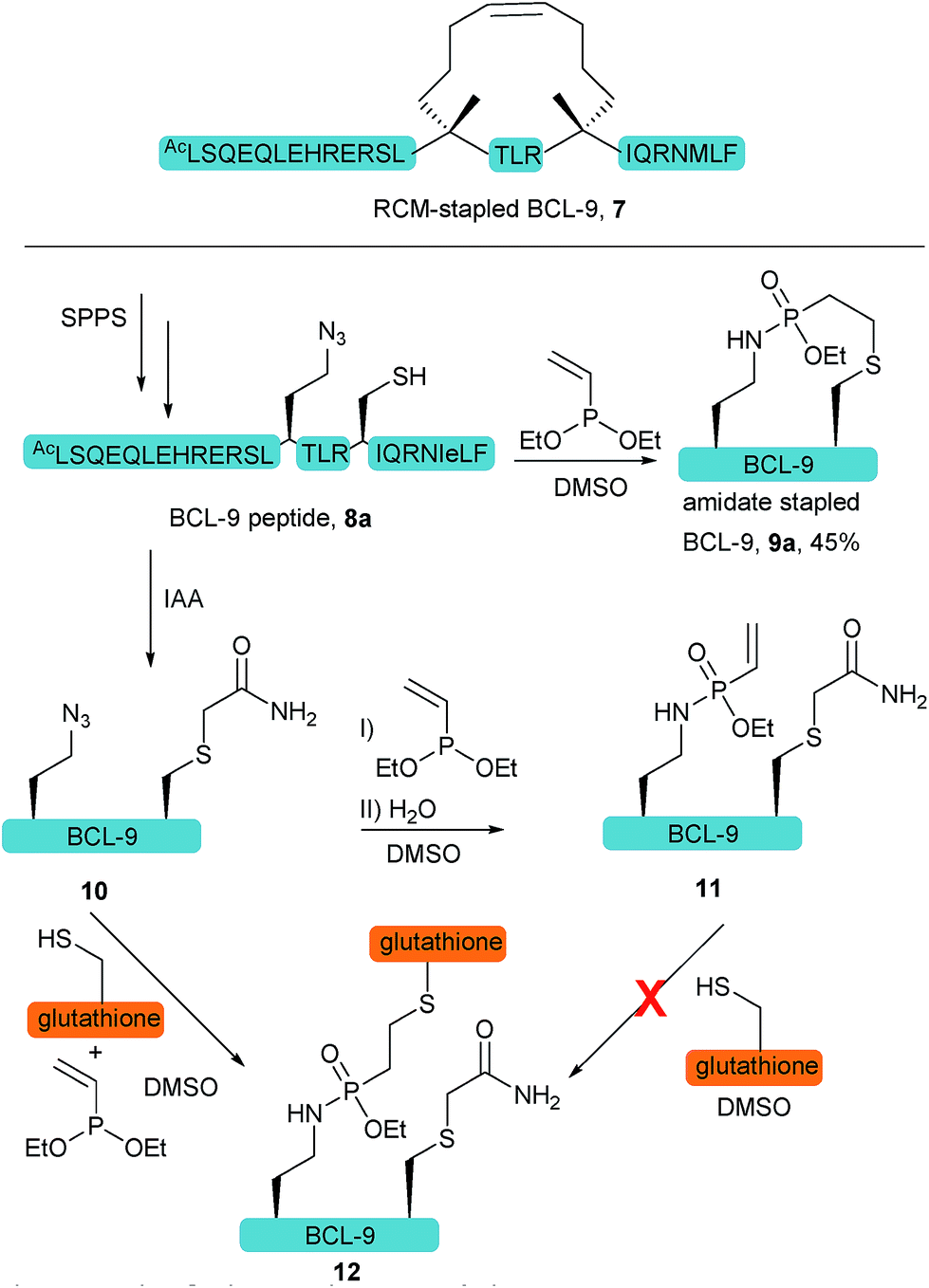 | ||
| Scheme 2 Principle of the intramolecular peptide cyclization of an azido containing cysteine-BCL-9 derived peptide. Investigations with a capped cysteine peptide (9) revealed an accelerated thiol addition when SPhR is performed in the presence of thiols. SPhR on peptides is always carried out with the crude diethyl phosphonite, synthesized from diethyl chlorophosphite (route III). | ||
To gain more insight into the in situ intramolecular cyclization, we capped the cysteine of peptide 8a with iodoacetamide to form peptide 10. This peptide was reacted with glutathione under two different reaction conditions (Scheme 2). First, glutathione was added in a one-pot reaction with peptide 10 (1.65 mM) and 5 eq. diethyl vinylphosphonite. Here, the thiol addition product 12 could be observed by LC-MS as the major product (ESI Fig. S10‡). In the second experiment, we reacted peptide 10 with 5 eq. vinyl phosphonite to form the phosphonamidate 11 after hydrolysis, as observed by LC-MS. After lyophilization peptide 11 was incubated with 1 eq. glutathione (1.65 mM) in DMSO for 24 hours. Under these conditions, no product formation could be observed by MS analysis (ESI Fig. S10‡). This experiment again points towards the decreased reactivity of ethyl-substituted vinylphosphonamidates in intermolecular reactions with thiol substrates (see Fig. 2a and ref. 35), and furthermore implies that the in situ process benefits from the reaction of thiols with a more reactive SPhR intermediate.
With the phosphonamidate-cyclized BCL9 peptide 9a in hand we set out to investigate its stability and helicity. At first, 9a was incubated at different pH values over several days and the stability monitored by UPLC. Under acidic conditions below pH 2 the P–N-bond gets cleaved within 24 hours, which is in accordance with our previous observations.31 At neutral and basic pH, the phosphonamidate moiety proved to be stable over several days without any observable decomposition (Fig. 4a and b). Next, we evaluated the peptides' helical propensities by CD spectroscopy and could show a drastic increase in helicity upon cyclization from 20% to around 50% (Fig. 4d). To verify the ability to disrupt the PPI between purified BCL9 and β-catenin we used a homogeneous assay with time resolved fluorescence (HTRF), in which both interacting proteins were recognized by distinct antibodies via orthogonal tags and conjugated fluorophores suitable for energy transfer.45 Disturbance of the protein–protein interaction causes a decreased FRET signal (ESI Fig. S11‡). Using this assay, we could show that the phosphonamidate cyclized peptides can disrupt the PPI at concentrations in the nM range (Fig. 4e). The IC50 values are almost identical to those reported for the RCM stapled BCL9 peptide.43 Furthermore, we observed that the stereochemistry at cysteine seems to have an influence on the disruption behavior, with a superior inhibition constant observed for the D-enantiomer 9b compared to that of the L-enantiomer 9a.
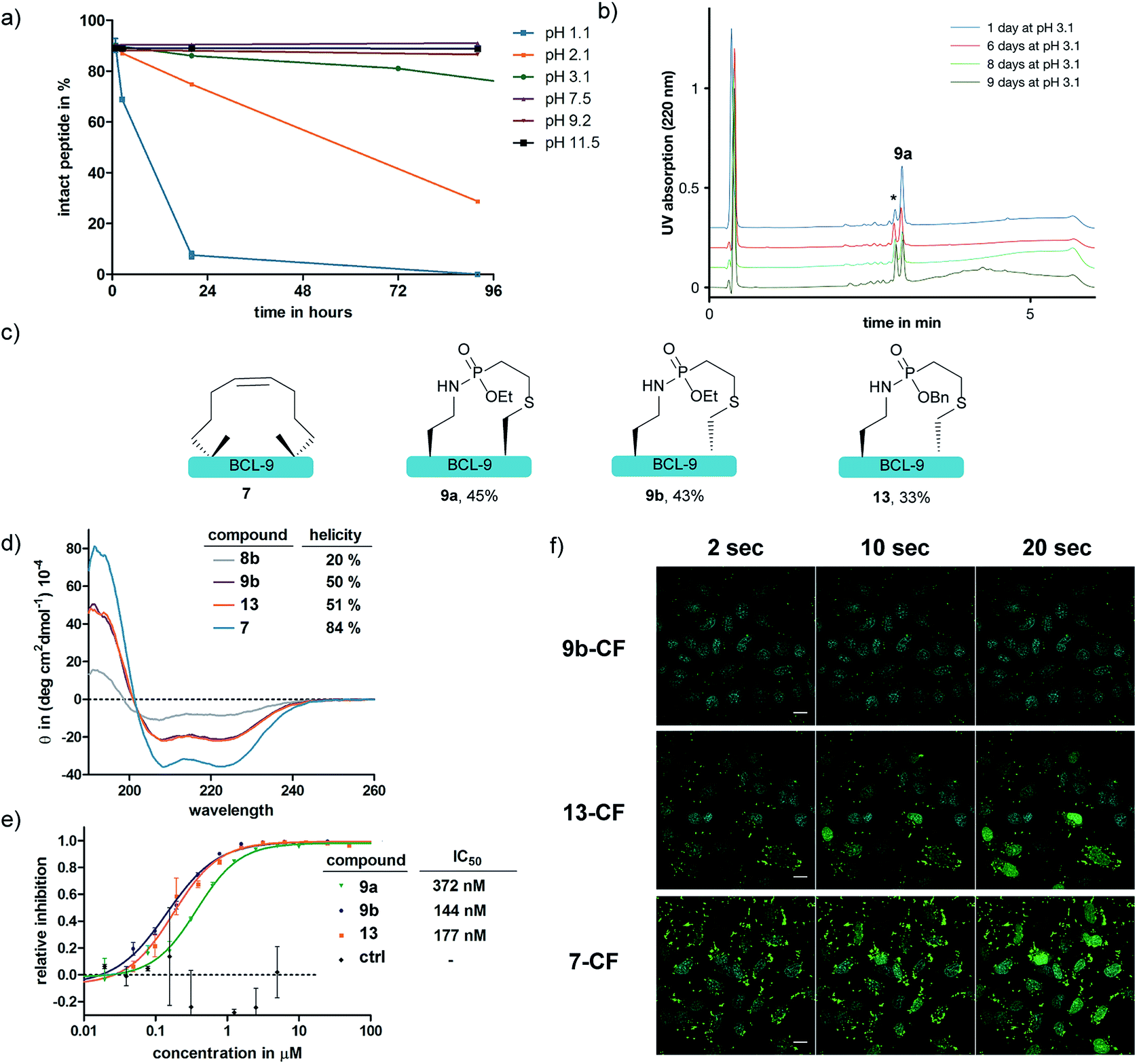 | ||
| Fig. 4 (a) Stability of 9a at different pH values. Shown are mean and error from three independent measurements, monitored by UPLC-UV. (b) UPLC-UV spectra of 9a upon incubation at pH 3.1 over several days. *P–N-bond cleavage is the only degradation product observed. (c) Structures of differently cyclized BCL-9 peptides. Stated are isolated yields from the cyclization reaction. (d) CD-measurement of peptides. (e) HTRF assay shows disruption of PPI by peptidic inhibitors. (f) Cellular uptake studies of 10 μM mixture of 10% carboxy-fluorescein (CF)-labelled and 90% non-labelled peptides in HeLa cells. Confocal images with scale bar = 20 μm. Depicted is the overlay of fluorescein-fluorescence and Hoechst nucleus stain at different time points of laser (405/488 nm) irradiation. | ||
Since peptide stapling is known to improve cellular uptake through endocytic vesicle trafficking46 and the BCL-9 and β-catenin interaction takes place intracellularly in the cytoplasm and the nucleus,45 we also tested the peptides' ability to penetrate cells. By treating HeLa cells with 10 μM mixture of 10% fluorescein labelled and 90% non-labelled peptides for three hours, we observed that peptide 9b shows almost no uptake, whereas the RCM stapled peptide 7 is taken up readily. We attributed this to a recent observation from the Futaki lab, in which they show the importance of net hydrophobicity for the cellular uptake of stapled peptides.47 In order to increase the hydrophobicity of our linkage, we synthesized peptide 13 with a more lipophilic O-benzyl-substituent at the phosphonamidate. For this, the SPhR of the azido peptide 8b with a benzyl phosphonite was carried out at slightly elevated temperatures to ensure high conversions. The peptide was only synthesized in the D-cysteine variant due to the aforementioned improved disruption behavior towards the BCL-9 and β-catenin interaction. Isolated 13 showed comparable helicity to the ethyl substituted 9b (Fig. 4d) and the benzyl-substituent did not hamper the peptides' ability to disrupt the PPI (Fig. 4e). In addition, we observed increased cellular uptake of benzyl substituted 13, compared to 9b, which nicely illustrates the modularity of our method that allows a facile exchange of the O-substituent of phosphonamidates to modulate the functional properties of the peptide ligation products.
In addition to the side-chain-stapled peptides, we further expanded our novel cyclization method to a more general macrocyclization technique for enhancing the cellular uptake properties of protein conjugates. Therefore, we aimed for the cyclization of an R10 peptide, which is, like other arginine-rich peptides, able to enhance the transduction of functional proteins into living cells when covalently attached to the protein cargo.48 We and others observed that the cellular transduction behavior is significantly improved for the cyclic version of cR10 when compared to its linear counterpart.22,49,50 As an alternative to the previously applied cyclized R10-CPP 14, we synthesized peptide 15 containing a cysteine and an azidohomoalanine residue to replace the former lactam bridge after our novel cyclization technique. The linear unprotected peptide was reacted with the crude diethylvinylphosphonite (route III) in dry DMSO overnight to yield 48% of the in situ macrocyclized peptide 16 after purification. For conjugation of 16 to a protein cargo, the N-terminus of the peptide was capped with an ethynylphosphonamidate-NHS building block 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 to yield 18 (Fig. 5a) for Cys-selective labeling reactions. Conjugation to eGFP that carries a single addressable cysteine proceeded quantitatively according to MALDI-TOF-MS (Fig. 5b). Cellular uptake of the protein was demonstrated by confocal fluorescence imaging after incubation of HeLa-cells with 30 μM conjugate for one hour at 37 °C in HEPES buffer and was compared to that of the standard lactam cyclized cR10-eGFP conjugate. Cytosolic and nucleolar GFP fluorescence was observed and both protein–peptide-conjugates showed the same amount of transduced cells, indicating the feasibility of using our novel cyclization method as a macrocyclization technique (Fig. 5c).
31 to yield 18 (Fig. 5a) for Cys-selective labeling reactions. Conjugation to eGFP that carries a single addressable cysteine proceeded quantitatively according to MALDI-TOF-MS (Fig. 5b). Cellular uptake of the protein was demonstrated by confocal fluorescence imaging after incubation of HeLa-cells with 30 μM conjugate for one hour at 37 °C in HEPES buffer and was compared to that of the standard lactam cyclized cR10-eGFP conjugate. Cytosolic and nucleolar GFP fluorescence was observed and both protein–peptide-conjugates showed the same amount of transduced cells, indicating the feasibility of using our novel cyclization method as a macrocyclization technique (Fig. 5c).
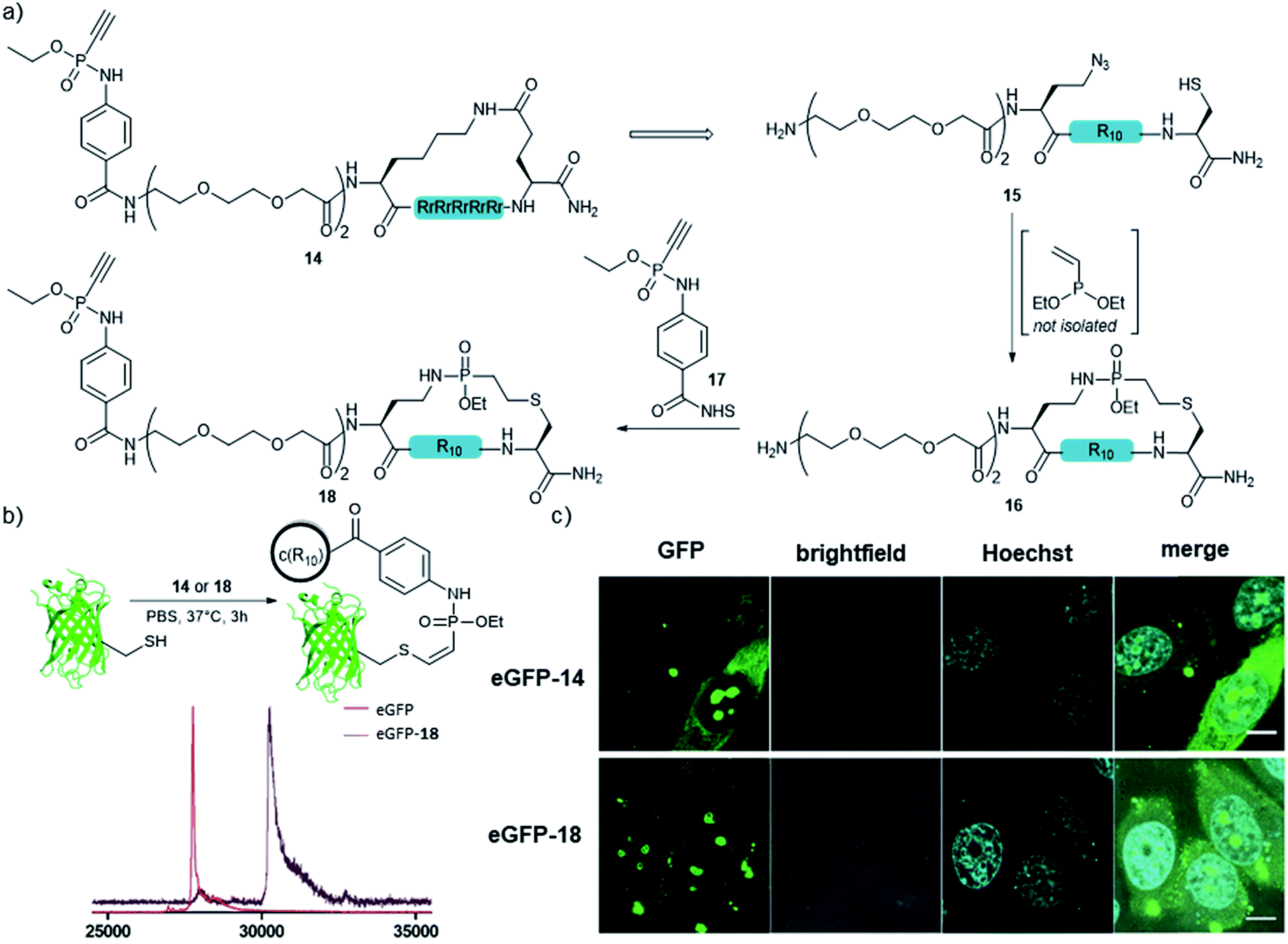 | ||
| Fig. 5 (a) Principle of the Staudinger induced peptide cyclization with R10 and subsequent attachment of a thiol reactive handle. (b) Conjugation of cR10 to eGFP C70M S147C. Shown are MALDI-TOF-MS spectra before and after the reaction with 23. (c) Cellular uptake studies of 30 μM eGFP-14 and eGFP-18 in HeLa cells. Confocal images with scale bar = 10 μm. | ||
Conclusions
In summary, we introduce the SPhR with vinylphosphonites as a versatile tool for the chemoselective cyclization of peptides. Furthermore, we demonstrate that variation of the phosphonite's O-substituent enables fine-tuning of the electrophilicity for intermolecular protein and antibody labeling. Additionally, this variation facilitates the incorporation of functional handles that can modulate the properties of the reaction product as demonstrated for the increased cellular uptake of phosphonamidate-cyclized BCL9 peptides. Finally, the high yielding peptide cyclization technique not only allows facile stapling of peptides but furthermore also facilitates a general macrocyclization strategy as shown for cCPPs. We believe that this tool will enable researchers to develop both novel cysteine reactive probes for applications where a narrow reactivity window is crucial and cyclic peptides with enhanced pharmacological properties that can be manipulated by phosphonamidate ester variation.Conflicts of interest
The authors declare competing financial interests. The technology described in the manuscript is part of a pending patent application by M.-A. K., M. G. and C. P. R. H.Acknowledgements
We thank H. Stephanowitz and Dr Fan Liu for performing tandem MS experiments, K. K. Hassanin for excellent technical assistance and Dr K. Licha for the provision of Cetuximab. This work was supported by grants from the Deutsche Forschungsgemeinschaft (SPP1623) to C. P. R. H. (HA 4468/9-1), the Einstein Foundation Berlin (Leibniz-Humboldt Professorship), the Boehringer-Ingelheim Foundation (Plus 3 award), the Leibniz Association with the Leibniz competition to C. P. R. H. and the Fonds der Chemischen Industrie to C. P. R. H.Notes and references
- S. B. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529–553 CrossRef CAS PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem.–Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- S. O. Doronina, B. E. Toki, M. Y. Torgov, B. A. Mendelsohn, C. G. Cerveny, D. F. Chace, R. L. DeBlanc, R. P. Gearing, T. D. Bovee, C. B. Siegall, J. A. Francisco, A. F. Wahl, D. L. Meyer and P. D. Senter, Nat. Biotechnol., 2003, 21, 778–784 CrossRef CAS PubMed.
- J. Bertran-Vicente, M. Penkert, O. Nieto-Garcia, J.-M. Jeckelmann, P. Schmieder, E. Krause and C. P. R. Hackenberger, Nat. Commun., 2016, 7, 12703 CrossRef CAS PubMed.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- J. E. Moore and W. H. Ward, J. Am. Chem. Soc., 1956, 78, 2414–2418 CrossRef CAS.
- S. Brocchini, S. Balan, A. Godwin, J.-W. Choi, M. Zloh and S. Shaunak, Nat. Protoc., 2006, 1, 2241–2252 CrossRef CAS PubMed.
- B. Bernardim, P. M. S. D. Cal, M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, I. S. Albuquerque, E. Perkins, F. Corzana, A. C. B. Burtoloso, G. Jiménez-Osés and G. J. L. Bernardes, Nat. Commun., 2016, 7, 13128 CrossRef CAS PubMed.
- D. R. Goddard and L. Michaelis, J. Biol. Chem., 1935, 112, 361–371 CAS.
- D. Abegg, R. Frei, L. Cerato, D. Prasad Hari, C. Wang, J. Waser and A. Adibekian, Angew. Chem., 2015, 127, 11002–11007 CrossRef.
- J. Václavík, R. Zschoche, I. Klimánková, V. Matoušek, P. Beier, D. Hilvert and A. Togni, Chem.–Eur. J., 2017, 23, 6490–6494 CrossRef PubMed.
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, Nat. Chem., 2015, 8, 120 CrossRef PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687 CrossRef CAS PubMed.
- M. J. Niphakis and B. F. Cravatt, Annu. Rev. Biochem., 2014, 83, 341–377 CrossRef CAS PubMed.
- R. Lonsdale and R. A. Ward, Chem. Soc. Rev., 2018, 47, 3816–3830 RSC.
- K. T. Barglow and B. F. Cravatt, Chem. Biol., 2004, 11, 1523–1531 CrossRef CAS PubMed.
- M. E. Otero-Ramirez, T. Passioura and H. Suga, Biomedicines, 2018, 6, 116 CrossRef PubMed.
- A. Celine and S. Claudio, Curr. Med. Chem., 2002, 9, 963–978 CrossRef.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- L. J. Walport, R. Obexer and H. Suga, Curr. Opin. Biotechnol., 2017, 48, 242–250 CrossRef CAS PubMed.
- G. Lättig-Tünnemann, M. Prinz, D. Hoffmann, J. Behlke, C. Palm-Apergi, I. Morano, H. D. Herce and M. C. Cardoso, Nat. Commun., 2011, 2, 453 CrossRef PubMed.
- S. Kalhor-Monfared, M. R. Jafari, J. T. Patterson, P. I. Kitov, J. J. Dwyer, J. M. Nuss and R. Derda, Chem. Sci., 2016, 7, 3785–3790 RSC.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502 CrossRef CAS PubMed.
- S. S. Kale, C. Villequey, X.-D. Kong, A. Zorzi, K. Deyle and C. Heinis, Nat. Chem., 2018, 10, 715–723 CrossRef CAS PubMed.
- N. A. Afagh and A. K. Yudin, Angew. Chem., Int. Ed., 2010, 49, 262–310 CrossRef CAS PubMed.
- L. Peng and P. R. Peter, Curr. Top. Med. Chem., 2002, 2, 325–341 CrossRef.
- M. Pelay-Gimeno, T. Bange, S. Hennig and T. N. Grossmann, Angew. Chem., Int. Ed., 2018, 57, 11164–11170 CrossRef CAS PubMed.
- M. R. J. Vallée, P. Majkut, I. Wilkening, C. Weise, G. Müller and C. P. R. Hackenberger, Org. Lett., 2011, 13, 5440–5443 CrossRef PubMed.
- M.-A. Kasper, M. Glanz, A. Stengl, M. Penkert, S. Klenk, T. Sauer, D. Schumacher, J. Helma, E. Krause, M. C. Cardoso, H. Leonhardt and C. Hackenberger, Angew. Chem., Int. Ed. DOI:10.1002/anie.201814715.
- R. Kleineweischede and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2008, 47, 5984–5988 CrossRef CAS PubMed.
- T. Fukuzumi, L. Ju and J. W. Bode, Org. Biomol. Chem., 2012, 10, 5837–5844 RSC.
- H. Y. Lam, Y. Zhang, H. Liu, J. Xu, C. T. T. Wong, C. Xu and X. Li, J. Am. Chem. Soc., 2013, 135, 6272–6279 CrossRef CAS.
- F. Gao, X. Yan and K. Auclair, Chem.–Eur. J., 2009, 15, 2064–2070 CrossRef CAS PubMed.
- K. D. Siebertz and C. P. R. Hackenberger, Chem. Commun., 2018, 54, 763–766 RSC.
- M. R. J. Vallée, L. M. Artner, J. Dernedde and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2013, 52, 9504–9508 CrossRef PubMed.
- F. Saito, H. Noda and J. W. Bode, ACS Chem. Biol., 2015, 10, 1026–1033 CrossRef CAS PubMed.
- Q. Luo, Y. Tao, W. Sheng, J. Lu and H. Wang, Nat. Commun., 2019, 10, 142 CrossRef PubMed.
- J. F. Ponte, X. Sun, N. C. Yoder, N. Fishkin, R. Laleau, J. Coccia, L. Lanieri, M. Bogalhas, L. Wang, S. Wilhelm, W. Widdison, J. Pinkas, T. A. Keating, R. Chari, H. K. Erickson and J. M. Lambert, Bioconjugate Chem., 2016, 27, 1588–1598 CrossRef CAS PubMed.
- M. L. Nielsen, M. Vermeulen, T. Bonaldi, J. Cox, L. Moroder and M. Mann, Nat. Methods, 2008, 5, 459–460 CrossRef CAS PubMed.
- C. F. Brewer and J. P. Riehm, Anal. Biochem., 1967, 18, 248–255 CrossRef CAS.
- K. Takada, D. Zhu, G. H. Bird, K. Sukhdeo, J.-J. Zhao, M. Mani, M. Lemieux, D. E. Carrasco, J. Ryan, D. Horst, M. Fulciniti, N. C. Munshi, W. Xu, A. L. Kung, R. A. Shivdasani, L. D. Walensky and D. R. Carrasco, Sci. Transl. Med., 2012, 4, 148ra117 Search PubMed.
- I. Wilkening, G. del Signore and C. P. R. Hackenberger, Chem. Commun., 2008, 2932–2934 RSC.
- A. Klaus and W. Birchmeier, Nat. Rev. Cancer, 2008, 8, 387 CrossRef CAS.
- G. L. Verdine and G. J. Hilinski, in Methods Enzymol, ed. K. D. Wittrup and G. L. Verdine, Academic Press, 2012, vol. 503, pp. 3–33 Search PubMed.
- K. Sakagami, T. Masuda, K. Kawano and S. Futaki, Mol. Pharm., 2018, 15, 1332–1340 CrossRef CAS PubMed.
- N. Nischan, H. D. Herce, F. Natale, N. Bohlke, N. Budisa, M. C. Cardoso and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2015, 54, 1950–1953 CrossRef CAS PubMed.
- H. D. Herce, D. Schumacher, A. F. L. Schneider, A. K. Ludwig, F. A. Mann, M. Fillies, M.-A. Kasper, S. Reinke, E. Krause, H. Leonhardt, M. C. Cardoso and C. P. R. Hackenberger, Nat. Chem., 2017, 9, 762–771 CrossRef CAS PubMed.
- A. F. L. Schneider, A. L. D. Wallabregue, L. Franz and C. P. R. Hackenberger, Bioconjugate Chem., 2019, 30, 400–404 CrossRef CAS PubMed.
Footnotes |
| † This work is dedicated to Prof. Ron Raines on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01345h |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |