DOI:
10.1039/C9SC00931K
(Edge Article)
Chem. Sci., 2019,
10, 6855-6862
Chemical strategies to modify amyloidogenic peptides using iridium(III) complexes: coordination and photo-induced oxidation†
Received
23rd February 2019
, Accepted 3rd June 2019
First published on 5th June 2019
Abstract
Amyloidogenic peptides are considered central pathological contributors towards neurodegeneration as observed in neurodegenerative disorders [e.g., amyloid-β (Aβ) peptides in Alzheimer's disease (AD)]; however, their roles in the pathologies of such diseases have not been fully elucidated since they are challenging targets to be studied due to their heterogeneous nature and intrinsically disordered structure. Chemical approaches to modify amyloidogenic peptides would be valuable in advancing our molecular-level understanding of their involvement in neurodegeneration. Herein, we report effective chemical strategies for modification of Aβ peptides (i.e., coordination and coordination-/photo-mediated oxidation) implemented by a single Ir(III) complex in a photo-dependent manner. Such peptide variations can be achieved by our rationally designed Ir(III) complexes (Ir-Me, Ir-H, Ir-F, and Ir-F2) leading to significantly modulating the aggregation pathways of two main Aβ isoforms, Aβ40 and Aβ42, as well as the production of toxic Aβ species. Overall, we demonstrate chemical tactics for modification of amyloidogenic peptides in an effective and manageable manner utilizing the coordination capacities and photophysical properties of transition metal complexes.
Introduction
A substantial amount of research effort has been dedicated towards identifying the association of amyloidogenic peptides with the pathologies of neurodegenerative diseases. Among these amyloidogenic peptides, amyloid-β (Aβ), a proteolytic product of the amyloid precursor protein found in the AD-affected brain with a self-aggregation propensity, has been implicated as a pathological factor in Alzheimer's disease (AD).1–4 As the main component of senile plaques, Aβ accumulation is a major pathological feature of AD.1–3,5 Recent developments in Aβ research (e.g., clinical failures of Aβ-directed therapeutics) have led to the re-evaluation of the amyloid cascade hypothesis.6 Aβ pathology, however, remains a pertinent facet of the disease with indications of Aβ oligomers as toxic species responsible for disrupting neuronal homeostasis.1–3,7 Furthering our elucidation of Aβ pathology presents an investigative challenge arising from its heterogeneous nature and intrinsically disordered structure.1,2 To overcome this obstacle and advance our understanding of the Aβ-related contribution towards AD, in this study, we illustrate chemical approaches to modify Aβ peptides at the molecular level using transition metal complexes.
Transition metal complexes have been reported to harness their ability to induce peptide modifications (e.g., hydrolytic cleavage and oxidation), inhibit the activities of enzymes, and image cellular components.8–43 In particular, the ability of transition metal complexes to alter peptides stems from their properties, such as the capacity for peptide coordination.17–29,36,37 Herein, we report effective chemical strategies for modification of Aβ peptides using a single Ir(III) complex in a photo-dependent manner (Fig. 1). Aβ modifications, achieved by our rationally engineered Ir(III) complexes, include two events: (i) complexation with Aβ in the absence of light; (ii) Aβ oxidation upon coordination and photoactivation, which can significantly regulate their aggregation and toxicity. Through our multidisciplinary studies, presented in this work, we demonstrate the development of new chemical tactics for modification of amyloidogenic peptides using transition metal complexes, useful for identifying their properties, such as aggregation, at the molecular level.
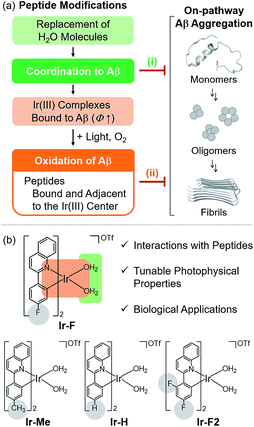 |
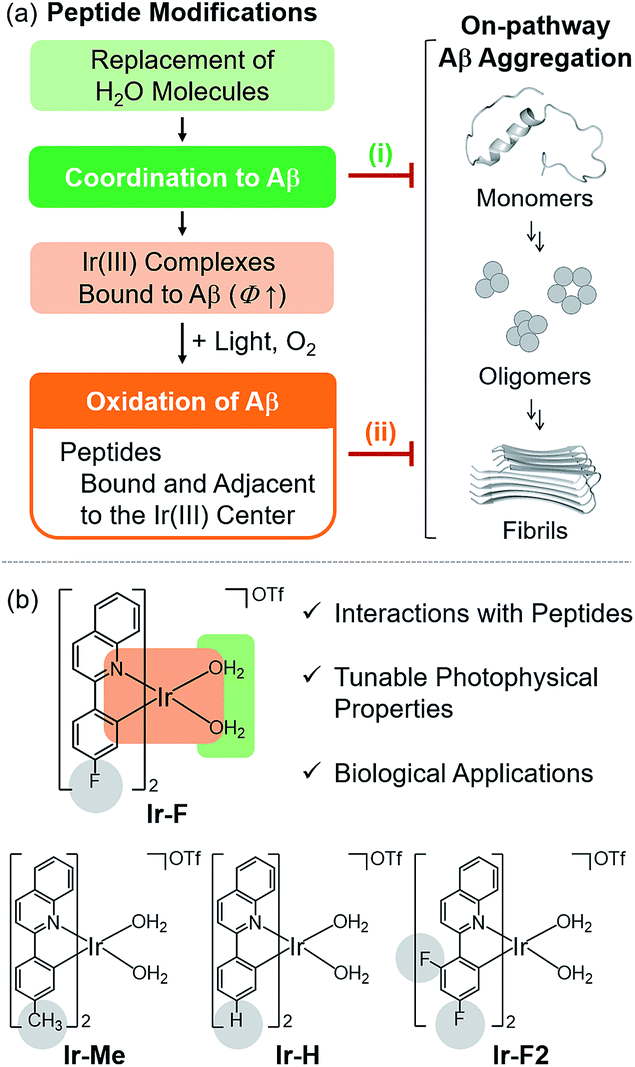
| | Fig. 1 Chemical approaches to modifying Aβ peptides using rationally designed Ir(III) complexes. (a) Two events of modifying Aβ peptides using Ir(III) complexes for controlling of Aβ aggregation: (i) coordination to Aβ peptides and (ii) oxidation of Aβ peptides mediated by coordination and photoactivation (Φ = emission quantum yield). (b) Design criteria and chemical structures of Ir-F, Ir-Me, Ir-H, and Ir-F2. Substituents are highlighted in gray. | |
Results and discussion
Rational strategies for peptide modification using Ir(III) complexes
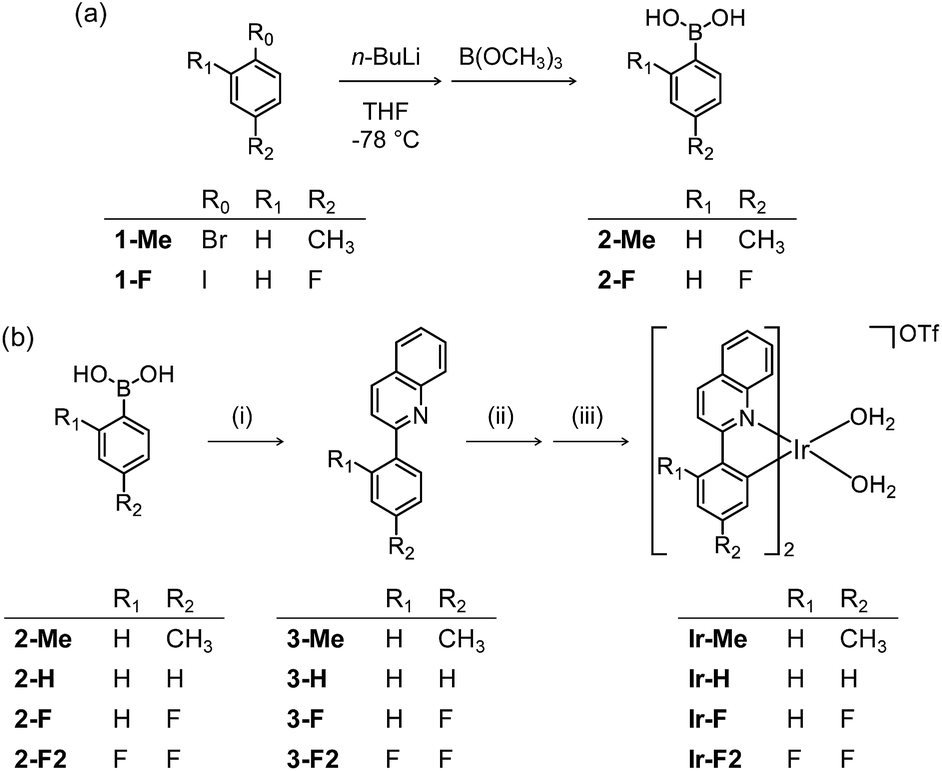
To chemically modify Aβ peptides in a photoirradiation-dependent manner (Fig. 1a), four Ir(III) complexes (Ir-Me, Ir-H, Ir-F, and Ir-F2; Fig. 1b) were rationally designed and prepared. Iridium is a third row transition metal exhibiting strong spin–orbit coupling at the center of Ir(III) complexes with facile electronic transitions.44,45 This spin–orbit coupling can be further strengthened by fine-tuning the ancillary ligands of Ir(III) complexes. As a result, Ir(III) complexes confer notable photophysical properties upon excitation by relatively low energy irradiation in the visible range, including their ability to generate reactive oxygen species [ROS; e.g., singlet oxygen (1O2) and the superoxide anion radical (O2˙−)] via electron or energy transfer.46–48 In addition, Ir(III) complexes with octahedral geometry are relatively stable upon light activation.48 Incorporation of 2-phenylquinoline derivatives as ligands yielded high emission quantum yield (Φ) and robust ROS generation.46 Therefore, ancillary ligands of four complexes were constructed based on the 2-phenylquinoline backbone by applying simple structural variations to provide appropriate structural and electronic environments to promote the photochemical activity of the corresponding Ir(III) complexes.46 Moreover, fluorine atoms were introduced into the ancillary ligand framework affording Ir-F and Ir-F2 to chemically impart the ability to interact with Aβ through hydrogen bonding, alter photophysical properties of the complexes, and enhance the molecules' biocompatibility.49–51 Two water (H2O) molecules were incorporated as ligands to enable covalent coordination to Aβ via replacement with amino acid residues of the peptide, e.g., histidine (His).20,52,53 The four Ir(III) complexes were synthesized following previously reported procedures with modifications (Scheme 1 and Fig. S1–S3†).20,54–56 As depicted in Fig. S4 and S5,† these Ir(III) complexes were confirmed to coordinate to His or Aβ in both H2O and an organic solvent [i.e., dimethyl sulfoxide (DMSO)] under our experimental conditions.
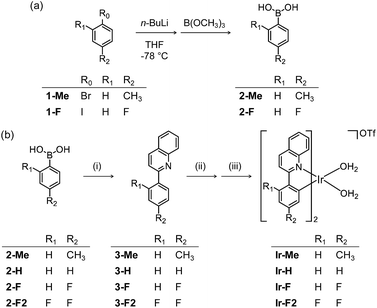 |
| | Scheme 1 Synthetic routes to Ir(III) complexes. Reagents and conditions: (i) 2-chloroquinoline, Pd(PPh3)4, THF/K2CO3 (aq), Δ; (ii) IrCl3·nH2O, 2-methoxyethanol/H2O, Δ; (iii) AgOTf, CH3OH/CH2Cl2. | |
Coordination-dependent photophysical properties and ROS production of Ir(III) complexes
Photophysical properties of the prepared Ir(III) complexes were investigated by UV-vis and fluorescence spectroscopy. As shown in Table 1 and Fig. S6,† in the absence of His or Aβ, low Φ values of the four Ir(III) complexes were observed, along with relatively poor 1O2 generation with photoactivation. Note that a solar simulator (Newport IQE-200) was used to irradiate the samples at a constant intensity (1 sun light; 100 mW cm−2). Upon addition of His, the Φ values of the four Ir(III) complexes drastically increased (e.g., ΦIr-F = 0.0071 versus ΦIr-F+His = 0.26; Table 1), indicating His coordination of the complexes, which was further confirmed by electrospray ionization-mass spectrometry (ESI-MS) (Fig. S5a†). The Φ values and 1O2 formation of the four Ir(III) complexes with His binding exhibited trends similar to their binding affinity with His (Ir-F > Ir-H > Ir-Me > Ir-F2). Ir-F, indicating the strongest binding affinity with His (Fig. S5b†), among the four Ir(III) complexes, showed notable binding affinities towards different Aβ species (for monomers, Kd = 1.6 × 10−4 M; for oligomers, Kd = 2.6 × 10−4 M; for fibrils, Kd = 7.1 × 10−4 M; Fig. S7†). Ir-F, exhibiting a relatively high value of Φ upon His binding, also produced significant amounts of 1O2 and O2˙− in the presence of His with photoactivation (Fig. S6 and S8†). Based on these properties, we selected Ir-F as a representative candidate of our Ir(III) complexes and illustrated its ability to modify Aβ peptides in detail (vide infra).
Table 1 Photophysical properties of Ir(III) complexes
|
|
Ir-Me
|
Ir-H
|
Ir-F
|
Ir-F2
|
| −His |
+His |
−His |
+His |
−His |
+His |
−His |
+His |
|
λ
ex,max (nm), (ε, ×103 M−1 cm−1) |
280 (±27), 336 (±14), 449 (±3) |
274 (±29), 343 (±16), 449 (±4) |
275 (±29), 339 (±15), 452 (±3) |
268 (±32), 338 (±15), 445 (±3) |
274 (±34), 336 (±17), 446 (±4) |
269 (±33), 335 (±16), 433 (±4) |
277 (±36), 350 (±21), 441 (±7) |
272 (±37), 348 (±19), 432 (±6) |
|
|
|
λ
em,max (nm) |
587 |
592 |
587 |
593 |
589 |
573 |
573 |
578 |
|
|
|
Φ
|
0.0038 (±0.0007) |
0.19 (±0.01) |
0.0037 (±0.0006) |
0.31 (±0.03) |
0.0071 (±0.001) |
0.26 (±0.03) |
0.0027 (±0.0002) |
0.081 (±0.007) |
|
|
|
τ (ns) |
5.8 (±1.8) |
601 (±20) |
11 (±1) |
619 (±61) |
4.8 (±2.0) |
810 (±23) |
4.4 (±0.4) |
484 (±41) |
|
|
|
k
r (×105 s−1) |
6.5 |
3.2 |
3.3 |
5.0 |
15 |
3.3 |
6.1 |
1.7 |
|
|
|
k
nr (×105 s−1) |
1.7 × 103 |
13 |
0.88 × 103 |
11 |
2.1 × 103 |
9.1 |
2.2 × 103 |
19 |
Photoirradiation-dependent peptide modification using Ir(III) complexes
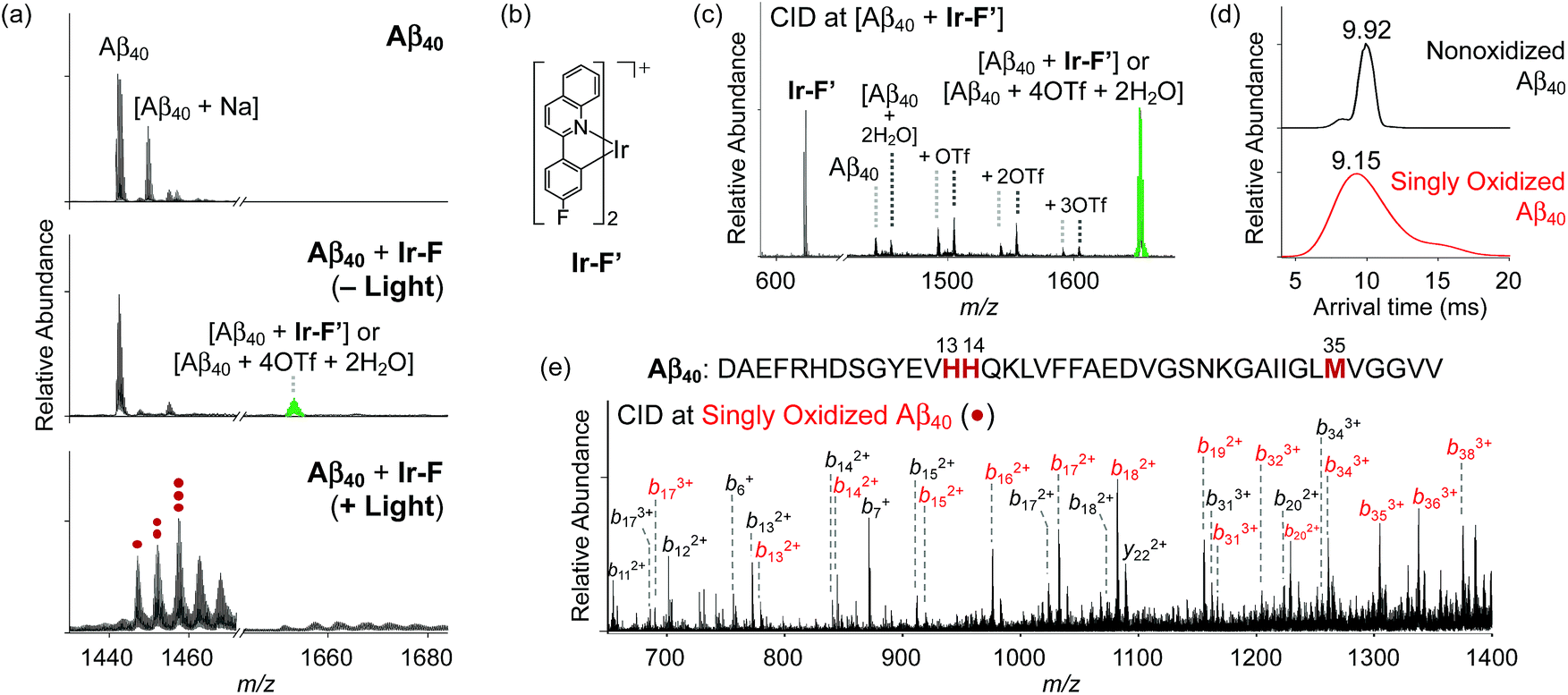
Modification of Aβ peptides upon treatment with Ir-F was monitored via mass spectrometric techniques [i.e., ESI-MS, ESI-MS2, and ion mobility-mass spectrometry (IM-MS)]. The ESI-MS analysis of Ir-F-treated Aβ samples revealed the complex formation between Aβ and Ir-F′ [the Ir-F form that does not have two H2O molecules bound to the Ir(III) center; Fig. 2b] in the absence of light as an indication at 1653 m/z (Fig. 2a, middle; green). To identify the molecular species corresponding to 1653 m/z, the peak was further analyzed via ESI-MS2 in conjunction with collision-induced dissociation (CID; Fig. 2c). The detected ion fragments exhibited m/z values attributed to Aβ40 and Ir-F′. Therefore, our MS results demonstrate the complexation between Aβ and Ir-F with the loss of two H2O molecules from the Ir(III) center (Ir-F′; Fig. 2b). Note that the m/z value of the Ir-F′–Aβ40 complex is equal to that of [Aβ40 + 4OTf + 2H2O]; thus, we cannot rule out the co-existence of the complex and an OTf adduct.
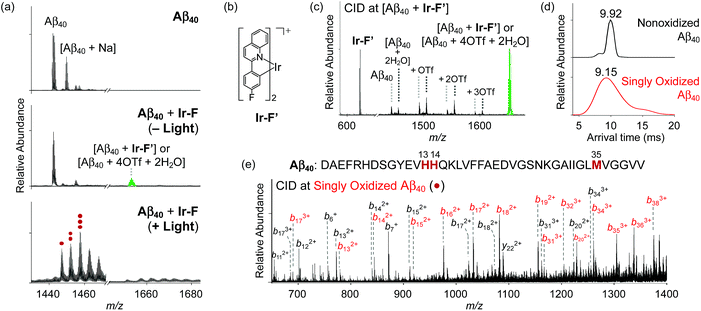 |
| | Fig. 2 Analysis of Aβ40 species generated upon treatment with Ir-F. (a) ESI-MS spectra of Ir-F-incubated +3-charged Aβ40 with and without light. The peak indicated in green corresponds to a complex of Aβ40 and Ir-F′ [structure shown in (b)]. The peaks corresponding to oxidized Aβ40 species are indicated with red dots. The number of red dots represents the number of oxygen atoms incorporated into Aβ40. (c) Collision-induced dissociation (CID) spectrum at 1653 m/z [green peak from (a)]. (d) Arrival time distributions (ATDs) between nonoxidized and singly oxidized Aβ40 monomers. (e) Sequence of Aβ40 and CID spectrum of the singly oxidized Aβ40 found in (a). Monooxidized b fragments are denoted in red. Charges are omitted in the MS spectra. Conditions: [Aβ40] = 100 μM; [Ir-F] = 500 μM; 37 °C; 1 h; no agitation; 1 sun light for 10 min (for the samples treated with light); aerobic conditions. | |
Upon photoirradiation, the ESI-MS analysis of Ir-F-treated Aβ40 samples led to the detection of oxidized Aβ40 (Fig. 2a, bottom). Aβ40 oxidation manifested a conformational change as probed by IM-MS (Fig. 2d). The most dominant arrival time indicated a peak at 9.92 ms. These results suggest that Aβ40 oxidation induced by Ir-F can alter the structural distribution of Aβ40. Similar observations were observed with Ir-Me, Ir-H, and Ir-F2, where the complexes were able to oxidize Aβ40 and consequently vary its structural distribution (Fig. S9 and S10†). In order to determine the location of peptide oxidation, the Aβ fragment ions, generated by selectively applying collisional energy to singly oxidized Aβ, were analyzed by ESI-MS2 (Fig. 2e). All b fragments smaller than b13 were detected in their nonoxidized forms, while those larger than b34 were only monitored in their oxidized forms. The b fragments between b13 and b34 were indicated in both their oxidized and nonoxidized forms. Such observations, along with previous reports regarding Aβ oxidation,19,57 suggest His13, His14, and Met35 of Aβ as plausible oxidation sites. Collectively, our studies demonstrate that Aβ peptides can be modified upon treatment with Ir-F [(i) coordination to Aβ by replacing two H2O molecules with the peptide in the absence of light; (ii) coordination-mediated oxidation of Aβ at three possible amino acid residues (e.g., His13, His14, and Met35) upon photoactivation (Fig. 1a)]. Note that the Aβ samples produced by treatment of photoactivated Ir-F showed high fluorescence intensity and were relatively stable in both H2O and cell growth media (Fig. S11†).
Effects of peptide modifications triggered by Ir(III) complexes on Aβ aggregation
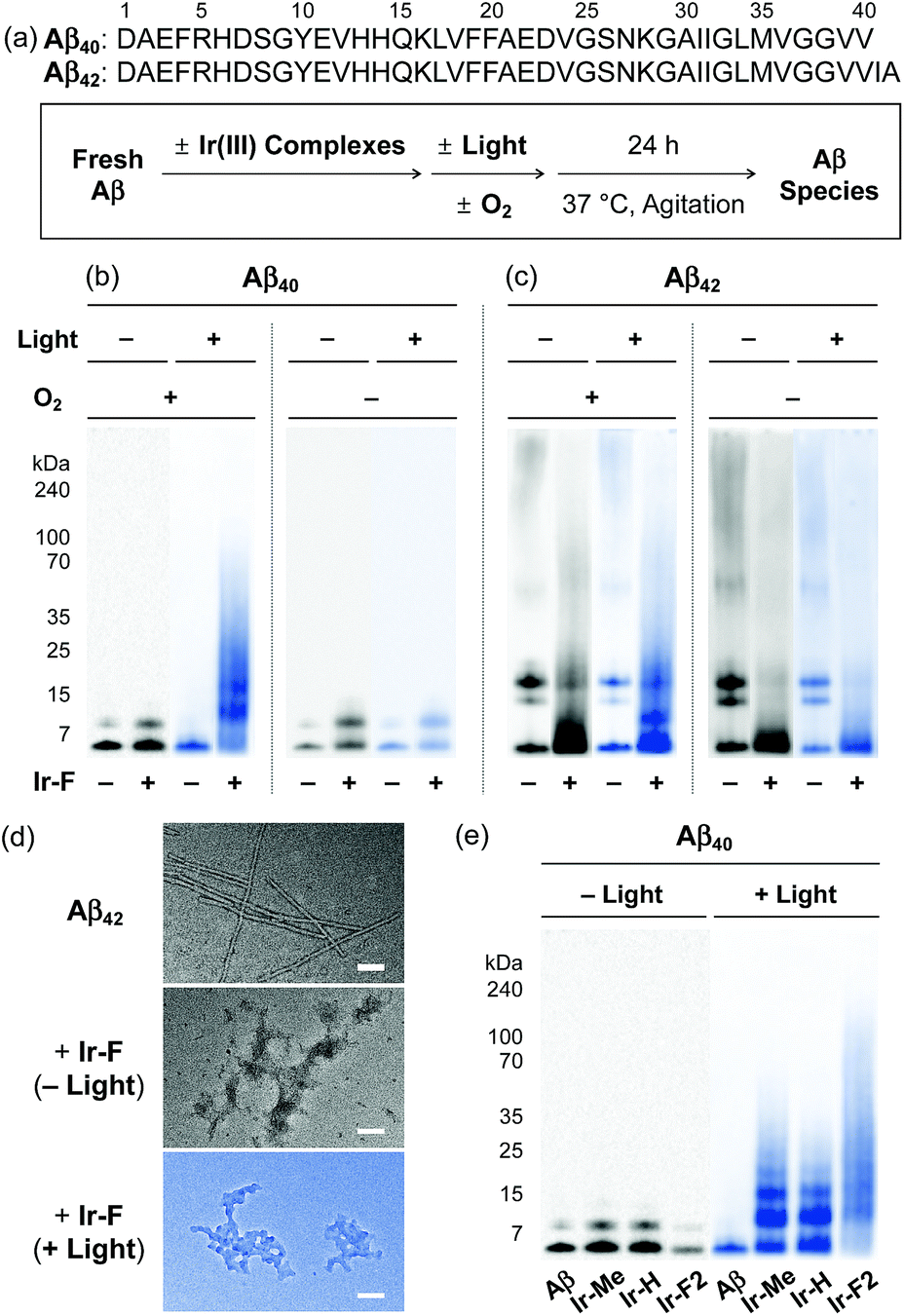
Based on the photoirradiation-dependent Aβ modifications by Ir(III) complexes, the impact of such variations on the aggregation of Aβ was determined employing Aβ40 and Aβ42, two main Aβ isoforms found in the AD-affected brain.2–4,58–62 For these experiments, freshly prepared Aβ solutions were treated with Ir(III) complexes with and without light under both aerobic and anaerobic conditions. The molecular weight (MW) distribution and the morphology of resultant Aβ species were analyzed by gel electrophoresis with Western blotting (gel/Western blot) using an anti-Aβ antibody (6E10) and transmission electron microscopy (TEM), respectively (Fig. 3a).
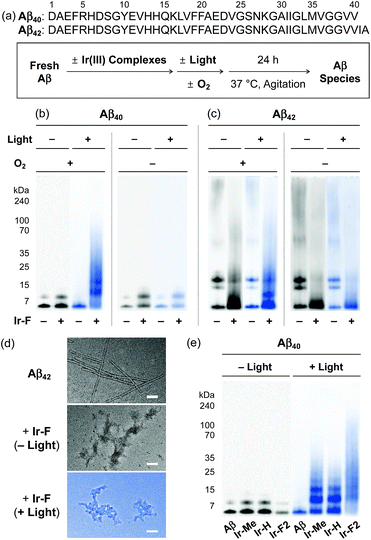 |
| | Fig. 3 Change in the formation of Aβ aggregates by incubation with Ir(III) complexes. (a) Sequences of Aβ40 and Aβ42 and scheme of the inhibition experiments. (b, c, and e) Analysis of the resultant Aβ40 and Aβ42 species generated under various conditions by gel/Western blot with an anti-Aβ antibody (6E10). (d) TEM images of the resultant Aβ42 aggregates (scale bar = 100 nm). Conditions: [Aβ] = 25 μM; [Ir(III) complexes] = 250 μM; 37 °C; 24 h; constant agitation; 1 sun light for 10 min (for the samples treated with light). | |
Under aerobic conditions (Fig. 3b, left), the aggregation of Aβ40 was affected by treatment with Ir-F prompting a shift in the MW distributions in the absence of light. Photoactivation of the Ir-F-treated Aβ40 sample resulted in a more diverse MW distribution compared to that of the corresponding sample without light (light, MW ≤ 100 kDa; no light, MW < 15 kDa). The distinct modulation of Aβ40 aggregation upon addition of Ir-F with photoirradiation is likely a consequence of the complex's ability to generate 1O2 and oxidize Aβ through photoactivation as observed in our spectrometric studies (vide supra; Fig. 2). Therefore, the same experiments were performed under anaerobic conditions to directly monitor the role of O2 in Ir-F′s modulative reactivity against Aβ40 aggregation. In the absence of O2 (Fig. 3b, right), Aβ40 aggregation was also altered by Ir-F regardless of light treatment. Our results suggest that both light and O2 are important in the regulation of Aβ40 aggregation through coordination-/photo-mediated peptide oxidation triggered by Ir-F. In addition, in the absence of light and O2, Aβ40 aggregation is directed by the covalent interactions between Ir-F and the peptide. Similar modulation of Aβ42 aggregation was observed upon incubation with Ir-F exhibiting different MW distributions compared to the Aβ42 samples without Ir-F in the absence and presence of light and O2 (Fig. 3c). Moreover, smaller amorphous aggregates of both Aβ40 and Aβ42, reported to be less toxic,63,64 were visualized by TEM from the samples containing Ir-F regardless of irradiation (Fig. 3d and S12c†).
Furthermore, preformed Aβ aggregates, generated at various preincubation time points (i.e., 2, 4, and 24 h), were disassembled and their aggregation pathways were altered when Ir-F was introduced (Fig. S13†). Such Ir-F-induced effects on preformed Aβ aggregates were observed to be dependent on photoirradiation. Moreover, the aggregation of both Aβ40 and Aβ42 was also changed with addition of the other Ir(III) complexes (i.e., Ir-Me, Ir-H, and Ir-F2) with and without light (Fig. 3e, S12 and S13†). In addition to Aβ, Ir-F was able to interact with and modify other amyloidogenic peptides [i.e., α-synuclein (α-Syn) and human islet amyloid polypeptide (hIAPP)] affecting their aggregation pathways (Fig. S14†).
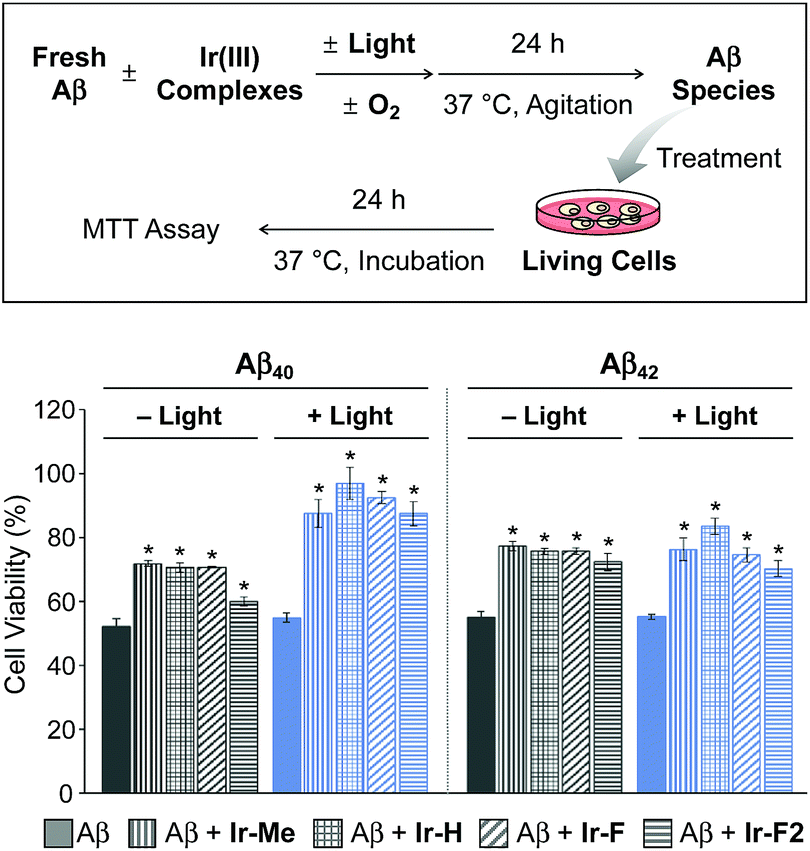
Cytotoxicity of Aβ species generated upon incubation with Ir(III) complexes
Prior to cytotoxicity measurements, the resultant species upon 24 h treatment of Aβ40 with Ir-F with light exposure were incubated with murine Neuro-2a (N2a) neuroblastoma cells in order to determine their cellular uptake. As depicted in Fig. S15,† the lysates of the cells added with the resultant species for 24 h, analyzed by inductively coupled plasma-mass spectrometry (ICP-MS), indicated an Ir concentration of 39 μg L−1, demonstrating the cellular uptake of the species containing Ir(III). Note that the Ir concentration (0.17 and 34 μg L−1) was measured from the lysates of the cells treated only with either Aβ40 or Ir-F, respectively. Moving forward, the toxicity of Aβ species produced by treatment with our Ir(III) complexes was monitored by the MTT assay [MTT = 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] (Fig. 4). The cytotoxicity of Aβ40 species incubated with our Ir(III) complexes was noticeably reduced in a photoirradiation-dependent manner. In the absence of light, the Aβ40 samples incubated with our Ir(III) complexes exhibited a decrease in cytotoxicity (ca. 20%) compared to the sample of the complex-free Aβ40. As for the photoirradiated samples, Aβ40-induced toxicity was lowered by ca. 35% by treatment with our Ir(III) complexes. This result suggests that modification of Aβ, such as oxidation, could attenuate Aβ-triggered toxicity in living cells.65 Furthermore, the cytotoxicity of Aβ42 species formed with Ir(III) complexes was also diminished by ca. 20% regardless of photoactivation. Note that the survival (≥80%) of cells treated with our Ir(III) complexes at the concentration used for cell studies with Aβ peptides was observed with and without light exposure (Fig. S16†).
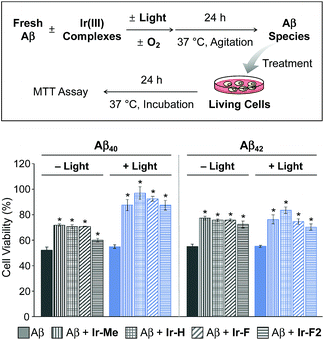 |
| | Fig. 4 Viability of N2a cells upon 24 h treatment with Aβ species produced by incubation with Ir(III) complexes for 24 h with and without light activation. Cell viability (%), measured by the MTT assay, was calculated and compared with that of samples treated with an equivalent amount of DMSO only. Conditions (final concentration): [Aβ] = 20 μM; [Ir(III) complexes] = 5 μM. Error bars represent the standard error of the mean from three independent experiments. *P < 0.05. | |
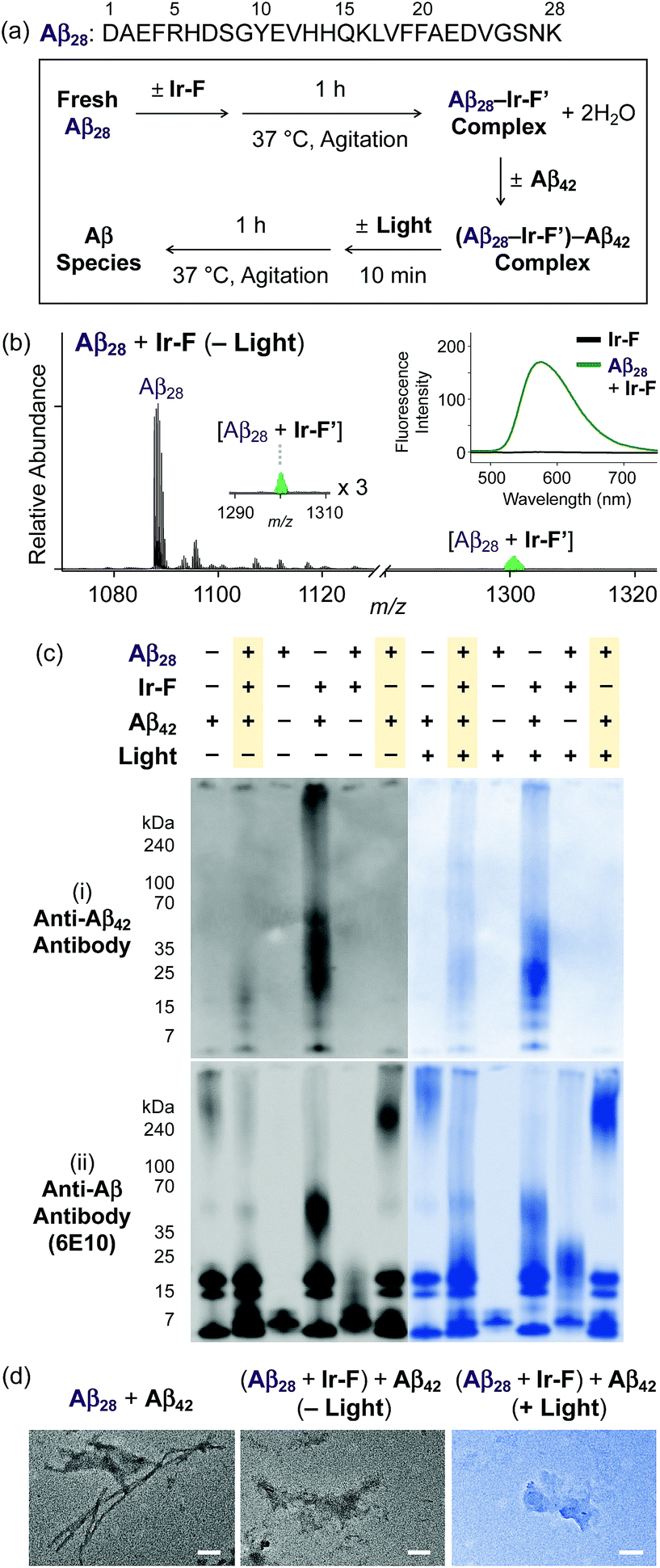
Ternary complexation with Aβ and intramolecular and intermolecular Aβ oxidation
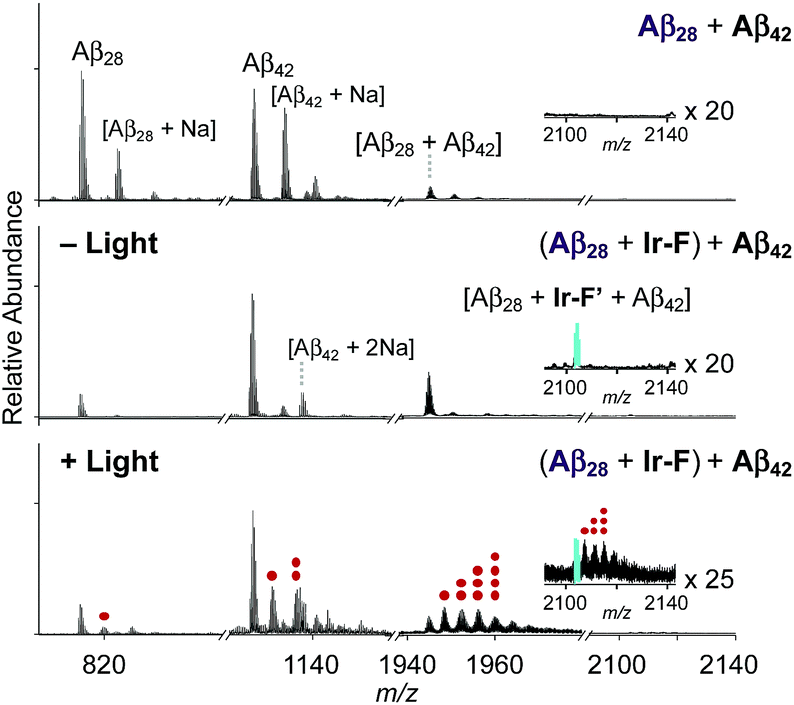
Premised on Ir-F′s covalent bond formation with Aβ and oxidation of Aβ (vide supra), additional studies regarding ternary complexation and promotion of intermolecular oxidation of Aβ were carried out employing Ir-F (Fig. 5). Aβ28, a fragment of Aβ equipped with the metal binding and self-recognition sites of the peptide with a relatively low propensity to aggregate than the full-length peptides, Aβ40 and Aβ42,1,66–68 was used to form a complex with Ir-F′ (Fig. 2b) as evidenced by ESI-MS (1301 m/z; Fig. 5b) and increased fluorescence (Fig. 5b, inset). As shown in Fig. 5a, following incubation, the sample of the Aβ28–Ir-F′ complex was treated with freshly prepared Aβ42 to monitor its effect on Aβ42 aggregation. Based on the gel/Western blot and TEM analyses, the aggregation of Aβ42 was modulated by the Aβ28–Ir-F′ complex (Fig. 5c and d). Such modulative reactivity of the Aβ28–Ir-F′ complex was also observed against Aβ40 aggregation (Fig. S17†). Our mass spectrometric studies confirmed that such control of Aβ42 aggregation by the Aβ28–Ir-F′ complex was a result of ternary complex formation with Aβ42, i.e., (Aβ28–Ir-F′)–Aβ42, and (ii) oxidation of Aβ, both intramolecular and intermolecular, upon photoactivation (Fig. 6). Based on previous reports detailing intermolecular interactions between Aβ peptides, hydrophobic interactions between the self-recognition sites (LVFFA; Fig. 3a and 5a) of Aβ are likely responsible for ternary complexation,1,2,69 consequentially altering the aggregation pathways of Aβ in the absence of photoirradiation. Furthermore, these studies indicate that intermolecular oxidation of Aβ can be promoted by Ir-F upon photoactivation (Fig. 6, S18, and S19†). This observation may explain the distinct difference between the modulation of Aβ aggregation with and without light as the intermolecular oxidation of Aβ by Ir(III) complexes could modify Aβ at sub-stoichiometric levels.
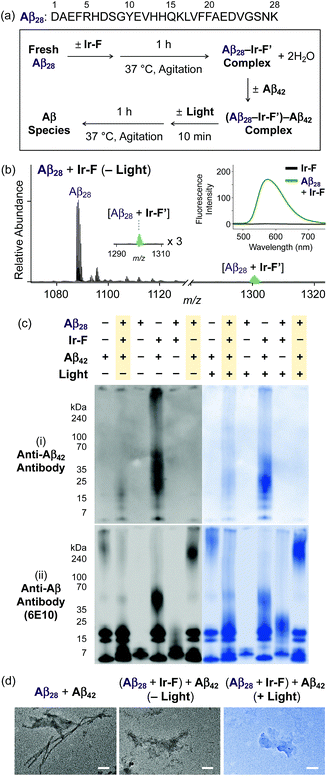 |
| | Fig. 5 Impact of Ir-F-preincubated Aβ28 on the aggregation of Aβ42. (a) Sequence of Aβ28 and scheme of the experiments. (b) ESI-MS spectrum of +3-charged Aβ28 upon incubation with Ir-F. The complex peak is indicated in green. (Inset) Fluorescence response of Ir-F to Aβ28 (λex = 433 nm). Charges are omitted in the MS spectra. Conditions: [Aβ28] = 100 μM; [Ir-F] = 100 μM; 37 °C; 1 h; no agitation; 1 sun light for 10 min (for the samples treated with light); aerobic conditions. (c) Analysis of the resultant Aβ species, obtained by addition of Aβ42 into Ir-F preincubated with Aβ28, by gel/Western blot with (i) anti-Aβ42 and (ii) anti-Aβ (6E10) antibodies. (d) TEM images of the resultant Aβ aggregates (scale bar = 100 nm). Conditions: [Aβ28] = 50 μM; [Ir-F] = 10 μM; [Aβ42] = 20 μM; 37 °C; 2 h; constant agitation; 1 sun light for 10 min (for the samples treated with light); aerobic conditions. | |
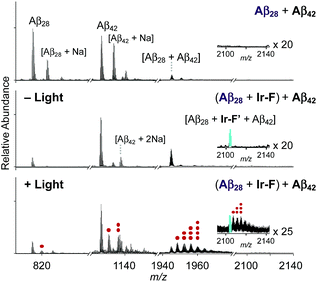 |
| | Fig. 6 ESI-MS spectra of Ir-F-incubated +4-charged Aβ28 and Aβ42 with and without light. The peak indicated in cyan refers to a ternary complex of Aβ28, Ir-F′, and Aβ42. The peaks corresponding to oxidized peptides (i.e., Aβ28, Aβ42, and Aβ28 with Aβ42) are indicated with red dots. The number of red dots represents the number of oxygen atoms incorporated into each peptide. Charges are omitted in the MS spectra. Conditions: [Aβ28] = 100 μM; [Ir-F] = 100 μM; [Aβ42] = 100 μM; 37 °C; 2 h; no agitation; 1 sun light for 10 min (for the samples treated with light); aerobic conditions. | |
Conclusions
Effective chemical strategies (i.e., coordination to Aβ and coordination-/photo-mediated oxidation of Aβ) for modification of Aβ peptides using a single Ir(III) complex were rationally developed. Such dual mechanisms (i.e., coordination and oxidation) exhibiting photo-dependency for altering Aβ peptides are novel and effective in controlling peptide aggregation and cytotoxicity. Our Ir(III) complexes can covalently bind to Aβ by replacing two H2O molecules bound to the Ir(III) center with Aβ regardless of light and O2 [coordination to Aβ; Fig. 1a(i)]. In the presence of light and O2, Ir(III) complexes bound to Aβ are capable of inducing the intramolecular and intermolecular oxidation of Aβ at His13, His14, and/or Met35 [oxidation of Aβ; Fig. 1a(ii)]. Taken together, our multidisciplinary studies demonstrate the feasibility of establishing new chemical approaches towards modification of amyloidogenic peptides (e.g., Aβ) using transition metal complexes designed based on their coordination and photophysical properties. In general, chemical modifications in peptides of interest can assist in furthering our understanding of principles of their properties, such as peptide assembly. Furthermore, peptide aggregation and cytotoxicity can be affected by biomolecules, including lipid membranes;70–73 thus, the regulatory reactivity of Ir(III) complexes towards amyloidogenic peptides in the presence of lipid membranes will be investigated in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government [NRF-2017R1A2B3002585 and NRF-2016R1A5A1009405 (to M. H. L.); NRF-2016R1A2B4009239 (to. T.-H. K.)]. J. K. acknowledges the Global Ph.D. Fellowship Program through the NRF funded by the Ministry of Education (NRF-2015HIA2A1030823). J. S. N. is grateful for the support from the ASAN Foundation Biomedical Science Scholarship. We thank Dr Shin Jung C. Lee, Yonghwan Ji, Eunju Nam, Mingeun Kim, and Jong-Min Suh for the help with MS and TEM measurements of Aβ, α-Syn, and hIAPP.
Notes and references
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221–1322 CrossRef CAS PubMed.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed.
- K. P. Kepp, Coord. Chem. Rev., 2017, 351, 127–159 CrossRef CAS.
- R. Jakob-Roetne and H. Jacobsen, Angew. Chem., Int. Ed., 2009, 48, 3030–3059 CrossRef CAS PubMed.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CrossRef CAS PubMed.
- G. P. Morris, I. A. Clark and B. Vissel, Acta Neuropathol., 2018, 136, 663–689 CrossRef CAS PubMed.
- S. J. C. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Chem. Soc. Rev., 2017, 46, 310–323 RSC.
- A. Zamora, G. Vigueras, V. Rodríguez, M. D. Santana and J. Ruiz, Coord. Chem. Rev., 2018, 360, 34–76 CrossRef CAS.
- K. L. Haas and K. J. Franz, Chem. Rev., 2009, 109, 4921–4960 CrossRef CAS PubMed.
- R. Wai-Yin Sun, D.-L. Ma, E. L.-M. Wong and C.-M. Che, Dalton Trans., 2007, 4884–4892 RSC.
- C.-H. Leung, H.-J. Zhong, D. S.-H. Chan and D.-L. Ma, Coord. Chem. Rev., 2013, 257, 1764–1776 CrossRef CAS.
- C.-H. Leung, S. Lin, H.-J. Zhong and D.-L. Ma, Chem. Sci., 2015, 6, 871–884 RSC.
- D. A. Fancy and T. Kodadek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6020–6024 CrossRef CAS PubMed.
- V. B. Kenche, L. W. Hung, K. Perez, I. Volitakes, G. Ciccotosto, J. Kwok, N. Critch, N. Sherratt, M. Cortes, V. Lal, C. L. Masters, K. Murakami, R. Cappai, P. A. Adlard and K. J. Barnham, Angew. Chem., Int. Ed., 2013, 52, 3374–3378 CrossRef CAS PubMed.
- W. Zhen, H. Han, M. Anguiano, C. A. Lemere, C.-G. Cho and P. T. Lansbury, J. Med. Chem., 1999, 42, 2805–2815 CrossRef CAS PubMed.
- G. S. Yellol, J. G. Yellol, V. B. Kenche, X. M. Liu, K. J. Barnham, A. Donaire, C. Janiak and J. Ruiz, Inorg. Chem., 2015, 54, 470–475 CrossRef CAS PubMed.
- G. Son, B. I. Lee, Y. J. Chung and C. B. Park, Acta Biomater., 2018, 67, 147–155 CrossRef CAS PubMed.
- L. He, X. Wang, D. Zhu, C. Zhao and W. Du, Metallomics, 2015, 7, 1562–1572 RSC.
- J. Kang, S. J. C. Lee, J. S. Nam, H. J. Lee, M.-G. Kang, K. J. Korshavn, H.-T. Kim, J. Cho, A. Ramamoorthy, H.-W. Rhee, T.-H. Kwon and M. H. Lim, Chem.–Eur. J., 2017, 23, 1645–1653 CrossRef CAS PubMed.
- B. Y.-W. Man, H.-M. Chan, C.-H. Leung, D. S.-H. Chan, L.-P. Bai, Z.-H. Jiang, H.-W. Li and D.-L. Ma, Chem. Sci., 2011, 2, 917–921 RSC.
- D. J. Hayne, S. Lim and P. S. Donnelly, Chem. Soc. Rev., 2014, 43, 6701–6715 RSC.
- H. Liu, Y. Qu and X. Wang, Future Med. Chem., 2018, 10, 679–701 CrossRef CAS PubMed.
- K. J. Barnham, V. B. Kenche, G. D. Ciccotosto, D. P. Smith, D. J. Tew, X. Liu, K. Perez, G. A. Cranston, T. J. Johanssen, I. Volitakis, A. I. Bush, C. L. Masters, A. R. White, J. P. Smith, R. A. Cherny and R. Cappai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6813–6818 CrossRef CAS PubMed.
- B. I. Lee, S. Lee, Y. S. Suh, J. S. Lee, A.-k. Kim, O.-Y. Kwon, K. Yu and C. B. Park, Angew. Chem., Int. Ed., 2015, 54, 11472–11476 CrossRef CAS PubMed.
- A. Aliyan, T. J. Paul, B. Jiang, C. Pennington, G. Sharma, R. Prabhakar and A. A. Martí, Chem, 2017, 3, 898–912 CAS.
- L. Messori, M. Camarri, T. Ferraro, C. Gabbiani and D. Franceschini, ACS Med. Chem. Lett., 2013, 4, 329–332 CrossRef CAS PubMed.
- X. Wang, X. Wang, C. Zhang, Y. Jiao and Z. Guo, Chem. Sci., 2012, 3, 1304–1312 RSC.
- A. Kumar, L. Moody, J. F. Olaivar, N. A. Lewis, R. L. Khade, A. A. Holder, Y. Zhang and V. Rangachari, ACS Chem. Neurosci., 2010, 1, 691–701 CrossRef CAS PubMed.
- J.-M. Suh, G. Kim, J. Kang and M. H. Lim, Inorg. Chem., 2019, 58, 8–17 CrossRef CAS PubMed.
- C.-Y. Wong, L.-H. Chung, L. Lu, M. Wang, B. He, L.-J. Liu, C.-H. Leung and D.-L. Ma, Curr. Alzheimer Res., 2015, 12, 439–444 CrossRef CAS PubMed.
- L. Lu, H.-J. Zhong, M. Wang, S.-L. Ho, H.-W. Li, C.-H. Leung and D.-L. Ma, Sci. Rep., 2015, 5, 14619 CrossRef CAS PubMed.
- J. Suh, S. H. Yoo, M. G. Kim, K. Jeong, J. Y. Ahn, M.-s. Kim, P. S. Chae, T. Y. Lee, J. Lee, J. Lee, Y. A. Jang and E. H. Ko, Angew. Chem., Int. Ed., 2007, 46, 7064–7067 CrossRef CAS PubMed.
- J. S. Derrick, J. Lee, S. J. C. Lee, Y. Kim, E. Nam, H. Tak, J. Kang, M. Lee, S. H. Kim, K. Park, J. Cho and M. H. Lim, J. Am. Chem. Soc., 2017, 139, 2234–2244 CrossRef CAS PubMed.
- W.-h. Wu, P. Lei, Q. Liu, J. Hu, A. P. Gunn, M.-s. Chen, Y.-f. Rui, X.-y. Su, Z.-p. Xie, Y.-F. Zhao, A. I. Bush and Y.-m. Li, J. Biol. Chem., 2008, 283, 31657–31664 CrossRef CAS PubMed.
- P. S. Donnelly, A. Caragounis, T. Du, K. M. Laughton, I. Volitakis, R. A. Cherny, R. A. Sharples, A. F. Hill, Q.-X. Li, C. L. Masters, K. J. Barnham and A. R. White, J. Biol. Chem., 2008, 283, 4568–4577 CrossRef CAS PubMed.
- I. Sasaki, C. Bijani, S. Ladeira, V. Bourdon, P. Faller and C. Hureau, Dalton Trans., 2012, 41, 6404–6407 RSC.
- D. Valensin, P. Anzini, E. Gaggelli, N. Gaggelli, G. Tamasi, R. Cini, C. Gabbiani, E. Michelucci, L. Messori, H. Kozlowski and G. Valensin, Inorg. Chem., 2010, 49, 4720–4722 CrossRef CAS PubMed.
- L.-J. Liu, W. Wang, S.-Y. Huang, Y. Hong, G. Li, S. Lin, J. Tian, Z. Cai, H.-M. D. Wang, D.-L. Ma and C.-H. Leung, Chem. Sci., 2017, 8, 4756–4763 RSC.
- K. Vellaisamy, G. Li, C.-N. Ko, H.-J. Zhong, S. Fatima, H.-Y. Kwan, C.-Y. Wong, W.-J. Kwong, W. Tan, C.-H. Leung and D.-L. Ma, Chem. Sci., 2018, 9, 1119–1125 RSC.
- V. Novohradsky, A. Zamora, A. Gandioso, V. Brabec, J. Ruiz and V. Marchán, Chem. Commun., 2017, 53, 5523–5526 RSC.
- F.-X. Wang, M.-H. Chen, Y.-N. Lin, H. Zhang, C.-P. Tan, L.-N. Ji and Z.-W. Mao, ACS Appl. Mater. Interfaces, 2017, 9, 42471–42481 CrossRef CAS PubMed.
- V. Novohradsky, A. Rovira, C. Hally, A. Galindo, G. Vigueras, A. Gandioso, M. Svitelova, R. Bresolí-Obach, H. Kostrhunova, L. Markova, J. Kasparkova, S. Nonell, J. Ruiz, V. Brabec and V. Marchán, Angew. Chem., Int. Ed., 2019, 58, 6311–6315 CrossRef CAS PubMed.
- D. Yu, Y. Guan, F. Bai, Z. Du, N. Gao, J. Ren and X. Qu, Chem.–Eur. J., 2019, 25, 3489–3495 CrossRef CAS PubMed.
- Y. You, Curr. Opin. Chem. Biol., 2013, 17, 699–707 CrossRef CAS PubMed.
- M. S. Lowry and S. Bernhard, Chem.–Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed.
- J. S. Nam, M.-G. Kang, J. Kang, S.-Y. Park, S. J. C. Lee, H.-T. Kim, J. K. Seo, O.-H. Kwon, M. H. Lim, H.-W. Rhee and T.-H. Kwon, J. Am. Chem. Soc., 2016, 138, 10968–10977 CrossRef CAS PubMed.
- R. Gao, D. G. Ho, B. Hernandez, M. Selke, D. Murphy, P. I. Djurovich and M. E. Thompson, J. Am. Chem. Soc., 2002, 124, 14828–14829 CrossRef CAS.
- Z. Liu and P. J. Sadler, Acc. Chem. Res., 2014, 47, 1174–1185 CrossRef CAS PubMed.
- A. Wragg, M. R. Gill, D. Turton, H. Adams, T. M. Roseveare, C. Smythe, X. Su and J. A. Thomas, Chem.–Eur. J., 2014, 20, 14004–14011 CrossRef CAS PubMed.
- J. Zhao, Y. Yu, X. Yang, X. Yan, H. Zhang, X. Xu, G. Zhou, Z. Wu, Y. Ren and W.-Y. Wong, ACS Appl. Mater. Interfaces, 2015, 7, 24703–24714 CrossRef CAS PubMed.
- F. S. Etheridge, R. J. Fernando, S. Pejić, M. Zeller and G. Sauvé, Beilstein J. Org. Chem., 2016, 12, 1925–1938 CrossRef CAS.
- D.-L. Ma, W.-L. Wong, W.-H. Chung, F.-Y. Chan, P.-K. So, T.-S. Lai, Z.-Y. Zhou, Y.-C. Leung and K.-Y. Wong, Angew. Chem., Int. Ed., 2008, 47, 3735–3739 CrossRef CAS PubMed.
- D.-L. Ma, H.-J. Zhong, W.-C. Fu, D. S.-H. Chan, H.-Y. Kwan, W.-F. Fong, L.-H. Chung, C.-Y. Wong and C.-H. Leung, PLoS One, 2013, 8, e55751 CrossRef CAS PubMed.
- N. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 767–768 CrossRef.
- B. Schmid, F. O. Garces and R. J. Watts, Inorg. Chem., 1994, 33, 9–14 CrossRef CAS.
- N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2008, 130, 210–217 CrossRef CAS PubMed.
- J. Han, H. J. Lee, K. Y. Kim, S. J. C. Lee, J.-M. Suh, J. Cho, J. Chae and M. H. Lim, ACS Chem. Neurosci., 2018, 9, 800–808 CrossRef CAS PubMed.
- G. Bitan, B. Tarus, S. S. Vollers, H. A. Lashuel, M. M. Condron, J. E. Straub and D. B. Teplow, J. Am. Chem. Soc., 2003, 125, 15359–15365 CrossRef CAS PubMed.
- L. Hou, I. Kang, R. E. Marchant and M. G. Zagorski, J. Biol. Chem., 2002, 277, 40173–40176 CrossRef CAS PubMed.
- S.-M. Liao, Q.-S. Du, J.-Z. Meng, Z.-W. Pang and R.-B. Huang, Chem. Cent. J., 2013, 7, 44 CrossRef PubMed.
- K. Brännström, T. Islam, L. Sandblad and A. Olofsson, FEBS Lett., 2017, 591, 1167–1175 CrossRef.
- V. Kadlcik, C. Sicard-Roselli, C. Houée-Levin, M. Kodicek, C. Ferreri and C. Chatgilialoglu, Angew. Chem., Int. Ed., 2006, 45, 2595–2598 CrossRef CAS PubMed.
- J. Bieschke, J. Russ, R. P. Friedrich, D. E. Ehrnhoefer, H. Wobst, K. Neugebauer and E. E. Wanker, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7710–7715 CrossRef CAS PubMed.
- S.-J. Hyung, A. S. DeToma, J. R. Brender, S. Lee, S. Vivekanandan, A. Kochi, J.-S. Choi, A. Ramamoorthy, B. T. Ruotolo and M. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3743–3748 CrossRef CAS PubMed.
- A. Taniguchi, Y. Shimizu, K. Oisaki, Y. Sohma and M. Kanai, Nat. Chem., 2016, 8, 974–982 CrossRef CAS.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Redox Biol., 2018, 14, 450–464 CrossRef CAS PubMed.
- M. A. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836–1846 RSC.
- B. Klajnert, T. Wasiak, M. Ionov, M. Fernandez-Villamarin, A. Sousa-Herves, J. Correa, R. Riguera and E. Fernandez-Megia, Nanomedicine, 2012, 8, 1372–1378 CrossRef CAS PubMed.
- M. G. Savelieff, S. Lee, Y. Liu and M. H. Lim, ACS Chem. Biol., 2013, 8, 856–865 CrossRef CAS PubMed.
- S. A. Kotler, P. Walsh, J. R. Brender and A. Ramamoorthy, Chem. Soc. Rev., 2014, 43, 6692–6700 RSC.
- M. F. M. Sciacca, S. A. Kotler, J. R. Brender, J. Chen, D.-k. Lee and A. Ramamoorthy, Biophys. J., 2012, 103, 702–710 CrossRef CAS PubMed.
- K. J. Korshavn, A. Bhunia, M. H. Lim and A. Ramamoorthy, Chem. Commun., 2016, 52, 882–885 RSC.
- K. J. Korshavn, C. Satriano, Y. Lin, R. Zhang, M. Dulchavsky, A. Bhunia, M. I. Ivanova, Y.-H. Lee, C. La Rosa, M. H. Lim and A. Ramamoorthy, J. Biol. Chem., 2017, 292, 4638–4650 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and Fig. S1–S19. See DOI: 10.1039/c9sc00931k |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a