
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective decontamination of the reactive air pollutant nitrous acid via node-linker cooperativity in a metal–organic framework†
Devon T.
McGrath
 a,
Michaela D.
Ryan
a,
John J.
MacInnis
a,
Trevor C.
VandenBoer‡
bc,
Cora J.
Young‡
ac and
Michael J.
Katz
*a
a,
Michaela D.
Ryan
a,
John J.
MacInnis
a,
Trevor C.
VandenBoer‡
bc,
Cora J.
Young‡
ac and
Michael J.
Katz
*a
aDepartment of Chemistry, Memorial University of Newfoundland, St. John's, NL, Canada. E-mail: mkatz@mun.ca
bDepartment of Earth Sciences, Memorial University of Newfoundland, St. John's, NL, Canada
cDepartment of Chemistry, York University, Toronto, ON, Canada
First published on 29th April 2019
Abstract
Nitrous acid (HONO) is a reservoir of NOx and an emerging pollutant having direct impacts on air quality, both in- and outdoors, as well as on human health. In this work, the amine-functionalized metal–organic framework (MOF), UiO-66-NH2, was investigated due to its potential to selectively decontaminate nitrous acid at environmentally relevant concentrations. UiO-66-NH2 proved to be effective in the removal of nitrous acid from a continuous gaseous stream. This is observed via the formation of an aryl diazonium salt that subsequently converts to a phenol with a concomitant release of nitrogen gas. This process is preceded via the formation of the nitrosonium cation (likely protonation from an acidic proton on the node). Thus, UiO-66-NH2 is capable of selectively converting the pollutant nitrous acid to benign products.
1 Introduction
Nitrogen dioxide (NO2) and nitric oxide (NO) are generated primarily from combustion processes in the transportation and energy sectors and are present in both indoor and outdoor air.1 Given the fact that nitrogen dioxide and nitric oxide are able to quickly interchange in the daytime atmosphere, they are often summed together as NOx.2 Environmentally, NOx catalyze the deterioration of air quality via the formation of photochemical smog.3 The resultant poor air quality can negatively impact visibility, vegetation, and human health.4–6 The presence of nitrogen dioxide in the atmosphere has a direct negative impact on both the environment (e.g., pollution)7,8 and human health (e.g., cardiovascular and respiratory conditions).4,9–11 Indirectly, nitrogen dioxide can react further to form secondary pollutants that can be equally, if not more, dangerous to human health and the environment.12 One of the well-known secondary pollutants produced by nitrogen dioxide is nitrous acid (HONO).Nitrous acid is a gas-phase pollutant that is emitted directly and formed indoors and outdoors via the reaction of nitrogen dioxide with surface-bound water (R1) and via the reaction of nitric oxide and hydroxyl radicals ((R2), reverse).13,14 Additionally, despite being advertised for the decontamination of nitrogen dioxide, titanium dioxide (TiO2), a common additive in building surfaces, windows, sidewalks, and paints,15,16 is implicated in the photochemical formation of nitrous acid.12 Nitrous acid subsequently photodissociates (R2) to form nitric oxide. In these regards, nitrous acid is a reservoir for NOx that may be released at a later time, unless properly mitigated.17,18
| 2NO2 + H2O (surface) → HONO + HNO3 | (R1) |
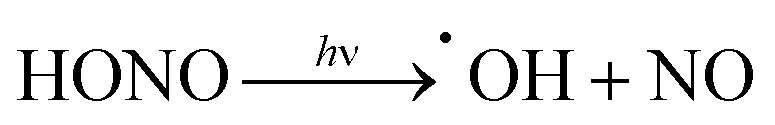 | (R2) |
Nitrous acid has direct negative impacts on air quality.12,19 As a dominant radical source at the surface of the Earth, photolysis of nitrous acid (via indoor lighting or solar irradiation) releases the highly reactive hydroxyl radical (R2). These radicals are able to react directly with tissues in the respiratory tract or take part in atmospheric oxidation to produce other dangerous pollutants such as ground-level ozone and particulate matter smaller than 2.5 μm (PM2.5).12,19
Indoor concentrations of nitrous acid can be over 10 times higher than those present outdoors.20–23 Although nitrous acid is harmful to human health on its own,15,24 it will also react with directly-emitted gaseous amines (R3) or those found in cigarette or cooking smoke, resulting in the formation of carcinogenic nitrosamines, a component of third hand smoke.25–28
R2NH + HONO → R2N–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + H2O O + H2O | (R3) |
It is important to find an efficient method by which nitrous acid can be rapidly sequestered due to its highly reactive nature, the associated health risks, as well as its contribution to poor air quality. Given that nitrous acid is derived from, and a reservoir for, NOx, the deactivation of nitrous acid could be a more advantageous method to reduce NOx emissions compared to direct sorption processes.
To address these challenges, we have turned our attention to the reactivity of nitrous acid within metal–organic frameworks (MOFs). MOFs are a class of porous materials composed of metal centers (nodes) and bridging organic ligands (linkers).29–33 The presence of different node geometries, as well as the synthetic tunability of linkers, via traditional organic chemistry routes, affords a high degree of tunability of the pore size, aperture, and reactivity.34 To that end, researchers have explored the utility of MOFs in applications such as chemical sensing, gas storage, chemical separations, and solar energy generation.35–40 Beyond these applications, specific MOFs have been featured in environmental decontamination strategies.41–48
The present work illustrates a chemisorption solution (Fig. 1) for nitrous acid (and ultimately NOx) where capacity, reactivity, and selectivity are greater than under physisorption conditions. In strongly-acidic aqueous solutions, it is known that the nitrite anion (NO2−) readily converts to nitrous acid and subsequently to the nitrosonium cation (NO+). The nitrosonium cation further reacts with aryl amines to form a diazonium salt, which in solution can be quenched to form aryl-halides, cyanides, and hydroxides.49,50 Herein, we demonstrate for the first time that similar chemistry is possible under heterogeneous conditions between nitrous acid gas and an appropriate MOF substrate, UiO-66-NH2 (Fig. 1).51–54 The Zr6O4(OH)412+ cluster node of the UiO family (Fig. 1a) contains acidic protons (pKa1 = 3.5)55–57 on the μ3-bridging –OH groups. We hypothesize that these acidic groups are able to produce the nitrosonium cation inside the pores. The 2-aminoterephthalate linker (BDC-NH2) of UiO-66-NH2 contains the requisite aryl amine unit to react with the nitrosonium cation. Under humid conditions, the aryl diazonium is able to produce an aryl hydroxide, releasing nitrogen gas (Fig. 1d). Thus, UiO-66-NH2 is uniquely able to rapidly and selectively (i.e., only nitrous acid is expected to undergo this chemistry) decontaminate gas-phase nitrous acid at environmentally-relevant concentrations (Fig. 1d).
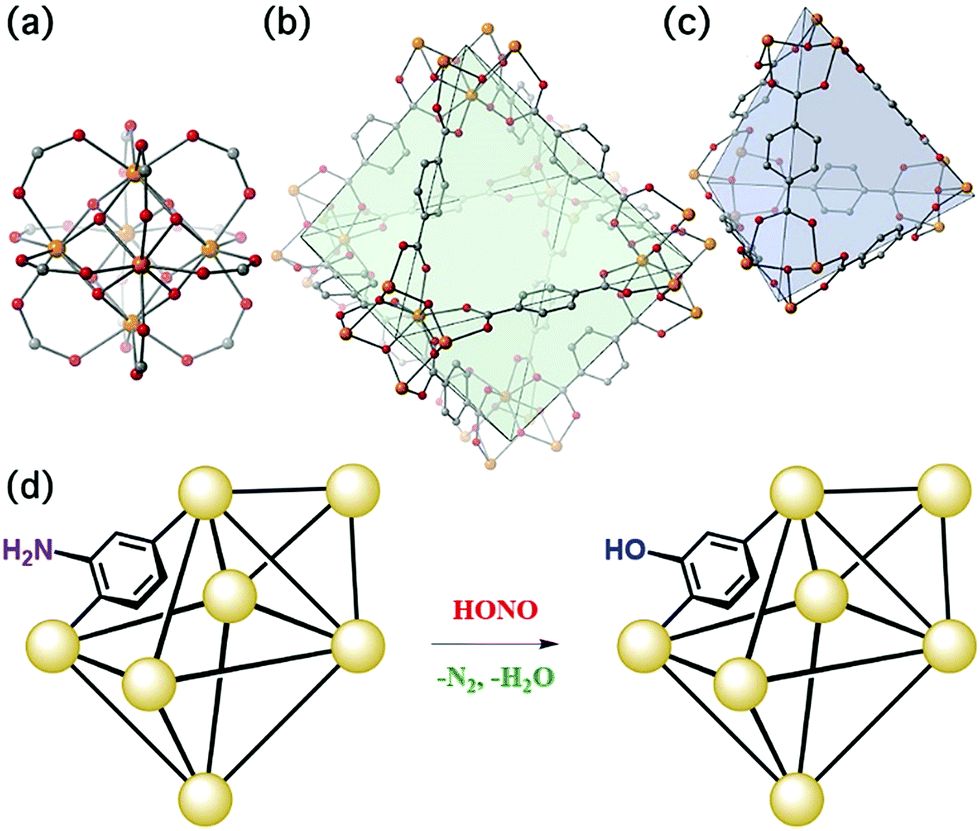 | ||
| Fig. 1 (a) The Zr6O4(OH)412+ node of UiO-66 coordinated to 12 carboxylate anions. (b) The octahedral pore of UiO-66. (c) The tetrahedral pore of UiO-66. (d) Schematic representation of UiO-66-NH2 and the proposed reaction. For (a), (b), and (c): Zr – orange, C – gray, O – red. | ||
2 Results and discussion
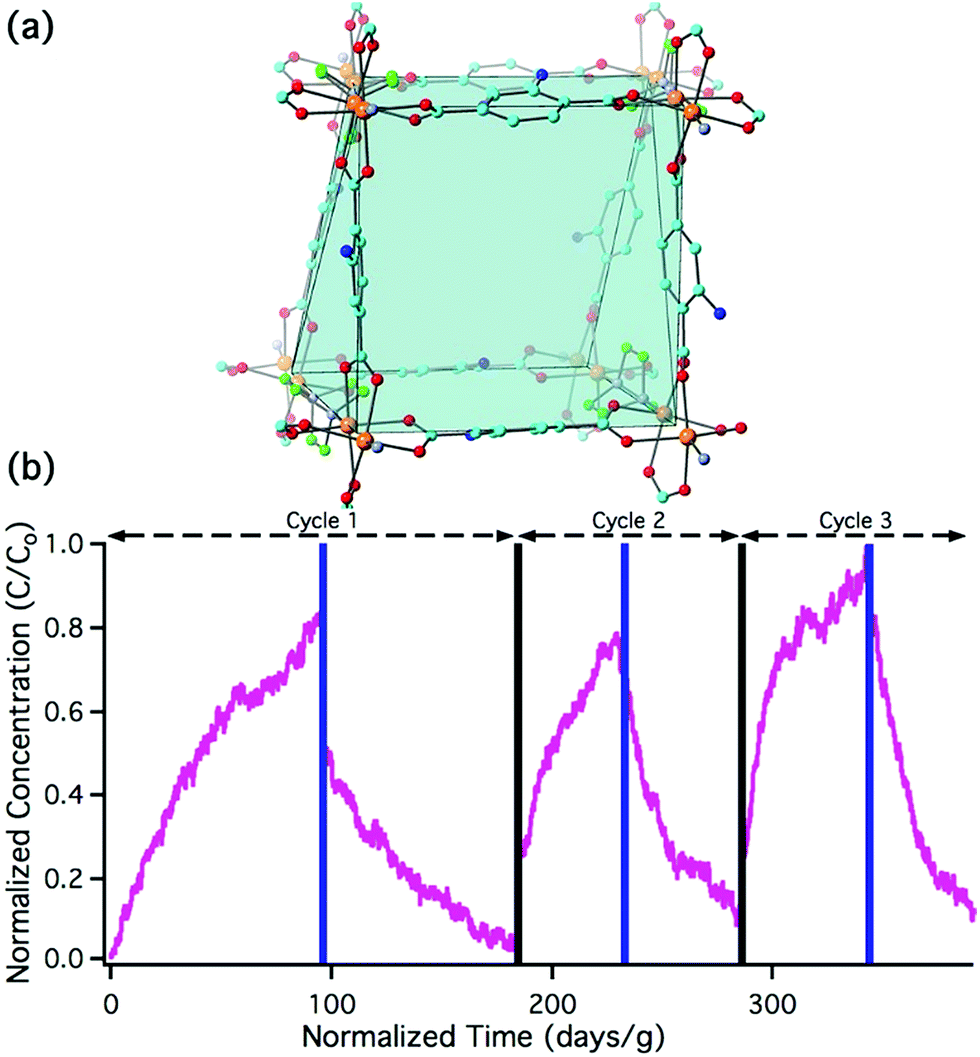
The sorption and desorption kinetics of nitrous acid were analyzed using a custom-built breakthrough experiment system (Fig. S2 and Section S3 in the ESI†). Briefly, nitrous acid was generated in situ by flowing dilute HCl gas at 50% relative humidity (RH) over a bed of solid sodium nitrite. The acid-displacement reaction produces gas-phase nitrous acid and solid sodium chloride; controlling concentration of the HCl and temperature results in a tunable nitrous acid source.58,59 For the purpose of these experiments, nitrous acid concentrations were maintained between 74 and 179 pg cm−3 (equivalent to 35 and 85 parts per billion by volume (ppbv)) at 50% RH; this is on the order of indoor and outdoor environmental concentrations.20,60 Approximately 10 mg of MOF was packed into a 1/4′′ perfluoroalkoxy (PFA) tube. Nitrous acid was flowed through the MOF and the downstream (post-MOF) concentrations were measured as a function of time on an American Ecotech oxides of nitrogen analyzer.Breakthrough exposure experiments were initially conducted on UiO-66. UiO-66 contains the acidic, Zr6O4(OH)412+, node and the benzenedicarboxylate (BDC) linker. Fig. 2a illustrates the downstream nitrous acid concentration as a function of time over three sorption and desorption cycles (Fig. S3† contains replicate data). Initially after exposure of UiO-66 to nitrous acid, the downstream concentration drops to zero. However, over a short period of time, the concentration of nitrous acid, post-UiO-66, begins to increase. This indicates that UiO-66 is not efficient at sequestering nitrous acid; 1 gram of this MOF would take approximately 15 days for saturation under these conditions (i.e., concentration and flow rate). This is equivalent to 0.44% of the acidic protons or 1.8% of the nodes reacting with HONO (Table S2† compares various breakthrough units).
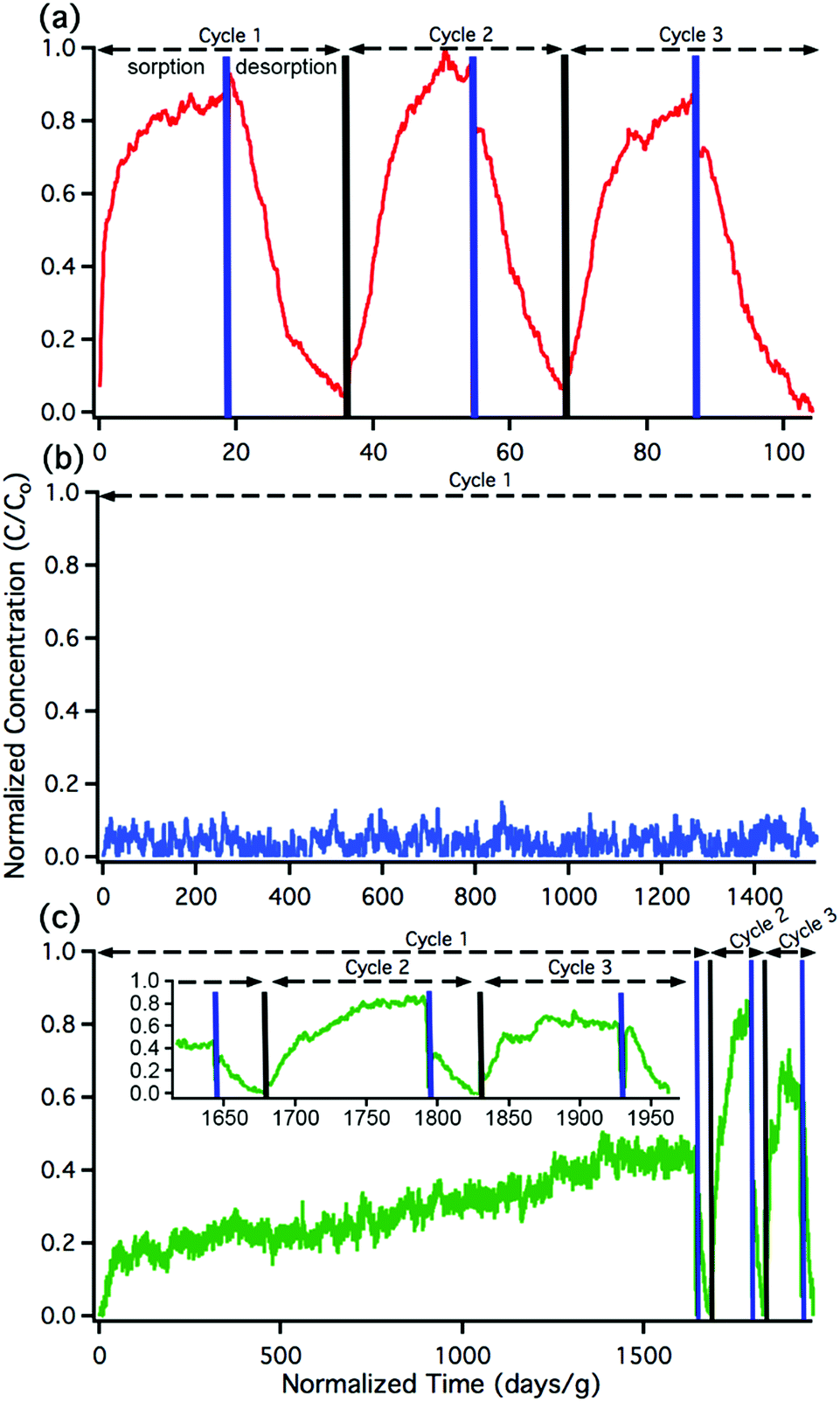 | ||
| Fig. 2 Nitrous acid breakthrough curves for (a) UiO-66, (b) UiO-66-NH2, and (c) UiO-66-(NH2)1/6; the inset illustrates an expanded view of cycles 2 and 3. The x-axis units are the same for all the plots. Note the difference in x-axis scale between (a), (b), and (c). | ||
Desorption performed under 50% RH released approximately equal amounts of nitrous acid as was sorbed (evident in the sorption/desorption traces). This would suggest physisorption. However, when desorption is performed at 0% RH (Fig. S4†), then only a nominal amount of nitrous acid is desorbed. This indicates that nitrous acid is being chemically trapped in the pores of the MOF and water vapour is necessary for the desorption process. This is proposed to be due to the formation of the nitrosonium cation and a concomitant loss of water (vide infra).
Nitrogen gas adsorption studies, performed at 77 K, before and after three cycles of nitrous acid exposure indicate the Brunauer–Emmett–Teller (BET) surface area (SA) decreased by 20% (1770 m2 g−1 decreased to 1416 m2 g−1 respectively; Fig. S11†). Furthermore, the powder X-ray diffractogram (PXRD; Fig. S12†) of UiO-66 post nitrous acid exposure does not show signs of peak broadening or crystallinity loss. Thus, UiO-66 has not degraded over the course of the experiment.
With the sorptive nature of UiO-66 identified, we turned our attention to the amine-functionalized MOF, UiO-66-NH2. This MOF contains the acidic node and the aryl-amine-containing BDC-NH2 linker that is expected to react with the nitrosonium cation. As shown in Fig. 2b, the lack of downstream nitrous acid clearly illustrates that UiO-66-NH2 is highly reactive. Even after 1400 days per g (equivalent to 17 mg of HONO per g of MOF, or 11% of the linkers and 16% of the hydroxides converted; Table S2†), UiO-66-NH2 does not show any measurable quantity of nitrous acid downstream.61 Attempts to desorb nitrous acid from UiO-66-NH2 were unsuccessful; no nitrous acid or NOx is observed post-MOF. This indicates that UiO-66-NH2 is acting as a highly reactive scavenger for nitrous acid. This data is consistent with chemisorption behaviour.
To verify that the MOF is not being degraded, the PXRD (Fig. S12†), and BET SA was measured before/after exposure to nitrous acid; a 14% reduction in overall BET SA was observed (Fig. S11†) after 30 days with no change in crystallinity (Fig. S12†). The lack of any notable change demonstrates that the MOF is stable under the experimental conditions. Even under high concentrations (>1 ppmv) of nitrous acid (Fig. S11†), only a 54% loss in BET surface area (1655 m2 g−1 decreased to 765 m2 g−1) is observed after a week-long exposure.
Replicate nitrous acid exposure cycles for UiO-66-NH2 could not be conducted because even with a small amount of MOF (10 mg), a full cycle for UiO-66-NH2 is expected to take upwards of 100 days. This issue was addressed by testing the sorption abilities of mixed linker MOF, UiO-66-(NH2)1/6 that contains 5/6 BDC and 1/6 BDC-NH2 linkers. By reducing the –NH2 content, the experimental time was reduced. As shown in Fig. 2c, the first sorption cycle shows a slow increase in downstream nitrous acid concentration as a function of time. This is inconsistent with the sorption data observed for UiO-66 but is consistent with the chemisorption data shown for UiO-66-NH2. In comparison, this breakthrough time is equivalent to 35 mg of HONO per gram of MOF and approximately 21% of the linkers and 31% of the acidic protons reacting (Table S2†). The difference between the first cycle of UiO-66-(NH2)1/6 and UiO-66-NH2 is due to the change in rate (i.e., a decrease in contact time) associated with a six-fold decrease in BDC-NH2. The first desorption cycle for UiO-66-(NH2)1/6 shows that the amount of gas sorbed (the area above the sorption curve) is much larger than the amount of gas desorbed (the area below the desorption curve). This observation is consistent with chemisorption. Beyond the first cycle, the second and third cycles (Fig. 2c) illustrate data that is consistent with the sorption behaviour of UiO-66. We propose that during the first nitrous acid exposure cycle all (17%) of the BDC-NH2 sites are exhausted in UiO-66-(NH2)1/6. In cycles 2 and 3, the MOF is only capable of sorption behavior akin to that observed for UiO-66 (i.e., the formation of the nitrosonium cation; vide infra). Similarly to both UiO-66 and UiO-66-NH2, UiO-66-(NH2)1/6 exhibits stable behaviour under reaction conditions with only a 15% decrease in BET SA before/after exposure experiments (1568 m2 g−1 decreased to 1338 m2 g−1 respectively; Fig. S11†).
To further elucidate the mechanism, solution phase NMR and solid-state IR spectroscopy were examined. However, under environmentally-relevant concentrations, the spectroscopy of UiO-66-NH2 was speculative. To address this, the nitrous acid concentration was increased ca. ten-fold; the breakthrough curve of UiO-66-NH2 resembled that of UiO-66-(NH2)1/6 indicating that at these concentrations the reaction kinetics are slower than the contact time.
Starting with UiO-66, at moderate (650 ppbv mixing ratio) nitrous acid concentrations (Fig. S8†), the NMR showed no change before/after nitrous acid exposure. The NMR of UiO-66-NH2 post-nitrous acid exposure (Fig. 3a) shows that three different mono-substituted BDC linkers are now present in solution. Considering the sorption profile of UiO-66-NH2 (Fig. 2b) did not begin to show nitrous acid breakthrough, remnant BDC-NH2 (Fig. 3b vs. a) is expectedly present (34% by NMR). Most interesting is the presence of a strongly deshielded BDC-X from 8.4 ppm to 9.6 ppm (21% by NMR). We propose that this is BDC-N2+, the electron withdrawing (i.e., deshielding) product of the rapid reaction between nitrosonium and an aryl amine.42 The NMR (Fig. 3) illustrates that once the node reacts with nitrous acid to form the nitrosonium cation, then BDC-NH2 rapidly reacts with the cation to form BDC-N2+ (Fig. 5). Given that diazonium salts are inherently unstable, and a 50% RH gas stream is used in the experiments (Fig. 2), the third moiety in the NMR is BDC-OH (Fig. 3a vs. c; 45% by NMR). At even higher (>1 ppmv) nitrous acid concentration (Fig. S7†),62 NMR analysis of the MOF post-nitrous acid exposure only shows signs of the formation of BDC-N2+. This demonstrates that BDC-OH forms as a result of the formation of BDC-N2+.
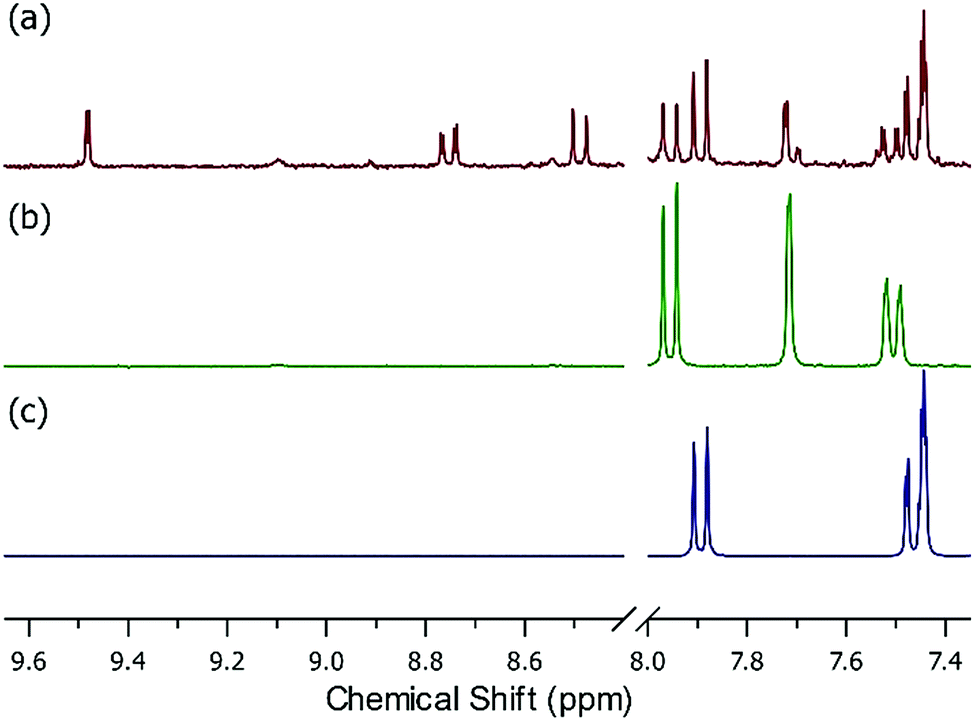 | ||
Fig. 3 Solution phase NMR measured in DMSO-d6 (lock solvent) with 2 drops of D2SO4 of (a) UiO-66-NH2 post-nitrous acid exposure, (b) UiO-66-NH2, and (c) BDC-OH. Note the presence of strongly deshielded protons between 8.4 and 9.6 ppm (21% by NMR) as well as the presence of resonance consistent with BDC-OH (45% by NMR) and the parent BDC-NH2 (34% by NMR) in (a). Each component integrated to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio as expected. :1:1 ratio as expected. | ||
The presence of BDC-N2+ is further corroborated by IR spectroscopy. Unlike pristine UiO-66-NH2, after exposure to nitrous acid, a new vibration at 2283 cm−1 is observed (Fig. S10†). This vibration is consistent with the stretching frequency of a diazonium; this does not appear in the spectra for nitrous acid-exposed UiO-66 (Fig. S10†).62,63
Breakthrough experiments were repeated with the pillared paddlewheel MOF, Zn2(BDC-NH2)2(DABCO) (Fig. 4a, where DABCO is 1,4-diazobicyclo[2.2.2]octane).29 This MOF contains the BDC-NH2 reactive site without the acidic node of UiO-66-NH2. As shown in Fig. 4b, the downstream concentration of nitrous acid as a function of time is consistent with physisorption behaviour rather than the chemisorption behaviour of UiO-66-NH2 (Fig. 2bvs.4b).64 NMR analysis of dissolved Zn2(BDC-NH2)2(DABCO) shows the presence of BDC-NH2 and DABCO with no notable BDC-N2+ or BDC-OH (Fig. S9†). The BDC-NH2 moiety is non-reactive to nitrous acid in the absence of the Zr6O4(OH)412+ node. This confirms the importance of the UiO-66 node to the heterogenous reaction.
 | ||
| Fig. 4 (a) Crystal structure of Zn2(BDC-NH2)2(DABCO) showing how 2D paddlewheel sheets of Zn2(BDC-NH2)2 are pillared via DABCO units: Zn – orange, C – cyan/green for BDC/DABCO, N – light blue/blue for BDC/DABCO, O – red. (b) Nitrous acid breakthrough curves for three adsorption/desorption cycles of Zn2(BDC-NH2)2(DABCO); these breakthrough experiments were run between 100 and 160 ppbv nitrous acid mixing ratios to ensure that the MOF has optimal interaction time with nitrous acid. Stoichiometric equivalents for the first breakthrough can be found in Table S2.† | ||
As illustrated in Fig. 5, UiO-66-NH2 operates in a cooperative method. We propose that the node is responsible for producing pore-bound nitrosonium cations (with the node acting as the counterion), via loss of water. The nitrosonium cation subsequently reacts with BDC-NH2 to form UiO-66-(N2+)−, once again releasing water. The nitrosonium-containing MOF further reacts with the gaseous water to produce UiO-66-OH.
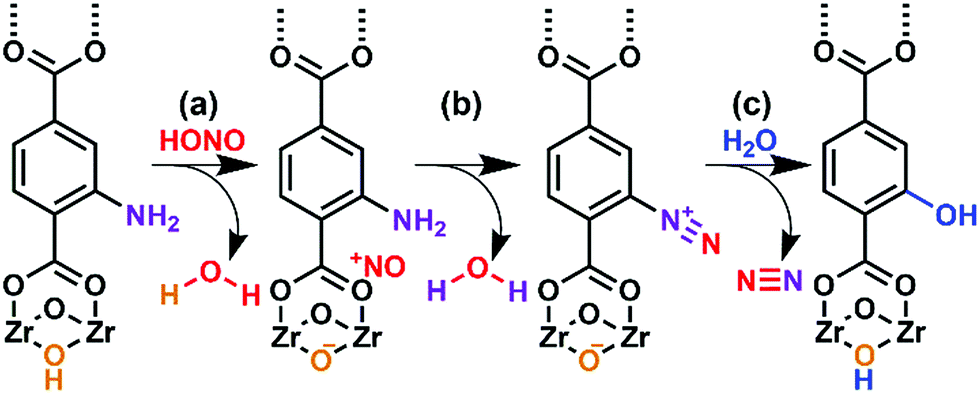 | ||
| Fig. 5 Hypothesized mode of action for the reaction between nitrous acid and UiO-66-NH2. (a) The reaction of gas-phase nitrous acid with –OH group on Zr-based node producing the nitrosonium cation and water. (b) The formation of the diazonium intermediate with concomitant loss of water. (c) The diazonium intermediate reacting with water producing UiO-66-OH with nitrogen gas as a benign reaction byproduct. | ||
3 Conclusions
In conclusion, we have demonstrated that UiO-66-NH2 is a rapid and selective scrubber of nitrous acid. Although UiO-based MOFs have been shown by others to excel at nitrogen dioxide sequestration,42,48 the mechanisms of interaction of UiO-66-NH2 with nitrogen dioxide and nitrous acid are different (i.e., physisorption vs. chemisorption). The reactivity and selectivity of UiO-66-NH2 to nitrous acid should not be affected by the presence of nitrogen dioxide. The products of this newly-demonstrated heterogeneous reaction are nitrogen gas and water. Considering that nitrous acid is a reservoir for NOx, then scrubbing nitrous acid acts as a denoxification method. That is, it returns inert nitrogen gas as a product instead of cycling oxidized species. The denoxification reaction performed by this material is unusual in environmental remediation. Unlike a vehicle catalytic converter, this material can be effectively applied post-emission at dilute concentrations and low temperatures.Considering the substantial amounts of nitrous acid found indoors, UiO-66-NH2 can be utilized in removal of nitrous acid from homes, particularly those where combustion devices such as gas stoves, fireplaces, and candles are used. By implementing MOF filters in oven range hoods and air exchange systems there is the potential for the removal of indoor nitrous acid in homes and businesses.
Furthermore, given that the chemistry presented in this work is selective to nitrous acid, UiO-66-NH2 is a promising candidate for environmental monitoring of nitrous acid in real world scenarios.20,65 Present techniques, albeit very well established, are cumbersome. UiO-66-NH2 can be used to get a better idea of sources and sinks of nitrous acid in the environment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge The Natural Sciences and Engineering Research Council of Canada (NSERC) for funding via the Discovery Grant and Research Tools and Instruments Grant. The Research and Development Corporation of Newfoundland (RDC; now TCII) is gratefully acknowledged for an Ignite and Leverage Grant. Additionally, the authors acknowledge Memorial University for a Seed Grant. TV would like to acknowledge the Canadian government for a Banting Postdoctoral Fellowship. We thank the Core Research Equipment & Instrument Training Network (CREAIT) at Memorial University for the use of their NMR and PXRD facilities within the Centre for Chemical Analysis, Research and Training (C-CART) and The Earth Resources Research and Analysis Facility (TERRA) respectively. A special thanks to Dr Wanda Aylward for performing PXRD analysis.Notes and references
- E. Boyle, Science, 2017, 356, 700–701 CrossRef CAS PubMed
.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed
- C. Muilwijk, P. J. C. Schrijvers, S. Wuerz and S. Kenjereš, Atmos. Environ., 2016, 147, 470–484 CrossRef CAS
- J. Lelieveld, J. S. Evans, M. Fnais, D. Giannadaki and A. Pozzer, Nature, 2015, 525, 367–371 CrossRef CAS PubMed
- D. P. Chock, A. M. Dunker, S. Kumar and C. S. Sloane, Environ. Sci. Technol., 1981, 15, 933–939 CrossRef CAS PubMed
- O. Ranjan, J. Menon and S. Nagendra, J. Hazard., Toxic Radioact. Waste, 2016, 20, A4016002 CrossRef
- Y. Cui, J. Lin, C. Song, M. Liu, Y. Yan, Y. Xu and B. Huang, Atmos. Chem. Phys., 2016, 16, 6207–6221 CrossRef CAS
- M. Grundström, H. W. Linderholm, J. Klingberg and H. Pleijel, Atmos. Environ., 2011, 45, 883–888 CrossRef
- M. Rao, L. A. George, T. N. Rosenstiel, V. Shandas and A. Dinno, Environ. Pollut., 2014, 194, 96–104 CrossRef CAS PubMed
- P. Collart, D. Dubourg, A. Levêque, N. B. Sierra and Y. Coppieters, Data in brief, 2018, 17, 172 CrossRef PubMed
- K. Larabee and W. Phipatanakul, Pediatrics, 2012, 130, S10 CrossRef
- J. M. Langridge, R. J. Gustafsson, P. T. Griffiths, R. A. Cox, R. M. Lambert and R. L. Jones, Atmos. Environ., 2009, 43, 5128–5131 CrossRef CAS
- B. J. Finlayson-Pitts, L. M. Wingen, A. L. Sumner, D. Syomin and K. A. Ramazan, Phys. Chem. Chem. Phys., 2003, 5, 223–242 RSC
- R. Forster, M. Frost, D. Fulle, H. F. Hamann, H. Hippler, A. Schlepegrell and J. Troe, J. Chem. Phys., 1995, 103, 2949–2958 CrossRef CAS
- A. Gandolfo, V. Bartolomei, E. Gomez Alvarez, S. Tlili, S. Gligorovski, J. Kleffmann and H. Wortham, Appl. Catal., B, 2015, 166–167, 84–90 CrossRef CAS
- T. Maggos, A. Plassais, J. Bartzis, C. Vasilakos, N. Moussiopoulos and L. Bonafous, Environ. Monit. Assess., 2008, 136, 35–44 CrossRef CAS PubMed
- T. C. Watling, P. D. Bolton and D. Swallow, Chem. Eng. Sci., 2018, 178, 312–323 CrossRef CAS
- J. Stutz, E. S. Kim, U. Platt, P. Bruno, C. Perrino and A. Febo, J. Geophys. Res.: Atmos., 2000, 105, 14585–14592 CrossRef CAS
- E. Couzo, B. Lefer, J. Stutz, G. Yarwood, P. Karamchandani, B. Henderson and W. Vizuete, Atmos. Environ., 2015, 112, 344–355 CrossRef CAS
- S. Zhou, C. J. Young, T. C. VandenBoer, S. F. Kowal and T. F. Kahan, Environ. Sci. Technol., 2018, 52, 8355–8364 CrossRef CAS PubMed
- S. Gligorovski, J. Photochem. Photobiol., A, 2016, 314, 1–5 CrossRef CAS
- K. Lee, J. Xue, A. S. Geyh, H. Ozkaynak, B. P. Leaderer, C. J. Weschler and J. D. Spengler, Environ. Health Perspect., 2002, 110, 145–150 CrossRef CAS PubMed
- E. Gómez Alvarez, D. Amedro, C. Afif, S. Gligorovski, C. Schoemaecker, C. Schoemacker, C. Fittschen, J.-F. Doussin and H. Wortham, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13294–13299 CrossRef PubMed
- W. Beckett, M. B. Russi, A. D. Haber, R. M. Rivkin, J. R. Sullivan Jr, Z. Tameroglu, V. Mohsenin and B. Leaderer, Environ. Health Perspect., 1995, 103, 372–375 CrossRef CAS PubMed
- P. Hanst, J. Spence and M. Miller, Environ. Sci. Technol., 1977, 11, 403–405 CrossRef CAS
- J. Pitts, C. Van, D. Grosjean, J. Schmid and D. Fitz, Environ. Sci. Technol., 1978, 12, 946 CrossRef CAS
- B. Hang, A. H. Sarker, C. Havel, S. Saha, T. K. Hazra, S. Schick, P. Jacob, 3rd, V. K. Rehan, A. Chenna, D. Sharan, M. Sleiman, H. Destaillats and L. A. Gundel, Mutagenesis, 2013, 28, 381–391 CrossRef CAS PubMed
- M. Sleiman, L. A. Gundel, J. F. Pankow, P. Jacob, B. C. Singer and H. Destaillats, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6576–6581 CrossRef CAS PubMed
- D. T. McGrath, V. A. Downing and M. J. Katz, CrystEngComm, 2018, 20, 6082–6087 RSC
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed
- A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC
- S. Ma and H.-C. Zhou, Chem. Commun., 2010, 46, 44–53 RSC
- Y. Sun and H.-C. Zhou, Sci. Technol. Adv. Mater., 2015, 16, 054202 CrossRef PubMed
-
J. Čejka, Metal-Organic Frameworks. Applications from Catalysis to Gas Storage, Wiley-VCH, Weinheim, 2011 Search PubMed
- X. Zhao, Y. Wang, D. S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef PubMed
- R. Ifraemov, R. Shimoni, W. He, G. Peng and I. Hod, J. Mater. Chem. A, 2018, 7, 3046–3053 RSC
- R.-B. Lin, S.-Y. Liu, J.-W. Ye, X.-Y. Li and J.-P. Zhang, Adv. Sci., 2016, 3, 1500434 CrossRef PubMed
- A. Indra, T. Song and U. Paik, Adv. Mater., 2018, 30, 1705146 CrossRef PubMed
- M. Schulz, A. Gehl, J. Schlenkrich, H. A. Schulze, S. Zimmermann and A. Schaate, Angew. Chem., Int. Ed., 2018, 57, 12961–12965 CrossRef CAS PubMed
- J. E. Mondloch, M. J. Katz, W. C. Isley, 3rd, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. DeCoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Nat. Mater., 2015, 14, 512–516 CrossRef CAS PubMed
- G. W. Peterson, J. J. Mahle, J. B. DeCoste, W. O. Gordon and J. A. Rossin, Angew. Chem., Int. Ed., 2016, 55, 6235–6238 CrossRef CAS PubMed
- J. B. DeCoste and G. W. Peterson, Chem. Rev., 2014, 114, 5695–5727 CrossRef CAS PubMed
- H. Jasuja, G. W. Peterson, J. B. Decoste, M. A. Browe and K. S. Walton, Chem. Eng. Sci., 2015, 124, 118–124 CrossRef CAS
- J. Aguilera-Sigalat, A. Fox-Charles and D. Bradshaw, Chem. Commun., 2014, 50, 15453–15456 RSC
- Y. Liu, A. J. Howarth, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 9001–9005 CrossRef CAS PubMed
- J. Zhang, J. B. DeCoste and M. J. Katz, Can. J. Chem., 2018, 96, 139–143 CrossRef CAS
- A. M. Ebrahim, B. Levasseur and T. J. Bandosz, Langmuir, 2013, 29, 168–174 CrossRef CAS PubMed
- H. H. Hodgson, Chem. Rev., 1947, 40, 251–277 CrossRef CAS
- F. Y. Mo, D. Qiu, Y. Zhang and J. Wang, Acc. Chem. Res., 2018, 51, 496–506 CrossRef CAS PubMed
- V. Guillerm, F. Ragon, M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vimont, G. Clet, Q. Yang, G. Maurin, G. Férey, A. Vittadini, S. Gross and C. Serre, Angew. Chem., Int. Ed., 2012, 51, 9267–9271 CrossRef CAS PubMed
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed
- Y. Bai, Y. Dou, L. H. Xie, W. Rutledge, J. R. Li and H. C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, K. P. Lillerud, C. Larabi, E. A. Quadrelli and F. Bonino, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 RSC
- S. Ling and B. Slater, Chem. Sci., 2016, 7, 4706–4712 RSC
- V. R. Bakuru, S. R. Churipard, S. P. Maradur and S. B. Kalidindi, Dalton Trans., 2019, 48, 843–847 RSC
- T. C. VandenBoer, C. J. Young, R. K. Talukdar, M. Z. Markovic, S. S. Brown, J. M. Roberts and J. G. Murphy, Nat. Geosci., 2015, 8, 55–60 CrossRef CAS
- J. Roberts, P. Veres, C. Warneke, J. Neuman, R. Washenfelder, S. S. Brown, M. Baasandorj, J. Burkholder, I. R. Burling, T. Johnson, R. Yokelson and J. de Gouw, Atmos. Meas. Tech., 2010, 3, 981–990 CrossRef CAS
- The oxides of nitrogen analyzer employed in this work required a minimum flow rate of 1400 sccm. Given that the MOF was exposed to ca. 120 sccm, a dilution flow was necessary to ensure an adequate flow rate. Thus, the concentration that are used in this work are as low as experimentally possible.
- No NOx is observed by the oxides of nitrogen analyzer downstream from the UiO-66-NH2 sample.
- Experiments with even higher concentrations of nitrous acid (>1 ppmv), in which the concentration of nitrous acid was a less-stable and likely contains unreacted HCl and unwanted NOx, were examined. IR analysis of the post-exposed material shows a stronger vibration at 2283 cm−1, consistent with the formation of BDC-N2+ (Fig. S10 in the ESI†).
-
G. Socrates, Infrared Characteristic Group Frequencies: Tables and Charts, Wiley, New York, NY, 2nd edn, 1994 Search PubMed
- Zn2(BDC-NH2)2(DABCO) has a higher capacity (3.5 mmoles per gram of MOF) for nitrous acid than UiO-66 (0.5 mmoles per gram of MOF). This equates to roughly 2.5 times more per formula unit or nearly 10x more HONO per linker/metal centre. The exact origin of this is unknown.
- D. B. Collins, R. F. Hems, S. Zhou, C. Wang, E. Grignon, M. Alavy, J. A. Siegel and J. P. D. Abbatt, Environ. Sci. Technol., 2018, 52, 12419–12427 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01357a |
| ‡ All the work was performed at Memorial University. Over the course of the research, the authors moved to the Department of Chemistry at York University, Toronto, ON, Canada. |
| This journal is © The Royal Society of Chemistry 2019 |