DOI:
10.1039/C9SC00876D
(Edge Article)
Chem. Sci., 2019,
10, 5582-5588
Metal vs. ligand protonation and the alleged proton-shuttling role of the azadithiolate ligand in catalytic H2 formation with FeFe hydrogenase model complexes†
Received
20th February 2019
, Accepted 30th April 2019
First published on 2nd May 2019
Abstract
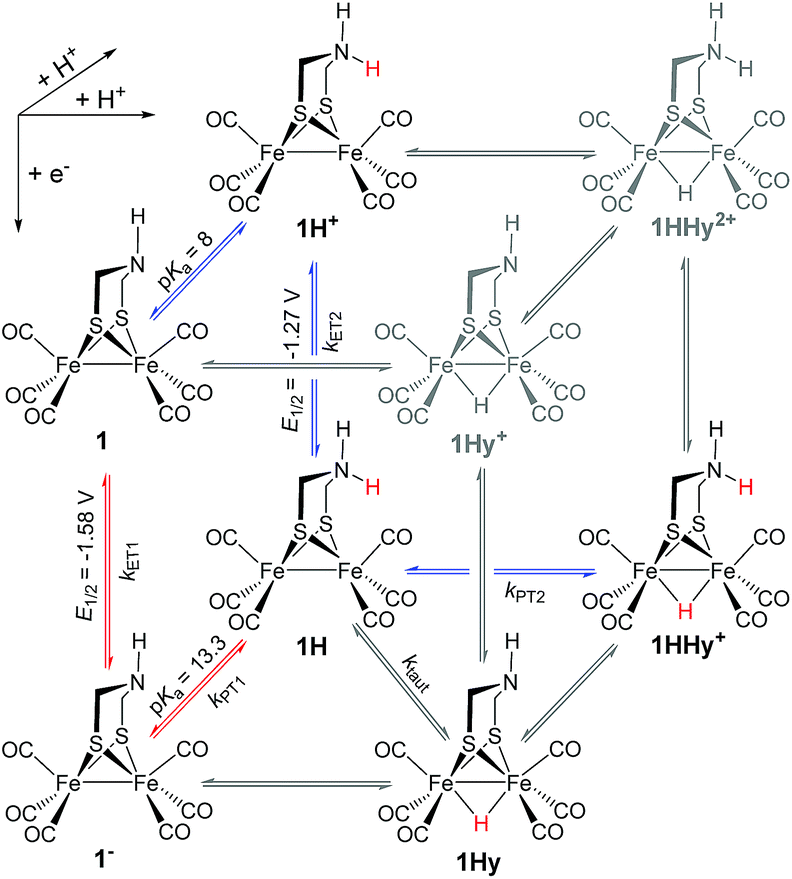
Electron and proton transfer reactions of diiron complexes [Fe2adt(CO)6] (1) and [Fe2adt(CO)4(PMe3)2] (4), with the biomimetic azadithiolate (adt) bridging ligand, have been investigated by real-time IR- and UV-vis-spectroscopic observation to elucidate the role of the adt-N as a potential proton shuttle in catalytic H2 formation. Protonation of the one-electron reduced complex, 1−, occurs on the adt-N yielding 1H and the same species is obtained by one-electron reduction of 1H+. The preference for ligand vs. metal protonation in the Fe2(I,0) state is presumably kinetic but no evidence for tautomerization of 1H to the hydride 1Hy was observed. This shows that the adt ligand does not work as a proton relay in the formation of hydride intermediates in the reduced catalyst. A hydride intermediate 1HHy+ is formed only by protonation of 1H with stronger acid. Adt protonation results in reduction of the catalyst at much less negative potential, but subsequent protonation of the metal centers is not slowed down, as would be expected according to the decrease in basicity. Thus, the adtH+ complex retains a high turnover frequency at the lowered overpotential. Instead of proton shuttling, we propose that this gain in catalytic performance compared to the propyldithiolate analogue might be rationalized in terms of lower reorganization energy for hydride formation with bulk acid upon adt protonation.
Introduction
Proton binding sites in the second coordination sphere of catalytic metal centres are widely considered as an important design principle for facilitating proton coupled electron transfer in molecular catalysts for e.g. water splitting,1–5 H2 formation and activation6–12 or CO2 reduction.13–21 For the directed design of synthetic catalysts it is, however, important to note that improvements to catalytic performance (TOF, overpotential) brought about by basic sites in the second coordination sphere are not necessarily arising from a proton relay activity. Instead, the modulation of redox and acid/base properties of the metal centre upon changes in protonation state in the second coordination sphere may be the actual origin of improved catalytic performance.14–17
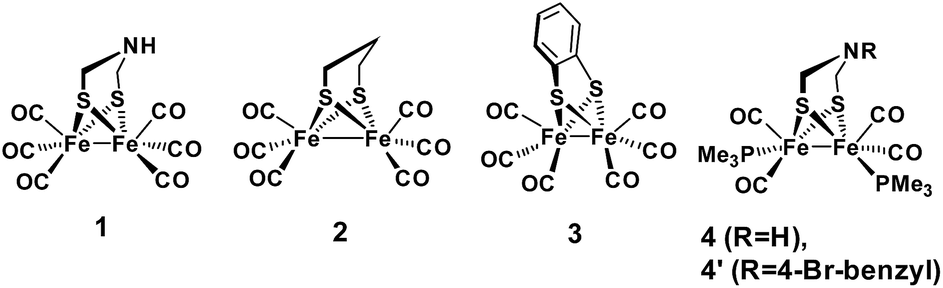
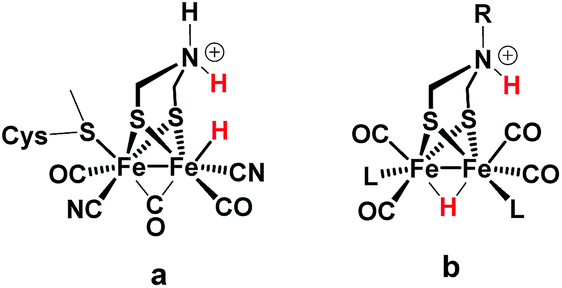
Regarding catalysts for H2 formation, the N2P2 ligands introduced by DuBois and co-workers,6,7,12 and the azaditiolate (adt) bridging ligand in diiron complexes of the general formula Fe2adt(CO)4X2 (X = e.g. CO, R3P) modelled after the FeFe-Hase active site,9,22–24 are prominent examples of ligand motifs with basic sites in the second coordination sphere. The amine function of the adt ligand is believed to assist enzymatic H2 formation and activation by shuttling of protons to or from the distal iron centre of the active site where the crucial terminal hydride intermediate is formed (Scheme 1a).25,26 A similar role of the adt ligand in catalysis by the synthetic models considered here seems less likely given the preferred bridging coordination of the hydride (Scheme 1b).23 The superior catalytic performance of Fe2(adt)(CO)6, 1, over its propyldithiolate (pdt) analogue 2 in electrochemical H2 formation has however been attributed to proton shuttling, involving specifically terminal protonation of the metal centre by tautomerization of a adt-NH+ precursor upon one-electron reduction of the catalyst.27 We were therefore intrigued whether the proposed role of the adt ligand as proton shuttle and the formation of terminal hydride intermediates in these model complexes could be inferred from direct spectroscopic observation. For this purpose we have combined laser flash induced reduction as well as rapid chemical reduction of the catalyst with both UV-vis and IR detection to elucidate structure and reactivity of reduced and reduced-protonated intermediates derived from 1 (ref. 28) and Fe2(adt)(CO)4(PMe3)2 (4).29 This approach enabled us to exclude any proton shuttling role of the adt bridging ligand in catalytic H2 formation while the kinetics of ligand and metal protonation steps suggest an alternative rationale for the improved performance.
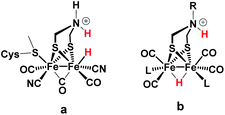 |
| | Scheme 1 FeFe Hase active site (a) and synthetic model (b) bearing a hydride and a proton. | |
Results and discussion
Generally, complex 1 catalyses H2 formation via an initial ET-PT sequence with weaker acids, or a PT-ET sequence when stronger acids are employed that initially protonate the adt-N with a pKa of 8 in acetonitrile30 (Scheme 2, potentials from ref. 27).
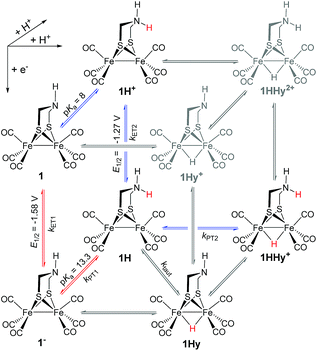 |
| | Scheme 2 Metal and ligand protonation reactions of 1 (Fe2(I,I)) and 1− (Fe2(I,0)). | |
Reduction of 1
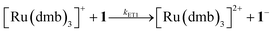
To investigate the ET-PT route, the one-electron reduced catalyst 1− was obtained by rapid electron transfer from laser flash generated [Ru(dmb)3]+ according to eqn (1) and (2) (kET1 = 3 × 109 M−1 s−1, eqn (2)). The electronic absorption bands of 1− (570, 700 nm, see ESI†) and the carbonyl region of its IR spectrum (1915, 1945, 2005 cm−1, Fig. 1a) resemble closely the spectra reported for the singly reduced pdt analogue 2−.31,32 The 700 nm band and the uniform shift of the three νC–O bands by 80–90 cm−1 towards lower wavenumbers are distinct markers of the intact (μ2,κ2-adt)Fe2(CO)6 core in the reduced complex 1−, in contrast to the dissociation of a sulphur–iron bond in the reduced bdt analogue (bdt = benzene-1,2-dithiolate) 3−.33 In the absence of proton sources, decay of 1− leads to complete recovery of 1 by diffusion controlled charge recombination with the electron donor radical TTF+ (krec,1 = 6 × 1010 M−1 s−1, eqn (3)).| | 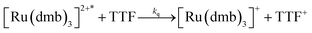 | (1) |
| |  | (2) |
| |  | (3) |
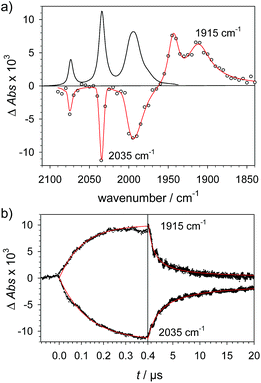 |
| | Fig. 1 (a) Transient IR spectrum 1 μs after excitation (○,  ) and normalized IR spectrum of the starting state ( ) and normalized IR spectrum of the starting state (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) for the reduction of 1 (1.3 mM) by flash-quench generated [Ru(dmb)3]+ in acetonitrile. (b) Kinetic traces (⋯) and fits ( ) for the reduction of 1 (1.3 mM) by flash-quench generated [Ru(dmb)3]+ in acetonitrile. (b) Kinetic traces (⋯) and fits ( ) monitoring the pseudo-first order formation of 1− (1915 cm−1) and bleach of 1 (2035 cm−1) followed by second order charge recombination with TTF+. ) monitoring the pseudo-first order formation of 1− (1915 cm−1) and bleach of 1 (2035 cm−1) followed by second order charge recombination with TTF+. | |
Metal vs. ligand protonation of 1−
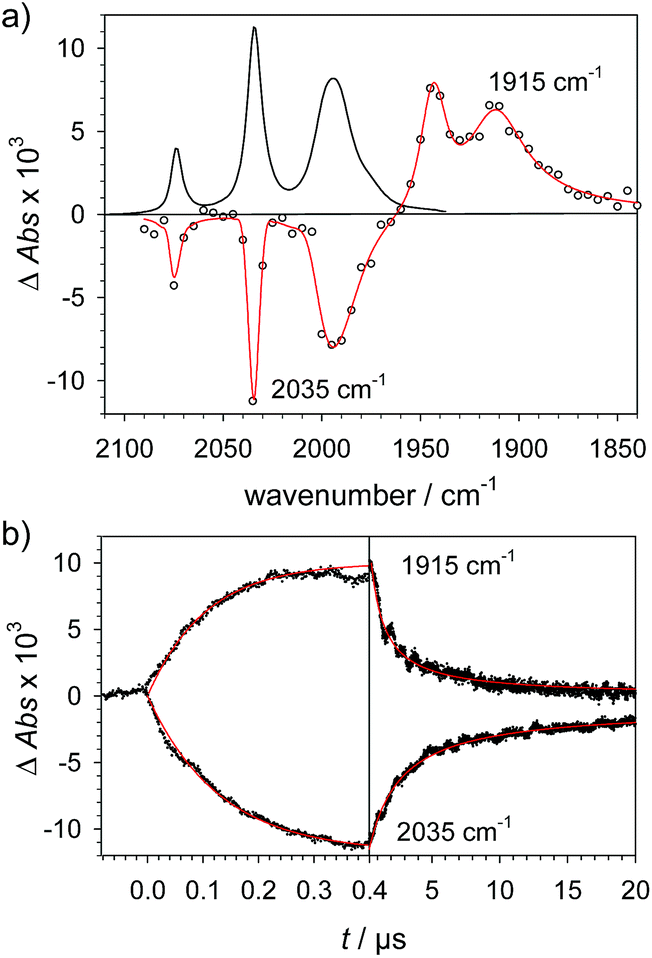
Protonation of the laser flash generated 1− (eqn (4)) with weak acids (Cl3CCOOH, pKa = 10.6 or ClCH2COOH, pKa = 15.3),34 which avoids protonation of the neutral parent complex, shifts all three νC–O bands by about 20 cm−1 to higher wavenumbers (2025, 1965 and 1935 cm−1), as shown in Fig. 2. The magnitude of the shift is similar to what is observed upon protonation of the neutral parent complex with stronger acids (cf.Fig. 3a) and characteristic for protonation of the adt-N.29| |  | (4) |
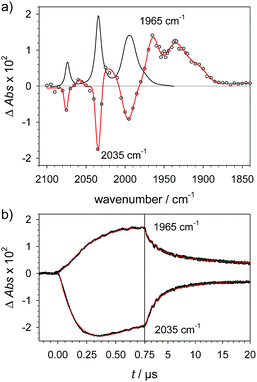 |
| | Fig. 2 (a) Transient IR spectrum 1 μs after excitation (○,  ) and normalized IR spectrum of the starting state () for formation of 1H by protonation of 1− with Cl3CCOOH (13 mM) following reduction of 1 (1.3 mM) by flash-quench generated [Ru(dmb)3]+ in acetonitrile. (b) Kinetic traces (⋯) and fits ( ) and normalized IR spectrum of the starting state () for formation of 1H by protonation of 1− with Cl3CCOOH (13 mM) following reduction of 1 (1.3 mM) by flash-quench generated [Ru(dmb)3]+ in acetonitrile. (b) Kinetic traces (⋯) and fits ( ) monitoring pseudo-first order formation of 1H (1965 cm−1) and bleach of 1 (2035 cm−1) followed by second order charge recombination with TTF+. ) monitoring pseudo-first order formation of 1H (1965 cm−1) and bleach of 1 (2035 cm−1) followed by second order charge recombination with TTF+. | |
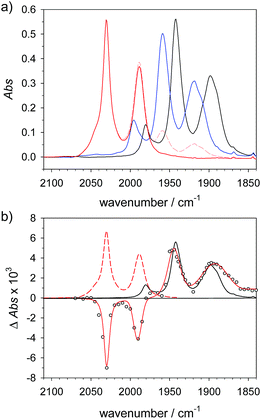 |
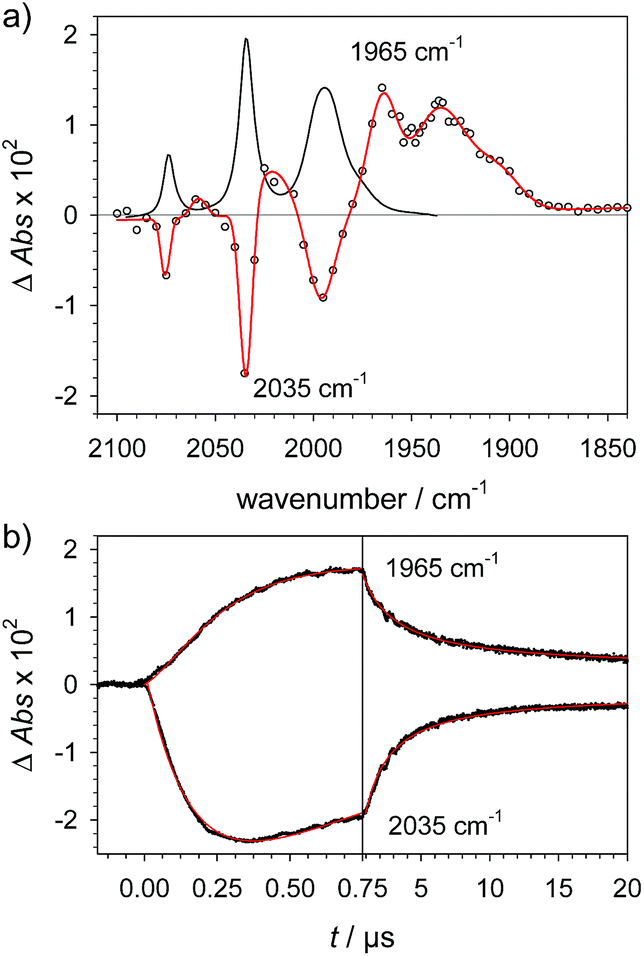
| | Fig. 3 (a) FTIR spectra of 4 (1.5 mM) () in acetonitrile, 4H+ ( ) obtained by protonation with toluenesulfonic acid (3 mM), and 4Hy+ ( ) obtained by protonation with toluenesulfonic acid (3 mM), and 4Hy+ ( ) obtained by protonation with toluenesulfonic acid (3 mM) in presence of tetrabutyl-ammonium chloride (1.5 mM) after correction of the raw spectrum ( ) obtained by protonation with toluenesulfonic acid (3 mM) in presence of tetrabutyl-ammonium chloride (1.5 mM) after correction of the raw spectrum ( ) for minor contribution from 4H+. (b) Transient IR spectrum upon reduction of 4Hy+ to 4Hy by electron transfer from flash-quench generated [Ru(dmb)3]+ (○, ) for minor contribution from 4H+. (b) Transient IR spectrum upon reduction of 4Hy+ to 4Hy by electron transfer from flash-quench generated [Ru(dmb)3]+ (○,  , 1 μs after excitation) and normalized IR spectra of 4Hy+ ( , 1 μs after excitation) and normalized IR spectra of 4Hy+ ( ) and 4 (). ) and 4 (). | |
Metal protonation of 1− is on the other hand expected to shift the carbonyl bands by about 80 cm−1 to higher wavenumbers. Shifts of this magnitude have been established for the bridging hydrides formed by protonation of Fe2(I,I) complexes with electron donor ligands like 4′ (ref. 35) as well as transiently generated Fe2(I,0) hexacarbonyl complexes 2− and 3−.32,33 Mutually cancelling shifts of the carbonyl bands upon one-electron reduction and metal protonation were further evidenced with help of complex 4 (vide infra). The transient IR spectrum of the protonation product hence proves that the adt ligand remains the preferred site of protonation, at least kinetically, also upon one-electron reduction of 1. This conclusion is corroborated by the UV-vis spectrum of 1H (ESI†), which is similar to its precursor 1−, while metal protonation of 2− was previously shown to cause complete bleach of its visible absorption bands that arise from transitions involving predominantly metal-based orbitals. The observed pseudo-first order rate constants for formation of 1H follow a linear dependence on acid concentration before becoming limited by the preceding electron transfer step (ESI†). The straightforward concentration dependence excludes any complications of the protonation kinetics due to dimerization of the acetic acids that was previously found to result in quadratic concentration dependence corresponding to an increase in effective acid strength.36 Interestingly, protonation of 1−, which occurs unambiguously at the adt-N, proceeds remarkably slowly with bimolecular rate constants kPT1 of 4 × 108 M−1 s−1 and 2 × 107 M−1 s−1 for Cl3CCOOH and ClH2CCOOH, respectively.
Notably, the protonation rate constant is almost two orders of magnitude below the diffusion controlled limit even for the strongly exergonic reaction with Cl3CCOOH (ΔpKa = 3). This is quite untypical for protonation of an amine base and indicative of significant reorganization associated with ligand protonation.
Spectral comparison against the one-electron reduced hydride 4Hy
The above assignment of the protonation reaction to ligand protonation builds to a large extent on the general notion of nearly perfectly cancelling shifts in carbonyl frequencies caused by one-electron reduction and formation of a bridging hydride. This expectation could previously only be supported by the vanishing transient absorption observed upon protonation of complexes 2− and 3−.32,33 Here, these assignments could be eventually verified by the positive observation of a one-electron reduced hydride 4Hy by laser flash induced reduction of hydride complex 4Hy+. The latter can be obtained by the Cl−-catalysed protonation of 4 that yields the thermodynamically stable hydride instead of the kinetically preferred ligand protonated 4H+ in analogy to the previously reported behaviour of 4′.37 Upon one electron reduction of 4Hy+ the carbonyl bands of the product 4Hy coincide almost perfectly with those of the parent complex 4 (Fig. 3) verifying the close spectral resemblance of the diiron carbonyl complexes and their bridging hydride derivatives in the Fe2(I,0) oxidation state.
Probing possible tautomerization of 1H
According to the above arguments, also formation of a possibly more stable hydride 1Hy by tautomerization (5) should lead to complete decay of all transient absorption given the predictable spectral similarity between 1 and 1Hy.| |  | (5) |
While the expected spectral changes are the same as for charge recombination between 1H and TTF+(6), tautomerization should nevertheless accelerate the decay of 1H if it proceeds with a rate that is at least comparable to charge recombination (6).
| | 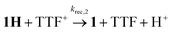 | (6) |
In flash photolysis experiments, 1H decays with second order kinetics on a time scale of about 10 μs in transient IR and about 100 μs with the lower transient concentrations in UV-vis experiments. These time scales are typical for diffusion controlled recombination (krec,2 = 1.5 × 1010 M−1 s−1) and put an upper limit on the order of 104 s−1 on the rate constant of a possibly competing tautomerization reaction.
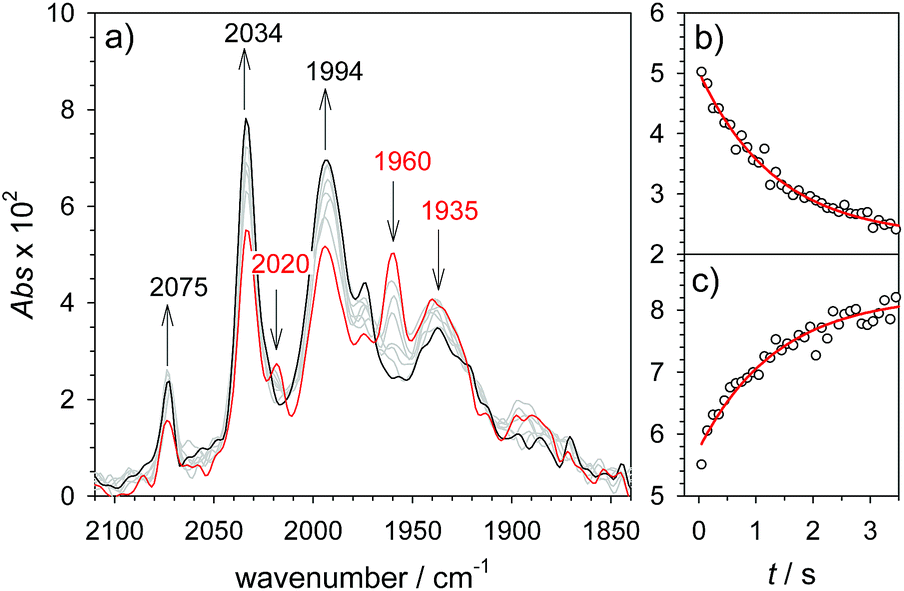
As a turnover frequency (TOF) of 103 to 104 s−1 has been proposed from electrocatalytic experiments (log(kcat/s−1) = 3.9 and 2.6 for 6 mM Cl3CCOOH and ClH2CCOOH, respectively),27 we wanted to probe the reactivity of 1H on a longer time scale than 100 μs. Thus, 1H+ was chemically reduced by cobaltocene in IR stopped flow experiments (rapid-scan FTIR). 1H+ was obtained by addition of two equivalents of 2,5-dichlorobenzenesulfonic acid (Cl2BSA, pKa = 6.7)34 to warrant quantitative protonation of 1. Reduction to 1H occurred on a shorter time scale than the time resolution of the experiment. Importantly, 1H was observed for several seconds (Fig. 4). Its decay with a lifetime of 1.3 s yields a product spectrum that can be assigned to the parent complex 1 or the tautomerization product 1Hy given the expected similarity of their spectra. The former could be generated by catalytic turnover that leads to depletion of acid and therefore regenerates unreduced, unprotonated catalyst. Whether the slow decay of 1H is predominantly due to turnover or tautomerization cannot be discriminated but the observed lifetime demonstrates in either case that the rate constant for tautomerization, if this occurs at all, has to be smaller than 1 s−1. We note that tautomerization would be an intramolecular process that is independent on external conditions, such as identity and concentration of acid and the method of reduction (chemical or electrochemical).
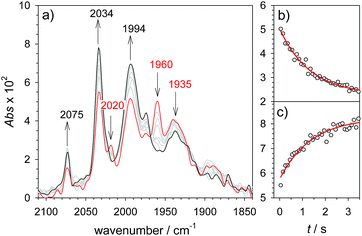 |
| | Fig. 4 Rapid scan FTIR spectra in acetonitrile between 50 ms ( ) and 3 s () after after mixing of 1H+ (1.3 mM 1, 2.6 mM Cl2BSA) with cobaltocene (20 mM) (a) and kinetic traces (○) with exponential fits ( ) and 3 s () after after mixing of 1H+ (1.3 mM 1, 2.6 mM Cl2BSA) with cobaltocene (20 mM) (a) and kinetic traces (○) with exponential fits ( ) monitoring decay of 1H at 1960 cm−1 (b) and formation of 1 at 2034 cm−1 (c). ) monitoring decay of 1H at 1960 cm−1 (b) and formation of 1 at 2034 cm−1 (c). | |
Reduction and subsequent protonation of 1H+
Alternative formation of 1Hvia the PT-ET route was studied by laser-flash induced reduction of 1H+(7).| | 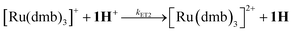 | (7) |
1H+ was obtained by protonation of 1 with Cl2BSA that shifts the IR peaks by about 20 cm−1 to higher wavenumbers (2090, 2051 and 2015 cm−1).29 The transient IR absorption spectrum after reduction of 1H+ (Fig. 5) shows the bleaching of all three νC–O peaks of 1H+ together with the same product absorption peaks previously attributed to 1H (2025, 1965, 1935 cm−1). While the ET-PT and PT-ET sequences lead expectedly to the same product 1H, its decay is much faster when generated in presence of the stronger acid required for the initial ligand protonation. The accelerated decay of 1H (Fig. 5) without appearance of new transient signals can be assigned to its metal protonation yielding 1HHy+(8).
| | 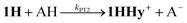 | (8) |
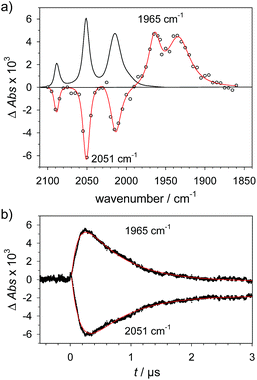 |
| | Fig. 5 (a) Transient IR spectrum 0.3 μs after excitation (○,  ) and normalized IR spectrum of the starting state () for formation of 1H by reduction of 1H+ (obtained by protonation of 1 with 2.6 mM Cl2BSA) with flash-quench generated [Ru(dmb)3]+ in acetonitrile. (b) Kinetic traces (⋯) and fits ( ) and normalized IR spectrum of the starting state () for formation of 1H by reduction of 1H+ (obtained by protonation of 1 with 2.6 mM Cl2BSA) with flash-quench generated [Ru(dmb)3]+ in acetonitrile. (b) Kinetic traces (⋯) and fits ( ) monitoring pseudo-first order formation of 1H (1965 cm−1) and bleach of 1H+ (2051 cm−1) followed by pseudo-first order decay of 1H by protonation with Cl2BSA and charge recombination with TTF+. ) monitoring pseudo-first order formation of 1H (1965 cm−1) and bleach of 1H+ (2051 cm−1) followed by pseudo-first order decay of 1H by protonation with Cl2BSA and charge recombination with TTF+. | |
This assignment is based on the expected IR absorption of 1HHy+ that is likely to cancel the bleach of absorption from the starting material 1H+ as previously found for hydrides 2Hy and 3Hy.32,33 Protonation is also manifested in a slower charge recombination of 1HHy+ with TTF+ (ESI†). Kinetic analysis of both 1H and TTF+ signals results in a protonation rate constant of kPT2 = 6 × 107 M−1 s−1 (see ESI†). The analysis eliminates the possible effect of accelerated recombination with accumulated TTF+ in the presence of acid. With a rate constant of 6 × 107 M−1 s−1 formation of the hydride 1HHy+ by metal protonation of 1H with Cl2BSA (pKa 6.7)34 occurs about as fast as metal protonation of the pdt analogue 2− (7 × 107 M−1 s−1)32 by TsOH (pKa 8–8.7).34 Both complexes are in the same Fe2(I,0) oxidation state but the pKa of the hydride should drop by about 5 units according to a shift of +330 mV in reduction potential for the 1H+/1H couple compared to 1/1− or 2/2−. Protonation of 1H, even by the stronger Cl2BSA, is therefore thermodynamically less favourable than protonation of 2− by TsOH, with ΔpKa lowered by about 3 for the former reaction. Applying the free energy relationship (Brønsted coefficient of 0.4) established for protonation of 2−,32 the protonation of 1H to 1HHy+ is hence one to two orders of magnitude faster than expected for protonation of its pdt analogue 2− with the same driving force. Intrinsically faster protonation of the adt-protonated 1H would prevent this step from becoming rate limiting when reducing weaker acids. Below, we discuss the possible origin of this favourable effect.
Implications for (electro)catalytic H2 formation
From previous electrochemical experiments comparing 1 and 2 it has been concluded that catalytic H2 formation from the same acids proceeds with similar second order catalytic rate constants despite the lower overpotential arising from the easier reduction of adt-protonated catalyst.27 Undiminished reactivity towards protonation despite the lowered driving force would imply that 1 breaks the usual scaling relation between overpotential and rate,38 an effect that has been attributed to the proton shuttling role of the adt27 ligand. By putting an upper limit of 1 s−1 on the first order rate constant for intramolecular tautomerization, our results exclude however that a mechanism involving hydride formation by proton shuttling at the Fe2(0,I) redox level could under any conditions result in the reported pseudo-first order catalytic rate constant on the order of 103 to 104 s−1, as previously proposed.27 It is important to note that the reported kinetics refer to the first catalytic wave (−1.4 V) that has been attributed to mechanisms where the hydride is formed prior to the second electron transfer, i.e. a CECE sequence with stronger acids or an ECCE sequence with weak acids. Therefore, possible formation of the hydride via tautomerization at the Fe2(0,0) redox level (1H− → 1Hy−) cannot explain the superior catalytic performance of the adt complex.
An alternative advantage of the adt ligand might instead arise from the lowered barrier for hydride formation due to the structural changes at the iron core induced by the preceding ligand protonation. This notion would be in line with the relative sluggishness of the latter reaction (see above) and is corroborated by structural data that has been previously obtained from EXAFS spectra39 of ligand- and metal-protonated states of the related Fe2(I,I) complex 4′.¶37 Specifically, protonation of the adt ligand in 4′ results in an elongated Fe–Fe bond distance in 4′H+ that is close to the distance found in the bridging hydride 4′HHy2+. Formation of the hydride from the ligand protonated complex requires hence much less reorganization of this coordinate than formation of hydride 4′Hy+ from 4′.
Generally, a proton shuttling ligand could accelerate not only the formation of the hydride intermediate but also its subsequent coupling with a proton. In case of adt complex 1, with its bridging hydride intermediate, neither of the two steps is, however, likely to involve the adt-N, for steric reasons. In contrast to the terminal hydride of the enzyme active site, the hydride is bridging in the model complex (cf.Scheme 1), as evidenced by the conserved symmetry shown by the IR spectra, and the absence of a bridging CO IR band. The superior catalytic performance of 1 over its pdt analogue 2 is instead attributed to the observed acceleration of hydride formation with bulk acid as described above.
Conclusions
In summary, we could demonstrate by real-time spectroscopic observation that the initial protonation of hydrogenase model complex 1, also in its one-electron reduced Fe2(I,0) state 1−, occurs on the adt ligand rather than the Fe2 core. The same ligand protonated intermediate 1H was alternatively generated by one-electron reduction of 1H+ and no evidence for tautomerization of 1H to a potentially more stable hydride 1Hy was observed in either case. 1H was stable on the time scale of 1 s, which shows that intramolecular proton shuttling cannot be responsible for the reported catalytic TOFs of 103 to 104 s−1.27 For catalytic H2 generation from weak acids this implies that no hydride intermediate is formed on the Fe2(I,0) level; instead, hydride formation may occur after further reduction, by direct protonation in the Fe2(0,0) state. Only with strong acids a doubly protonated intermediate 1HHy+ is formed in the Fe2(I,0) state, by direct metal protonation of 1H, not by ligand protonation of a hydride precursor. Generally, it can therefore be concluded that, in contrast to the enzymatic reaction, the adt ligand of model complex 1 has no proton relay function that enables rapid formation of the bridging hydride intermediates on the Fe2(I,0) level. The protonation kinetics reveal however an exceptionally high barrier for protonation of the adt ligand that we suggest in turn may lower the barrier for hydride formation, presumably due to rearrangements of the metal core upon ligand protonation. In this way the basic site in the second coordination sphere might allow the catalyst to overcome the usual trade-off between overpotential and rate without invoking any proton-shuttling role.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Swedish Research Council (grant no. 2016-04271) and the Foundation Olle Engkvist Byggmästare (granted 2016/3). S. W. gratefully acknowledges a grant from the Chinese Scholarship Council. G. B. acknowledges funding from the Swedish Research Council (grant no 621-2014-5670), The Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning, Formas (contract no. 213-2014-880) and the Wenner-Gren foundation (G. B. and C. E.).
Notes and references
- D. K. Dogutan, R. McGuire and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS PubMed.
- J. L. Boyer, D. E. Polyansky, D. J. Szalda, R. F. Zong, R. P. Thummel and E. Fujita, Angew. Chem., Int. Ed., 2011, 50, 12600–12604 CrossRef CAS PubMed.
- Y. M. Badiei, D. E. Polyansky, J. T. Muckerman, D. J. Szalda, R. Haberdar, R. Zong, R. P. Thummel and E. Fujita, Inorg. Chem., 2013, 52, 8845–8850 CrossRef CAS PubMed.
- W. A. Hoffert, M. T. Mock, A. M. Appel and J. Y. Yang, Eur. J. Inorg. Chem., 2013, 2013, 3846–3857 CrossRef CAS.
- H. L. Sun, Y. Z. Han, H. T. Lei, M. X. Chen and R. Cao, Chem. Commun., 2017, 53, 6195–6198 RSC.
- M. R. DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
- S. E. Smith, J. Y. Yang, D. L. DuBois and R. M. Bullock, Angew. Chem., Int. Ed., 2012, 51, 3152–3155 CrossRef CAS PubMed.
- M. O'Hagan, W. J. Shaw, S. Raugei, S. Chen, J. Y. Yang, U. J. Kilgore, D. L. DuBois and R. M. Bullock, J. Am. Chem. Soc., 2011, 133, 14301–14312 CrossRef PubMed.
- T. B. Rauchfuss, Acc. Chem. Res., 2015, 48, 2107–2116 CrossRef CAS PubMed.
- P. F. Huo, C. Uyeda, J. D. Goodpaster, J. C. Peters and T. F. Miller, ACS Catal., 2016, 6, 6114–6123 CrossRef CAS.
- S. Raugei, M. L. Helm, S. Hammes-Schiffer, A. M. Appel, M. O'Hagan, E. S. Wiedner and R. M. Bullock, Inorg. Chem., 2016, 55, 445–460 CrossRef CAS PubMed.
- B. Ginovska-Pangovska, A. Dutta, M. L. Reback, J. C. Linehan and W. J. Shaw, Acc. Chem. Res., 2014, 47, 2621–2630 CrossRef CAS PubMed.
- M. R. Dubois and D. L. Dubois, Acc. Chem. Res., 2009, 42, 1974–1982 CrossRef PubMed.
- J. T. Bays, N. Priyadarshani, M. S. Jeletic, E. B. Hulley, D. L. Miller, J. C. Linehan and W. J. Shaw, ACS Catal., 2014, 4, 3663–3670 CrossRef CAS.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2014, 136, 11821–11829 CrossRef CAS PubMed.
- M. L. Pegis, B. A. McKeown, N. Kumar, K. Lang, D. J. Wasylenko, X. P. Zhang, S. Raugei and J. M. Mayer, ACS Cent. Sci., 2016, 2, 850–856 CrossRef CAS PubMed.
- A. M. Lilio, M. H. Reineke, C. E. Moore, A. L. Rheingold, M. K. Takase and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 8251–8260 CrossRef CAS PubMed.
- C. W. Machan, J. Yin, S. A. Chabolla, M. K. Gilson and C. P. Kubiak, J. Am. Chem. Soc., 2016, 138, 8184–8193 CrossRef CAS PubMed.
- S. Roy, B. Sharma, J. Peaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- W. H. Wang, J. F. Hull, J. T. Muckerman, E. Fujita and Y. Himeda, Energy Environ. Sci., 2012, 5, 7923–7926 RSC.
- A. Chapovetsky, M. Welborn, J. M. Luna, R. Haiges, T. F. Miller and S. C. Marinescu, ACS Cent. Sci., 2018, 4, 397–404 CrossRef CAS PubMed.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270, 127–150 CrossRef.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-Wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 4306–4309 CrossRef CAS PubMed.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592–6599 RSC.
- M. Bourrez, R. Steinmetz and F. Gloaguen, Inorg. Chem., 2014, 53, 10667–10673 CrossRef CAS PubMed.
- H. Li and T. B. Rauchfuss, J. Am. Chem. Soc., 2002, 124, 726–727 CrossRef CAS PubMed.
- J. A. Wright, L. Webster, A. Jablonskytė, P. M. Woi, S. K. Ibrahim and C. J. Pickett, Faraday Discuss., 2011, 148, 359 RSC.
- J. L. Stanley, Z. M. Heiden, T. B. Rauchfuss, S. R. Wilson, L. De Gioia and G. Zampella, Organometallics, 2008, 27, 119–125 CrossRef CAS PubMed.
- S. J. Borg, T. Behrsing, S. P. Best, M. Razavet, X. Liu and C. J. Pickett, J. Am. Chem. Soc., 2004, 126, 16988–16999 CrossRef CAS PubMed.
- S. H. Wang, A. Aster, M. Mirmohades, R. Lomoth and L. Hammarström, Inorg. Chem., 2018, 57, 768–776 CrossRef CAS PubMed.
- M. Mirmohades, S. Pullen, M. Stein, S. Maji, S. Ott, L. Hammarström and R. Lomoth, J. Am. Chem. Soc., 2014, 136, 17366–17369 CrossRef CAS PubMed.
-
K. Izutsu, Acid-Base Dissociation Constants in Dipolar Aprotic Solvents, Blackwell, Oxford, 1990 Search PubMed.
- S. Tschierlei, S. Ott and R. Lomoth, Energy Environ. Sci., 2011, 4, 2340–2352 RSC.
- E. S. Rountree and J. L. Dempsey, Inorg. Chem., 2016, 55, 5079–5087 CrossRef CAS PubMed.
- G. Eilers, L. Schwartz, M. Stein, G. Zampella, L. de Gioia, S. Ott and R. Lomoth, Chem.–Eur. J., 2007, 13, 7075–7084 CrossRef CAS PubMed.
- C. M. Klug, A. J. P. Cardenas, R. M. Bullock, M. O'Hagan and E. S. Wiedner, ACS Catal., 2018, 8, 3286–3296 CrossRef CAS.
- S. Löscher, L. Schwartz, M. Stein, S. Ott and M. Haumann, Inorg. Chem., 2007, 46, 11094–11105 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, UV-vis transient absorption spectra of 1− and 1H. See DOI: 10.1039/c9sc00876d |
| ‡ Present address: Department of Physical Chemistry, University of Geneva, 30 quai Ernest Ansermet, CH-1211 Geneva, Switzerland. |
| § Present address: CNRS, LCC (Laboratoire de Chimie de Coordination), 205 route de Narbonne, BP 44099 31077 Toulouse cedex 4, France. |
| ¶ Protonation kinetics of the phosphine complexes 4 (ref. 29) and 4′ (ref. 37) have so far been studied only in the Fe2(I,I) state where hydride formation is extremely slow. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Shihuai
Wang
,
Mohammad
Mirmohades
,
Charlène
Esmieu§
,
Gustav
Berggren
,
Shihuai
Wang
,
Mohammad
Mirmohades
,
Charlène
Esmieu§
,
Gustav
Berggren
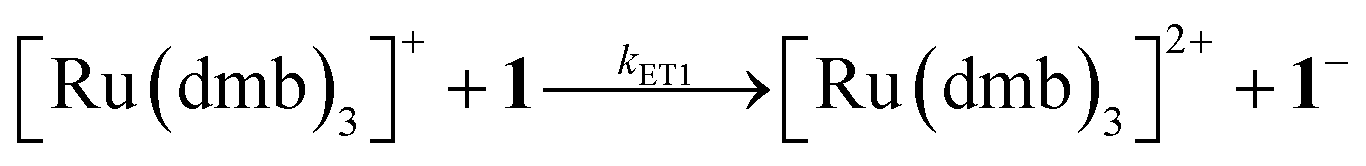

 ) and normalized IR spectrum of the starting state (
) and normalized IR spectrum of the starting state ( ) monitoring the pseudo-first order formation of 1− (1915 cm−1) and bleach of 1 (2035 cm−1) followed by second order charge recombination with TTF+.
) monitoring the pseudo-first order formation of 1− (1915 cm−1) and bleach of 1 (2035 cm−1) followed by second order charge recombination with TTF+.
 ) and normalized IR spectrum of the starting state (
) and normalized IR spectrum of the starting state ( ) monitoring pseudo-first order formation of 1H (1965 cm−1) and bleach of 1 (2035 cm−1) followed by second order charge recombination with TTF+.
) monitoring pseudo-first order formation of 1H (1965 cm−1) and bleach of 1 (2035 cm−1) followed by second order charge recombination with TTF+.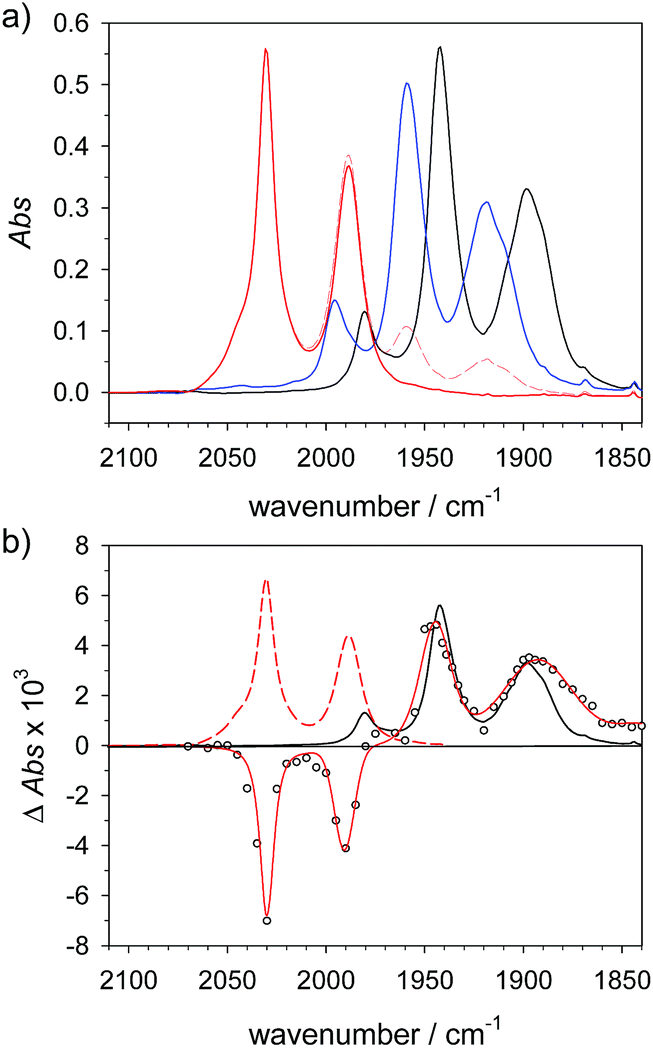
 ) obtained by protonation with toluenesulfonic acid (3 mM), and 4Hy+ (
) obtained by protonation with toluenesulfonic acid (3 mM), and 4Hy+ ( ) obtained by protonation with toluenesulfonic acid (3 mM) in presence of tetrabutyl-ammonium chloride (1.5 mM) after correction of the raw spectrum (
) obtained by protonation with toluenesulfonic acid (3 mM) in presence of tetrabutyl-ammonium chloride (1.5 mM) after correction of the raw spectrum ( ) for minor contribution from 4H+. (b) Transient IR spectrum upon reduction of 4Hy+ to 4Hy by electron transfer from flash-quench generated [Ru(dmb)3]+ (○,
) for minor contribution from 4H+. (b) Transient IR spectrum upon reduction of 4Hy+ to 4Hy by electron transfer from flash-quench generated [Ru(dmb)3]+ (○,  , 1 μs after excitation) and normalized IR spectra of 4Hy+ (
, 1 μs after excitation) and normalized IR spectra of 4Hy+ ( ) and 4 (
) and 4 (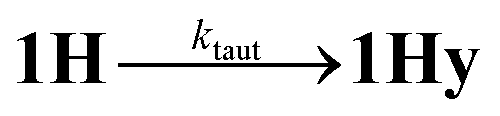
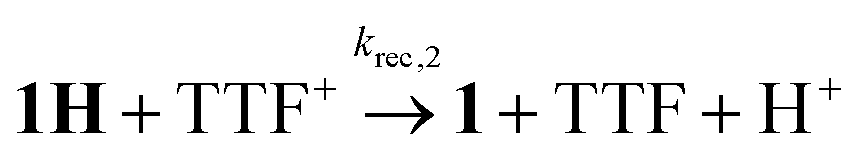
 ) and 3 s (
) and 3 s ( ) monitoring decay of 1H at 1960 cm−1 (b) and formation of 1 at 2034 cm−1 (c).
) monitoring decay of 1H at 1960 cm−1 (b) and formation of 1 at 2034 cm−1 (c).
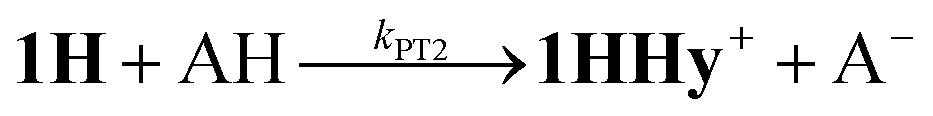
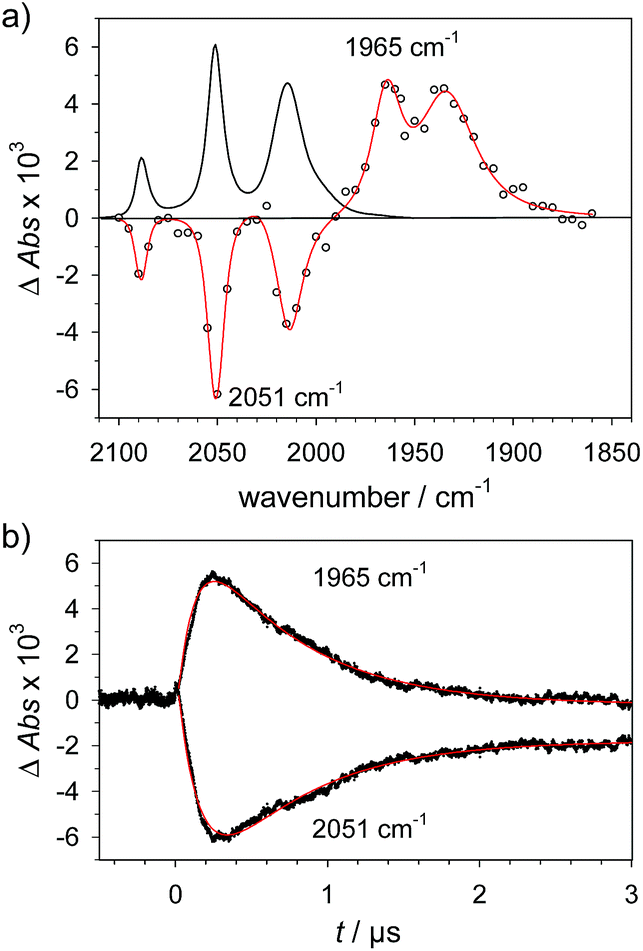
 ) and normalized IR spectrum of the starting state (
) and normalized IR spectrum of the starting state ( ) monitoring pseudo-first order formation of 1H (1965 cm−1) and bleach of 1H+ (2051 cm−1) followed by pseudo-first order decay of 1H by protonation with Cl2BSA and charge recombination with TTF+.
) monitoring pseudo-first order formation of 1H (1965 cm−1) and bleach of 1H+ (2051 cm−1) followed by pseudo-first order decay of 1H by protonation with Cl2BSA and charge recombination with TTF+.