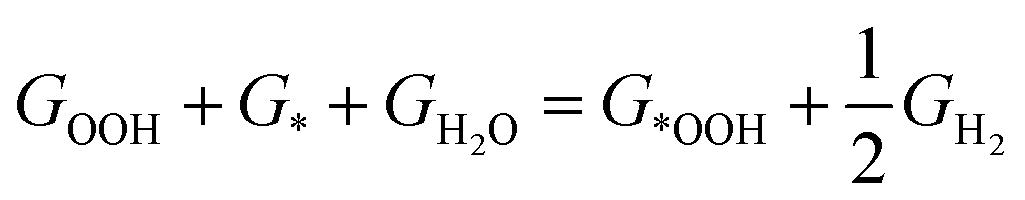DOI:
10.1039/C9SC01078E
(Edge Article)
Chem. Sci., 2019,
10, 5589-5595
Optimal coordination-site exposure engineering in porous platinum for outstanding oxygen reduction performance†
Received
4th March 2019
, Accepted 26th April 2019
First published on 8th May 2019
Abstract
In this study, we report that optimal coordination-site exposure engineering in porous platinum brings ultrahigh activity and durability for the fuel cell oxygen reduction reaction (ORR). The porous platinum with numerous grain boundaries (GBP-Pt) consisting of 3 nm crystals exhibits 7 times higher ORR activity than commercial Pt. For fuel-cell measurements, the GBP-Pt catalyst based MEA exhibits high power density (1.49 W cm−2, 0.71 A mg−1 Pt for mass activity) and stability (12.9% loss after 30 K cycles), all of which far surpass the U.S. DOE target in 2020 (0.44 A mg−1 Pt for mass activity and 40% loss for stability). Density Functional Theory (DFT) calculation and X-ray Absorption Fine Structure (XAFS) results suggest that proper Pt coordination site exposure in grain boundaries provides optimal adsorption energies for oxygen species and high stability in the ORR, even superior to Pt(111) sites. We anticipated that coordination-site exposure engineering would open a new avenue to offer robust electrocatalysts for the fuel-cell oxygen reduction reaction.
Introduction
Fuel cells, as an energy conversion system with high power density, offer an eco-friendly and efficient pathway to transfer the chemical energy of hydrogen to electric energy through electrochemical reactions.1–6 However, the oxygen reduction reaction (ORR) at the cathode, which involves a 4 electron transfer process, still impedes the efficiency of fuel cells.7–9 To mitigate this problem, platinum based materials have been widely used as electrocatalysts. Until now, noble metals have been playing an irreplaceable role in fuel cell technology, while their high cost and rarity limit their developments and practical applications.10–12 To decrease the quantity demanded for Pt, fabricating robust electrocatalysts with outstanding activity and durability has become crucial.13–15
To date, the synthesis and fabrication of nanoparticles have been regarded as an ideal attempt for the design of efficient Pt electrocatalysts.16,17 During the past few years, progress has been made in liquid half-cell ORR tests by alloying,16,18 chemical doping,14 and adjusting Pt morphology and crystal facets.17 Despite these various strategies, the activity and stability of Pt based electrocatalysts are still limited, especially in highly corrosive fuel cell environments.19 According to the Sabatier principle, this restricted activity could be mainly attributed to the unsatisfactory adsorption energy of reaction intermediates on Pt sites.20 It has been reported that adsorption energy for small species is well influenced by the coordination environment of the catalytic sites.21 However, almost all available electrocatalysts still suffer from the massive formation of low-coordination Pt sites in the synthesis or catalytic process,22,23 such as the edge exposure or dissolution of alloying metal in acid media.24,25 The bonding energy between low-coordination Pt sites and oxygen containing species is excessively strong, which reduces the free surface area for the ORR.26,27 These sites are also thermodynamically unstable and easier to dissolve,25,28 having a great negative impact on the durability of electrocatalysts. Thus, robust Pt electrocatalysts, with catalytic sites exploited to the maximum extent in activity and durability, are still highly desirable.
Herein, we report that optimal coordination-site exposure engineering in porous platinum could bring ultrahigh activity and durability for fuel cell oxygen reduction. The optimal Pt coordination-site exposure in grain boundaries provides suitable adsorption energies for oxygen species and high thermodynamic stability, resulting in high intrinsic activity and durability in the ORR, even superior to Pt(111) sites. Our as-obtained grain-boundary porous platinum (GBP-Pt) consisting of 3 nm crystals exhibits 7 times higher ORR activity than commercial Pt. For application in fuel cells, the GBP-Pt catalyst based MEA exhibits high power density (1.49 W cm−2, 0.71 A mg−1 Pt for mass activity) and stability (12.9% loss after 30 K cycles), all of which far surpass the U.S. DOE target in 2020 (0.44 A mg−1 Pt for mass activity and 40% loss for stability).
Results and discussion
Oxygen reduction reaction
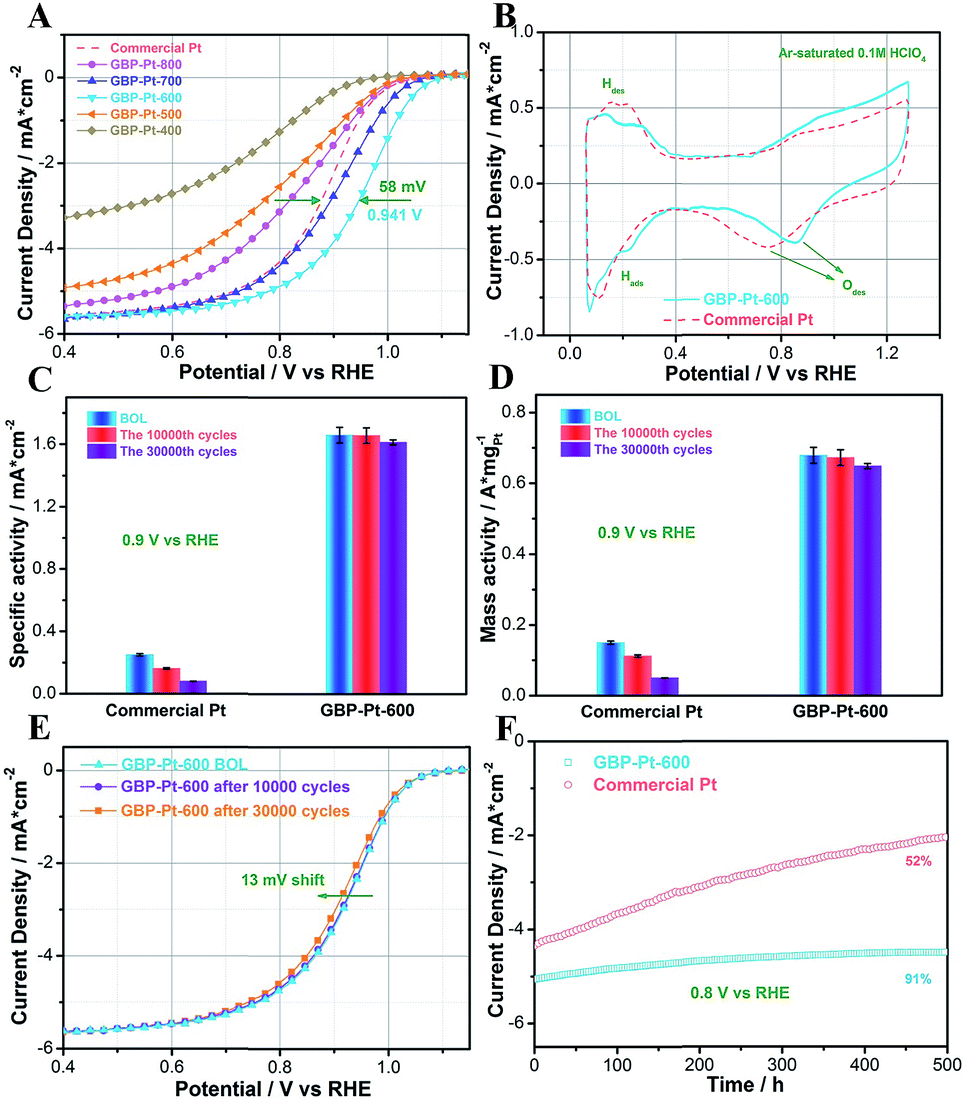
Electrochemical oxygen reduction tests in acidic media were first performed to show the high catalytic performance of the as-obtained catalysts. For comparison, commercial Pt (Hesen) with the same 3 nm grain size as GBP-Pt was also tested. All these samples were mixed with Vulcan XC-72R carbon with 20 wt% Pt loading and the Pt content was determined by ICP-AES to be about 20 μgPt/cm2 for each electrode. As shown in Fig. 1a, in O2-saturated HClO4 solution, GBP-Pt-600 exhibits remarkably excellent activity, and the half-wave potential of GBP-Pt-600 is 0.941 V, which is 58 mV more positive than that of commercial Pt. Fig. 1b shows the cyclic voltammograms (CVs) of the GBP-Pt-600 sample and commercial Pt at room temperature in Ar-saturated 0.1 M HClO4 at a sweep rate of 20 mV s−1. A more positive peak of oxygen species desorption was observed for GBP-Pt-600, which represents reduced adsorption energy on the Pt surface in favor of the acidic ORR. As is well known, the strong bonding of oxygen and the Pt surface seriously impedes intrinsic activity of Pt in ORR catalysis.29 To further investigate the intrinsic activity of each Pt site, specific activities of the catalysts were first calculated using the kinetic current densities and electrochemical surface area (ECSA) as shown in Fig. 1c. The ECSA of each electrocatalyst was obtained by calculating hydrogen under potential deposition (HUPD) charge from CV curves (Fig. S2†) between 0.1 V and 0.4 V (0.4–0.55 V background subtraction). The specific activity of GBP-Pt-600 was 1.70 ± 0.05 mA cm−2, which was almost 7 times higher than that of commercial Pt (0.263 ± 0.03 mA cm−2) at 0.9 vs. RHE, representing an ultra-high intrinsic activity of each Pt site in GBP-Pt-600. The mass activity quantified by ICP-AES for GBP-Pt-600 (0.68 ± 0.02 A mg−1 Pt) at 0.9 V vs. RHE is 4 times greater than that of commercial Pt (0.16 ± 0.01 A mg−1 Pt).
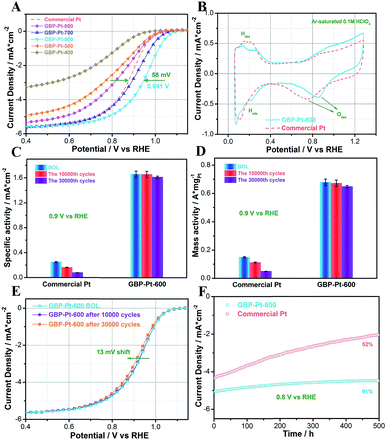 |
| | Fig. 1 (a) ORR polarization curves of GBP-Pt samples for different calcining temperatures and commercial Pt. (b) Cyclic voltammetry curves in Ar-saturated 0.1 M HClO4 for GBP-Pt-600 and commercial Pt. (c) Specific activities of the GBP-Pt-600 sample and commercial Pt at BOL, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, and 30000 cycles, respectively. (d) Mass activities of the GBP-Pt-600 sample and commercial Pt at BOL, 10000 cycles, and 50000 cycles, respectively. (e) Cycling stability test for GBP-Pt-600 BOL, after 10000 cycles and 30000 cycles. (f) Chronoamperometry measurement of the GBP-Pt-600 sample and commercial Pt. All these samples were mixed with Vulcan XC-72R carbon with 20 wt% Pt loading and the Pt content was 20 μgPt/cm2 for each electrode. Solution ohmic drop (iR-drop) was compensated and capacitive background current was corrected by subtracting the curve recorded in Ar-saturated solution. 000 cycles, and 30000 cycles, respectively. (d) Mass activities of the GBP-Pt-600 sample and commercial Pt at BOL, 10000 cycles, and 50000 cycles, respectively. (e) Cycling stability test for GBP-Pt-600 BOL, after 10000 cycles and 30000 cycles. (f) Chronoamperometry measurement of the GBP-Pt-600 sample and commercial Pt. All these samples were mixed with Vulcan XC-72R carbon with 20 wt% Pt loading and the Pt content was 20 μgPt/cm2 for each electrode. Solution ohmic drop (iR-drop) was compensated and capacitive background current was corrected by subtracting the curve recorded in Ar-saturated solution. | |
Stability is a more impressive key index for the GBP-Pt-600 electrocatalyst. The durability of GBP-Pt-600 was also measured through an accelerated durability test (ADT). As shown in Fig. 1e, inappreciable left shifts could be observed in the ORR polarization curves for the GBP-Pt-600 sample after 10000 cycles compared to the beginning of life (BOL). Even as the cycles further extend to 30000 cycles, only 13 mV of negative shift occurs. After the accelerated test for 30000 cycles, the specific activity and mass activity of GBP-Pt-600 still remain at 1.613 mA cm−2 (3.4% loss) and 0.649 A mg−1 Pt (4.7% loss), respectively. By contrast, commercial Pt catalysts exhibited 100 mV potential shifts and lost 62.4% of their initial mass activities, respectively (Fig. S3†). Chronoamperometry measurement (j–t) was also performed at a constant voltage of 0.8 V in 0.1 M HClO4. As shown in Fig. 1f, the current density of GBP-Pt-600 was maintained with 9% decay over 500 h of continuous operation, suggesting the unprecedented durability for our GBP-Pt-600 catalyst.
Fuel cell assembly
In terms of outstanding ORR performance, the GBP-Pt-600 sample was further used as the cathodic catalyst in fuel cell devices to meet the standard of the 2020 U.S. DOE target. The DOE–Fuel Cell Tech Team (DOE/FCTT) protocol was used in the study of fuel cell performance. Using O2 as the oxidant, membrane electrode assemblies (MEAs) with the electrocatalysts were prepared through a hot-pressing method. For comparison, the loading of the commercial Pt catalyst was also adjusted to 0.2 mg cm−2 Pt for the fuel cell test (Fig. 2a). As the cell voltage becomes 0.8 V, the H2/O2 fuel cell reaches a current density of 466 mA cm−2, which is 142 mA cm−2 higher than that of commercial Pt (324 mA cm−2). The maximum peak power density for GBP-Pt-600 was 1.49 W cm−2, which was 310 mW cm−2 higher than that of commercial Pt, reaching the highest power density among previous literature. The mass activity at 0.9 V for GBP-Pt-600 (0.71 ± 0.04 A mg−1 Pt) also exceeded the U.S. DOE target in 2020 (0.44 A mg−1 Pt) (Fig. 2e). For more practical application, the H2/air fuel cell was also tested for these samples. An obvious improvement of 150 mW cm−2 could also be observed in power density for the GBP-Pt-600 sample (Fig. 2b). To satisfy a power system, the long-term durability of GBP-Pt-600 needs to reach the standard for the fuel cell conditions. After ADT tests under working conditions of 358 K with scanning currents from 0 A to 5 A, only 12.9% loss in mass activity could be observed after 30 K cycles under H2/O2 conditions (Fig. 2c), absolutely meeting the DOE 2020 target on catalytic durability (smaller than 40% loss in mass activity). Moreover, in a constant-potential operation test at a high working current, the current density loss was negligible after 300 h (Fig. 2d), also suggesting the outstanding durability of GBP-Pt-600. Compared to remarkable electrocatalysts in reported literature, our GBP-Pt-600 catalyst possesses outstanding activity and stability (Fig. 2f).30–32
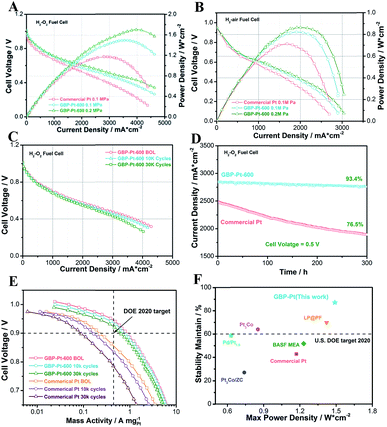 |
| | Fig. 2 (a) H2/O2 fuel cell polarization curves of the GBP-Pt-600 sample and commercial Pt at a back pressure of 0.1 Mpa and 0.2 Mpa. (b) H2/air fuel cell polarization curves of the GBP-Pt-600 sample and commercial Pt at a back pressure of 0.1 MPa. (c) Accelerated durability test (ADT) at 358 K with scanning currents from 0 A to 5 A at BOL, 10000 cycles, and 30000 cycles, respectively. (d) H2/O2 fuel cell test in a constant-potential operation. (e) Mass activities of the GBP-Pt-600 sample and commercial Pt under H2/O2 fuel cell conditions. (f) The comparison between GBP-Pt-600 and remarkable electrocatalysts in fuel cells. The loading of Pt content was 0.2 mg cm−2 for each sample and H2, O2, and air flow rates were 0.4 L min−1 at 100% RH. | |
Mechanism investigation
To better understand the correlation between the remarkable electrochemical performance and catalyst structure, detailed analyses of GBP-Pt-600 were further performed. The reduced adsorption energy of oxygen species on the Pt surface and high specific activity in Fig. 1 show the high intrinsic activity of each Pt site in GBP-Pt-600. Density Functional Theory (DFT) calculation was first employed to obtain atomic-scale insight into the high intrinsic activity. As stated in the Sabatier principle, the ORR activity appears as volcano-type plots against surface adsorption energies of key reaction intermediates. An optimal ORR catalyst must bind hydroxyl species (*OH) with a ΔG of ∼0.87 eV. In a geometric structure of a simple substance, trends in adsorption energies for small species on surfaces are well described by the coordination number of the surface sites. In our case, since abundant grain boundaries exist in porous platinum, the coordination number and adsorption energies in grain-boundary sites were investigated to explain the different intrinsic activities. The “generalized” coordination number 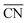 was calculated to eliminate “finite-size effects” by introducing a weight to each first-nearest neighbor atom j,33 corresponding to its own coordination number [cn(j)]. The formula to estimate
was calculated to eliminate “finite-size effects” by introducing a weight to each first-nearest neighbor atom j,33 corresponding to its own coordination number [cn(j)]. The formula to estimate 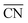 for a site is
for a site is 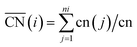 . As shown in Fig. 3a, taking the “a” site as an example, the
. As shown in Fig. 3a, taking the “a” site as an example, the  of the “a” site in a single Pt grain is 4.25. While two grains attach to form a grain boundary, the
of the “a” site in a single Pt grain is 4.25. While two grains attach to form a grain boundary, the  value of the “A” site in the grain boundary increases to 8. Based on the obtainment of the
value of the “A” site in the grain boundary increases to 8. Based on the obtainment of the 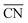 value, the adsorption energies of key intermediate species on Pt sites were further evaluated (Fig. 3b). The adsorption energies of intermediates present a linear relationship against the change of
value, the adsorption energies of key intermediate species on Pt sites were further evaluated (Fig. 3b). The adsorption energies of intermediates present a linear relationship against the change of 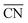 values in different Pt sites. The ΔG for hydroxyl species (*OH) is determined to be 0.111 eV in edge “a” sites while it changes to 0.821 eV in grain boundaries “A”, which approaches close to the optimal value 0.87 eV. Moreover, the two possible rate-determining steps in the ORR are also investigated in Fig. 3c. It can be seen that the “A” site in the grain boundary shows optimal Gibbs free energy and activity for the ORR compared to other sites, even better than Pt(111) sites. Based on these results, optimal adsorption energies of key intermediate species for the ORR could be realized by introducing grain boundary sites.
values in different Pt sites. The ΔG for hydroxyl species (*OH) is determined to be 0.111 eV in edge “a” sites while it changes to 0.821 eV in grain boundaries “A”, which approaches close to the optimal value 0.87 eV. Moreover, the two possible rate-determining steps in the ORR are also investigated in Fig. 3c. It can be seen that the “A” site in the grain boundary shows optimal Gibbs free energy and activity for the ORR compared to other sites, even better than Pt(111) sites. Based on these results, optimal adsorption energies of key intermediate species for the ORR could be realized by introducing grain boundary sites.
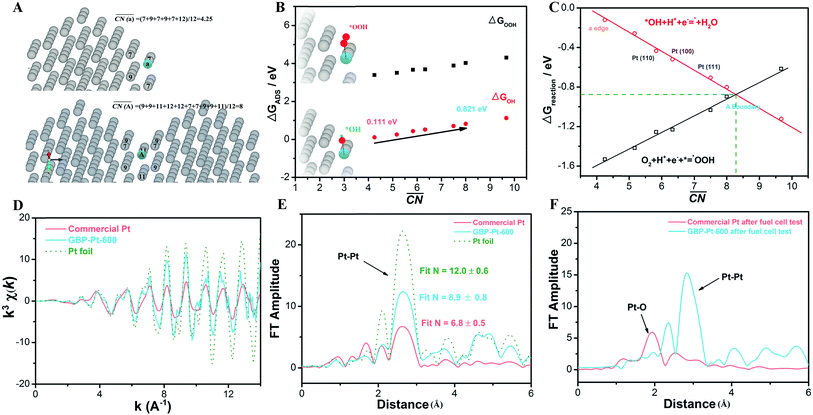 |
| | Fig. 3 The “generalized” coordination number 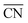 calculation for “a” single cluster and “A” grain boundary sites. (b) The adsorption energies of key intermediates species on different calculation for “a” single cluster and “A” grain boundary sites. (b) The adsorption energies of key intermediates species on different 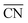 Pt sites. (c) The calculation for Gibbs free energy in two possible rate-determining steps for the ORR. (d) Pt L3-edge k3χ(k) oscillation curves for GBP-Pt-600, commercial Pt and Pt foil as the reference. (e) The Pt L3-edge EXAFS Fourier transformed curves for GB. Pt sites. (c) The calculation for Gibbs free energy in two possible rate-determining steps for the ORR. (d) Pt L3-edge k3χ(k) oscillation curves for GBP-Pt-600, commercial Pt and Pt foil as the reference. (e) The Pt L3-edge EXAFS Fourier transformed curves for GB. | |
To further verify the change of coordination number (cn) in grain boundaries, X-ray Absorption Fine Structure (XAFS) analysis of Pt L3-edge k3χ(k) oscillations in GBP-Pt-600 was performed as shown in Fig. 3d. As control samples, commercial Pt with the same grain diameter (∼3 nm) and Pt foil were also investigated to show the different cn values. It is found that the oscillation intensity of GBP-Pt-600 is higher than that of 3 nm nanoparticles in commercial Pt and lower than that of Pt foil. The Fourier transformed (FT) curves (Fig. 3e) further clarified this amplitude variation. The FT curves of GBP-Pt-600 and the foil are dominated by a strong peak at about 2.64 Å, associated with a satellite peak (at about 2.12 Å) ahead which usually exists in the FT-EXAFS at the K-edge of most heavy metals.34 This FT peak corresponds to the Pt–Pt bond. For commercial Pt with ∼ 3 nm grain diameter, a peak at 1.68 Å was detected besides the Pt–Pt pair. It is a typical interaction between Pt and light elements, attributed to the strong adsorption of Pt sites.35 In order to quantitatively extract the local structures, the EXAFS data were fitted in the R space using the IFEFFIT code. The quality and the result of the fits have been shown in Fig. S4† and Table S1,† respectively. The EXAFS fit of the Pt foil gave a standard local atomic structure, a Pt center coordinated by 12 Pt atoms at 2.76 Å with a slight distortion σ2 = 0.0047 Å2. The reduced EXAFS oscillation of the commercial 3 nm Pt sample mainly came from the low coordination number of Pt (average coordination number N is 6.8). These undercoordinated Pt atoms tend to adsorb oxygen to saturate the coordination environment so that a Pt–O peak could be observed. As for the GBP-Pt-600 sample, a relatively high peak for Pt–Pt could still be observed. This indicated that the high Pt coordination number (N = 8.9) was still retained in GBP-Pt-600, which is beneficial for the activity and stability of the ORR. It should be noticed that the coordination numbers measured by XAFS are average values, which would be higher than the surface value (DFT calculation). In addition, an increased coordination number could be observed by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). As shown in Fig. S5,† the blue and yellow balls represent the Pt atoms of two different grains. The Pt atoms in the grain boundary sites retained a relatively higher coordination number on the edge than on the isolated crystal. Ex situ EXAFS measurements were also performed to show the influence of coordination environments on electrocatalytic durability. The comparison of the R space for GBP-Pt-600 and commercial 3 nm Pt after the fuel cell test is provided in Fig. 3f where a significant increase of the Pt–O peak could be observed in commercial Pt. It has been reported that the dissolution of Pt mainly hinges on the quantity of Pt–O species and decreasing the formation of Pt oxide is vital for reducing Pt atom dissolution.36,37 But for our GBP-Pt-600 sample, Fourier transformed (FT) curves show no obvious change after the fuel cell test, which indicated high durability during the acidic oxygen reduction reaction (Fig. S6†).
Structure analyses
Besides the optimal coordination site exposure, plenty of grain boundaries in GBP-Pt-600 also brought a high surface area for enhanced electrocatalytic activity. The Brunauer–Emmett–Teller (BET) analysis (Fig. 4a) first provides solid evidence that the GBP-Pt-600 catalyst possesses a high surface area of 45.32 m2 g−1. Moreover, a pore distribution (Fig. 4b) of 1.53 nm micropores could also be observed, which could accelerate the gas diffusion during the catalytic process. This porous structure with high surface area was further observed in transmission electron microscopy (TEM) images (Fig. S7†). Each porous Pt consisted of crystalline grains with a size of about 3 nm, forming a 2D-foam-like structure with a large number of boundaries and pores. High-resolution transmission electron microscopy (HRTEM) as shown in Fig. 4c and S8† was further employed to investigate this structure. Numerous grain boundaries were revealed in GBP-Pt-600 and every three or four boundaries could bring pores and triple junctions. To study the effect of grain boundaries on the surface area of the GBP-Pt-600 sample, the volume fraction of grain boundaries was calculated using the formula f = (3 × Δ)/(d + Δ), where Δ means the thickness of grain boundaries and d represents the diameter. The diameter of the Pt crystal was calculated to be 3.12 nm using the Debye–Scherrer formula38 (Fig. S9†), which corresponds to the grain size distribution histogram for the GBP-Pt-600 sample in Fig. 4d. As the value of volume fraction f is calculated to be 42%, it could be identified that grain boundaries represent an important component on the surface of the GBP-Pt-600 sample. This grain-boundary-rich porous platinum was synthesized via a special topo-chemical template strategy using a unique 2D space-confined reaction (Fig. 4e and S10†). Isolated Pt atoms in the nitrogen pot of the carbon framework (denoted as Pt–CN) were used as precursors and calcination of the Pt–CN precursor was further aimed to reorganize Pt atoms to form this porous structure. Based on the above results, the porous structure with high surface area in GBP-Pt-600 could also contribute to the high electrochemical performance.
 |
| | Fig. 4 (a) The Brunauer–Emmett–Teller analysis of the GBP-Pt-600 sample. (b) Pore size distribution of GBP-Pt-600. (c) High-resolution transmission electron microscopy image of the GBP-Pt-600 sample. The scale bar is 2 nm. (d) Grain size distribution histogram for GBP-Pt-600. (e) Schematic illustration of the synthesis process for grain-boundary porous Pt. | |
Conclusions
In conclusion, we report that optimal coordination-site exposure in grain-boundary porous platinum brings excellent activity and durability for the oxygen reduction reaction in fuel cells. The GBP-Pt-600 electrocatalyst exhibits 7 times higher ORR activity than commercial Pt/C. For the fuel cell test, a high activity (1.49 W cm−2, 0.71 A mg−1 Pt for mass activity) and stability (12.9% loss after 30 K cycles) could also be observed, all of which far surpass the U.S. DOE target in 2020 (0.44 A mg−1 Pt for mass activity and 40% loss for stability). Density Functional Theory (DFT) calculation suggests that proper Pt coordination site exposure in grain boundaries provides optimal adsorption energies for oxygen species, which could result in high intrinsic activity in the ORR, even superior to Pt(111) sites. X-ray Absorption Fine Structure (XAFS) analysis and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) were further used to verify this change of coordination numbers and investigate the origin of high stability. We anticipated that coordination-site exposure engineering opens a new avenue toward novel robust electrocatalysts for the fuel-cell oxygen reduction reaction.
Experimental
Synthesis of isolated Pt in the nitrogen pot of the carbon framework (denoted as Pt–CN)
Typically, 2 g urea and 1 g of dicyanodiamide were dissolved in 20 mL water. Then 50 mg H2PtCl6·6H2O was added into another 10 mL solution containing 5.8 g urea. Then these two solutions were mixed. The mixed solution was freeze-dried in a bulk tray dryer and the obtained white powder was heated in a porcelain boat at 530 °C for 4 h in an Ar flow at a speed of 0.5 mL s−1. The resulting sample was denoted as Pt–CN.
Synthesis of grain-boundary porous Pt (denoted as GBP-Pt)
The direct calcination of Pt–CN in air at high temperature was selected to be an effective way to remove the CN template. In a typical experiment, 50 mg Pt–CN nanosheet precursor was placed in a tube and heated to 400 °C, 500 °C, 600 °C, 700 °C and 800 °C, respectively, with a heating rate of 5°C min−1. After heating for 1 h, the sample cooled down to room temperature naturally. The resulting powder was collected for further characterization and denoted as GBP-Pt.
Structure characterization
The as-prepared products were examined using an X-ray powder diffraction (Philips X'Pert Pro Super diffractometer) instrument with Cu Kα radiation (λ = 1.54178 Å). The size and morphology of the samples were analyzed by using a transmission electron microscope (JEM-2100F field-emission electron microscope) which was operated at an acceleration voltage of 200 kV. X-ray photoelectron spectra (XPS) were obtained on an ESCALAB MK II X-ray photoelectron spectrometer with Mg Kα as the excitation source. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and EDS mapping images were taken on a JEOL JEM-ARF200F atomic resolution analytical microscope. The loading of Pt was measured on an inductively coupled plasma-atomic emission spectrometer (ICP-AES) on an Optima 7300 DV (PerkinElmer Corporation). The X-ray absorption fine structure (XAFS) measurements at the Pt L-edge were performed at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF), China. The storage ring of the SSRF was operated at 3.5 GeV with a maximum current of 210 mA. The nitrogen adsorption–desorption isotherms and corresponding pore size distribution were evaluated using the Brunauer–Emmett–Teller (BET) equation in the Micromeritics ASAP 2000 system.
Electrochemical measurements
Vulcan carbon XC-72 was used as the support for these catalysts. Typically, 5 mg of catalysts with 20 mg XC-72 were dispersed in hexane under stirring and evaporation at 50 °C with a Pt loading content of 20%. The carbon supported catalysts were further used in electrochemical measurements. 1.5 mg of catalysts/XC-72 were first suspended in 0.6 mL of 2:1 water/isopropanol solution with 5% Nafion in a small bottle and the catalyst ink was prepared by ultrasonic treatment for 40 min. 8 μL of the catalyst ink was loaded onto a polished glassy carbon electrode with 5 mm diameter. The Pt loadings of GBP-Pt and commercial Pt were determined by ICP-AES to be 19.6, and 20 μgPt/cm2, respectively. The catalyst coated electrode was mounted onto a Pine ASR rotator and used as the working electrode in O2 saturated 0.1 M HClO4 at room temperature (pH = 1). The electrochemical measurements were performed in a conventional three-electrode system in the CHI760D electrochemical station. Ag/AgCl and the graphite rod were used as the reference and the counter electrodes. The Luggin capillary was used to decrease the ohmic resistance of the solution. Cathodic polarization curves were recorded at a sweep rate of 10 mV s−1 and the rotating speed is 1600 rpm. Solution ohmic drop (iR-drop) was compensated and capacitive background current was corrected by subtracting the curve recorded in Ar-saturated solution. The potentials in our work were calibrated to the reversible hydrogen electrode (RHE) through RHE calibration, according to the Nernst equation: ERHE = EAg/AgCl + 0.059 × pH + 0.1976. The rotating ring-disk electrode (RRDE) measurements were also performed with the same catalyst loading. A glassy carbon (GC, f = 5.61 mm) disk-Pt ring RRDE was used. To determine the H2O2 yield, the ring electrode was held at 1.30 V to oxidize H2O2 diffused from the disk electrode. For the ORR at a RDE, the relative mass or specific activity could be calculated with the Koutecky–Levich equation 1/j = 1/jK + 1/jL, where j is the measured current density; jK and jL are the kinetic and diffusion-limiting current densities, respectively. The accelerated durability test (ADT) was performed in the same potential window and electrolyte with a sweep rate of 50 mV s−1 for 10 K or 30 K cycles. Then the sweep rate was changed back to 10 mV s−1 and the curves were recorded.
Fuel cell measurements
The membrane electrode assembly (MEA) was prepared through a hot-pressing method. The catalyst “ink” was prepared by ultrasonic dispersion of catalysts/XC-72 (1.4 mg) and 5 wt% Nafion solutions into 1.0 mL deionized water for 1 hour. The ink was directly coated on PTFE-pretreated Toray 060 carbon paper (1.2 × 1.2 cm2) as the cathode, and no microporous layer was used. The loading of Pt content for all catalysts was 0.2 mg cm−2 Pt. The anode catalyst is 60 wt% Pt/C with a loading of 0.2 mg cm−2 Pt. Then, the MEA was prepared by hot-pressing the cathode, anode, NRE 211 Nafion membrane, and gasket at about 3 MPa for 180 s. The compression was 15–20%. The active area of the MEA was 1.2 × 1.2 cm2. Fuel cell performance was tested at 354 K on a Model 850e fuel cell test system (Scribner Associates Inc.). H2 and O2 flow rates were 0.4 L min−1 at 100% RH for polarization curve measurement, and a back pressure of 1 or 2 bar was applied.
DFT calculations
All first-principles calculations were performed with the density functional theory (DFT) method implemented in the Vienna Ab initio Simulation Package (VASP) package. Each grain was simulated by Pt201 in cubic boxes, while the grain boundaries were generated by attachments of two Pt grains. The two topmost layers of Pt and the adsorbates are allowed to relax. The projector augmented wave (PAW) pseudopotential and the Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional were used in the calculations with a 450 eV plane-wave cut-off energy. All calculations were spin-polarized and the lattice parameters were optimized until the convergence tolerance of force on each atom was smaller than 0.05 eV. All periodic slab models have a vacuum spacing of at least 15 Å. The “generalized” coordination number 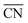 was calculated to eliminate “finite-size effects” by introducing a weight to each first-nearest neighbor atom j,33 corresponding to its own coordination number [cn(j)]. The formula to estimate
was calculated to eliminate “finite-size effects” by introducing a weight to each first-nearest neighbor atom j,33 corresponding to its own coordination number [cn(j)]. The formula to estimate  for a site is
for a site is 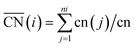 . In this work, the adsorption energies were calculated via the following equation to model the ORR.20,32,33
. In this work, the adsorption energies were calculated via the following equation to model the ORR.20,32,33| | | * + O2 + H+ + e− = = ∗OOH | (1) |
| | | ∗OOH + H+ + e− = = ∗O + H2O | (2) |
| | | ∗OH + H+ + e− = = ∗ + H2O | (4) |
It is well known that two RDS (potential-determining steps) are eqn (1) and (4), where the key reaction intermediates are free radicals *OOH and *OH. The ΔGO2 is calculated to be 4.92 eV at 298 K using the computational hydrogen electrode (CHE).
| | 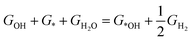 | (5) |
| | 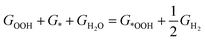 | (6) |
where
G∗OH and
G∗OOH are calculated using
eqn (7), where TS is gas-phase entropy correction, ZPE is the zero-point-energy correction,
Esolvation is the solvation energy granted and
EDFT is the total electronic energy.
| | | G∗ = ZPE + EDFT + Esolvation − TS | (7) |
Then the energies for reactions (1) and (4) could be calculated using eqn (8) and (9),
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank Dr Jie Tian at Engineering and Materials Science Experiment Centre for the help of HRTEM experiments. This work was financially supported by the National Basic Research Program of China (2015CB932302), Natural Science Foundation of China (No. 21890751, 91745113, 11621063, and 21601172), National Program for Support of Topnotch Young Professionals, and Fundamental Research Funds for the Central Universities (No. WK 2060190084). The authors also appreciate the support from the Major/Innovative Program of Development Foundation of Hefei Center for Physical Science and Technology.
Notes and references
- S. Chu and A. Majumdar, Nature, 2012, 488, 294 CrossRef CAS PubMed.
- R. F. Service, Science, 2009, 324, 1257–1259 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS.
- S. Krishnan and F. A. Armstrong, Chem. Sci., 2012, 3, 1015–1023 RSC.
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS.
- Q. Wang, Z.-Y. Zhou, Y.-J. Lai, Y. You, J.-G. Liu, X.-L. Wu, E. Terefe, C. Chen, L. Song and M. Rauf, J. Am. Chem. Soc., 2014, 136, 10882–10885 CrossRef CAS PubMed.
- K. Shimizu, L. Sepunaru and R. G. Compton, Chem. Sci., 2016, 7, 3364–3369 RSC.
- Y.-C. Wang, Y.-J. Lai, L. Song, Z.-Y. Zhou, J.-G. Liu, Q. Wang, X.-D. Yang, C. Chen, W. Shi, Y.-P. Zheng, M. Rauf and S.-G. Sun, Angew. Chem., Int. Ed., 2015, 127, 10045–10048 CrossRef.
- X. Huang, Y. Wang, W. Li and Y. Hou, Sci. China: Chem., 2017, 60, 1494–1507 CrossRef CAS.
- R. Wang, J. Liu, P. Liu, X. Bi, X. Yan, W. Wang, X. Ge, M. Chen and Y. Ding, Chem. Sci., 2014, 5, 403–409 RSC.
- L. Guo, W.-J. Jiang, Y. Zhang, J.-S. Hu, Z.-D. Wei and L.-J. Wan, ACS Catal., 2015, 5, 2903–2909 CrossRef CAS.
- C. Meng, T. Ling, T. Y. Ma, H. Wang, Z. Hu, Y. Zhou, J. Mao, X. W. Du, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2017, 29, 1604607 CrossRef.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS.
- M. del Carmen Gimenez-Lopez, A. Kurtoglu, D. A. Walsh and A. N. Khlobystov, Adv. Mater., 2016, 28, 9103–9108 CrossRef CAS.
- F. Saleem, Z. Zhang, B. Xu, X. Xu, P. He and X. Wang, J. Am. Chem. Soc., 2013, 135, 18304–18307 CrossRef CAS PubMed.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- M. Li, Z. Zhao, T. Cheng, A. Fortunelli, C.-Y. Chen, R. Yu, Q. Zhang, L. Gu, B. V. Merinov, Z. Lin, E. Zhu, T. Yu, Q. Jia, J. Guo, L. Zhang, W. A. Goddard, Y. Huang and X. Duan, Science, 2016, 354, 1414–1419 CrossRef CAS PubMed.
- D. Banham and S. Ye, ACS Energy Lett., 2017, 2, 629–638 CrossRef CAS.
- J. Greeley, I. Stephens, A. Bondarenko, T. P. Johansson, H. A. Hansen, T. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552 CrossRef CAS PubMed.
- F. Calle-Vallejo, M. T. Koper and A. S. Bandarenka, Chem. Soc. Rev., 2013, 42, 5210–5230 RSC.
- D. Li, C. Wang, D. S. Strmcnik, D. V. Tripkovic, X. Sun, Y. Kang, M. Chi, J. D. Snyder, D. van der Vliet and Y. Tsai, Energy Environ. Sci., 2014, 7, 4061–4069 RSC.
- D. Y. Chung, J. M. Yoo and Y.-E. Sung, Adv. Mater., 2018, 1704123 CrossRef PubMed.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- P. P. Lopes, D. Strmcnik, D. Tripkovic, J. G. Connell, V. Stamenkovic and N. M. Markovic, ACS Catal., 2016, 6, 2536–2544 CrossRef CAS.
- K. J. J. Mayrhofer, B. B. Blizanac, M. Arenz, V. R. Stamenkovic, P. N. Ross and N. M. Markovic, J. Phys. Chem. B, 2005, 109, 14433–14440 CrossRef CAS PubMed.
- M. Nesselberger, S. Ashton, J. C. Meier, I. Katsounaros, K. J. J. Mayrhofer and M. Arenz, J. Am. Chem. Soc., 2011, 133, 17428–17433 CrossRef CAS PubMed.
- D. Y. Chung, H. Shin, J. M. Yoo, K.-S. Lee, N.-S. Lee, K. Kang and Y.-E. Sung, J. Power Sources, 2016, 334, 52–57 CrossRef CAS.
- I. E. Stephens, A. S. Bondarenko, U. Grønbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744–6762 RSC.
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D.-J. Liu, Science, 2018, 362, 1276–1281 CrossRef CAS.
- Z. Liu, C. Yu, I. A. Rusakova, D. Huang and P. Strasser, Top. Catal., 2008, 49, 241–250 CrossRef CAS.
- K. Sasaki, H. Naohara, Y. Cai, Y. M. Choi, P. Liu, M. B. Vukmirovic, J. X. Wang and R. R. Adzic, Angew. Chem., Int. Ed., 2010, 49, 8602–8607 CrossRef CAS.
- F. Calle-Vallejo, J. I. Martínez, J. M. García-Lastra, P. Sautet and D. Loffreda, Angew. Chem., Int. Ed., 2014, 53, 8316–8319 CrossRef CAS.
- N. Ishiguro, S. Kityakarn, O. Sekizawa, T. Uruga, T. Sasabe, K. Nagasawa, T. Yokoyama and M. Tada, J. Phys. Chem. C, 2014, 118, 15874–15883 CrossRef CAS.
- N. Ishiguro, T. Saida, T. Uruga, O. Sekizawa, K. Nagasawa, K. Nitta, T. Yamamoto, S.-i. Ohkoshi, T. Yokoyama and M. Tada, Phys. Chem. Chem. Phys., 2013, 15, 18827–18834 RSC.
- O. Sekizawa, T. Uruga, K. Higashi, T. Kaneko, Y. Yoshida, T. Sakata and Y. Iwasawa, ACS Sustainable Chem. Eng., 2017, 5, 3631–3636 CrossRef CAS.
- H. Imai, K. Izumi, M. Matsumoto, Y. Kubo, K. Kato and Y. Imai, J. Am. Chem. Soc., 2009, 131, 6293–6300 CrossRef CAS.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01078e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Wangsheng
Chu
c,
Changzheng
Wu
b,
Wangsheng
Chu
c,
Changzheng
Wu

 was calculated to eliminate “finite-size effects” by introducing a weight to each first-nearest neighbor atom j,33 corresponding to its own coordination number [cn(j)]. The formula to estimate
was calculated to eliminate “finite-size effects” by introducing a weight to each first-nearest neighbor atom j,33 corresponding to its own coordination number [cn(j)]. The formula to estimate  for a site is
for a site is 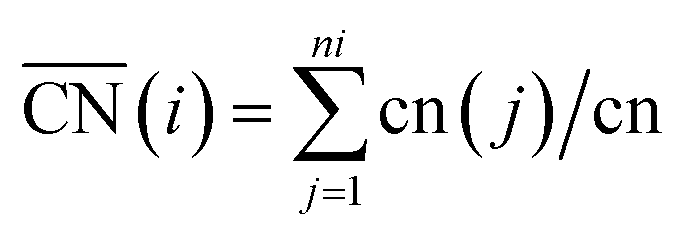 . As shown in Fig. 3a, taking the “a” site as an example, the
. As shown in Fig. 3a, taking the “a” site as an example, the  of the “a” site in a single Pt grain is 4.25. While two grains attach to form a grain boundary, the
of the “a” site in a single Pt grain is 4.25. While two grains attach to form a grain boundary, the  value of the “A” site in the grain boundary increases to 8. Based on the obtainment of the
value of the “A” site in the grain boundary increases to 8. Based on the obtainment of the  value, the adsorption energies of key intermediate species on Pt sites were further evaluated (Fig. 3b). The adsorption energies of intermediates present a linear relationship against the change of
value, the adsorption energies of key intermediate species on Pt sites were further evaluated (Fig. 3b). The adsorption energies of intermediates present a linear relationship against the change of  values in different Pt sites. The ΔG for hydroxyl species (*OH) is determined to be 0.111 eV in edge “a” sites while it changes to 0.821 eV in grain boundaries “A”, which approaches close to the optimal value 0.87 eV. Moreover, the two possible rate-determining steps in the ORR are also investigated in Fig. 3c. It can be seen that the “A” site in the grain boundary shows optimal Gibbs free energy and activity for the ORR compared to other sites, even better than Pt(111) sites. Based on these results, optimal adsorption energies of key intermediate species for the ORR could be realized by introducing grain boundary sites.
values in different Pt sites. The ΔG for hydroxyl species (*OH) is determined to be 0.111 eV in edge “a” sites while it changes to 0.821 eV in grain boundaries “A”, which approaches close to the optimal value 0.87 eV. Moreover, the two possible rate-determining steps in the ORR are also investigated in Fig. 3c. It can be seen that the “A” site in the grain boundary shows optimal Gibbs free energy and activity for the ORR compared to other sites, even better than Pt(111) sites. Based on these results, optimal adsorption energies of key intermediate species for the ORR could be realized by introducing grain boundary sites.

 calculation for “a” single cluster and “A” grain boundary sites. (b) The adsorption energies of key intermediates species on different
calculation for “a” single cluster and “A” grain boundary sites. (b) The adsorption energies of key intermediates species on different  Pt sites. (c) The calculation for Gibbs free energy in two possible rate-determining steps for the ORR. (d) Pt L3-edge k3χ(k) oscillation curves for GBP-Pt-600, commercial Pt and Pt foil as the reference. (e) The Pt L3-edge EXAFS Fourier transformed curves for GB.
Pt sites. (c) The calculation for Gibbs free energy in two possible rate-determining steps for the ORR. (d) Pt L3-edge k3χ(k) oscillation curves for GBP-Pt-600, commercial Pt and Pt foil as the reference. (e) The Pt L3-edge EXAFS Fourier transformed curves for GB.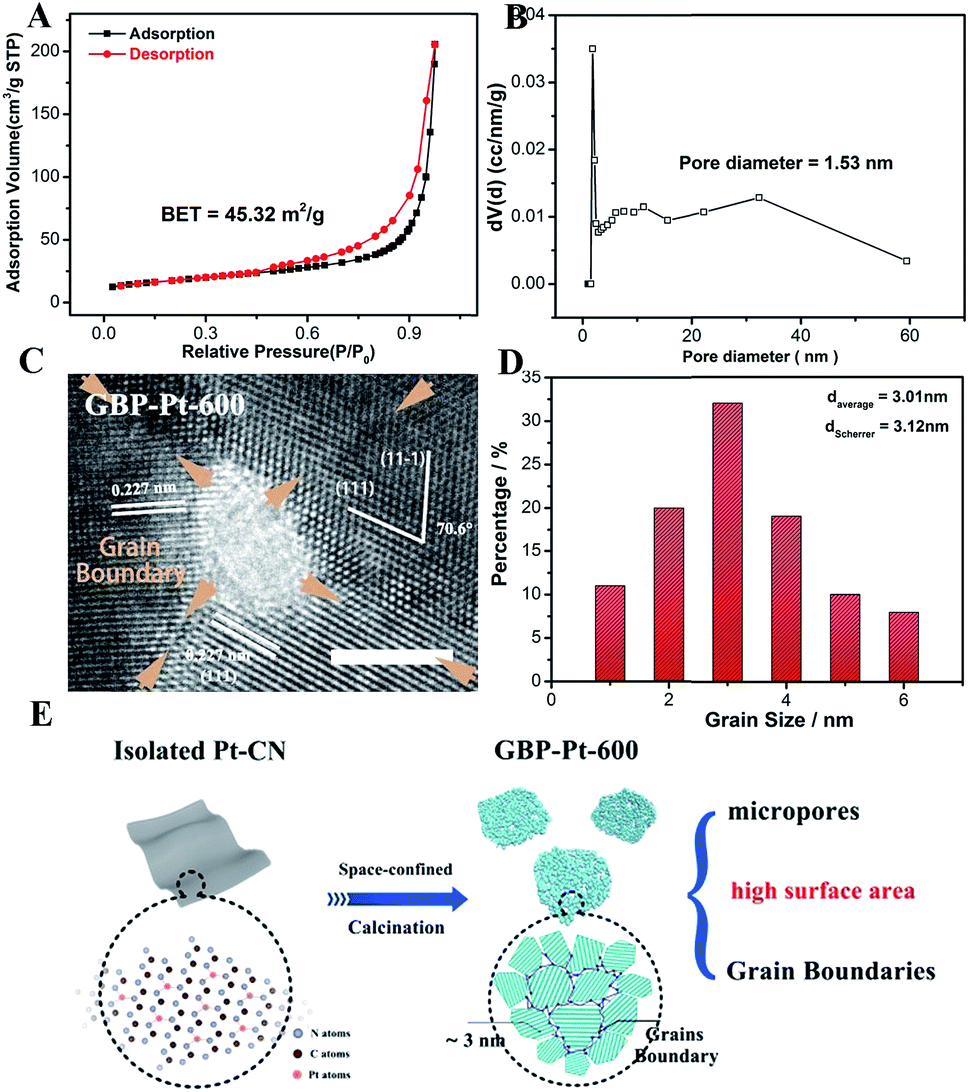
 was calculated to eliminate “finite-size effects” by introducing a weight to each first-nearest neighbor atom j,33 corresponding to its own coordination number [cn(j)]. The formula to estimate
was calculated to eliminate “finite-size effects” by introducing a weight to each first-nearest neighbor atom j,33 corresponding to its own coordination number [cn(j)]. The formula to estimate  for a site is
for a site is  . In this work, the adsorption energies were calculated via the following equation to model the ORR.20,32,33
. In this work, the adsorption energies were calculated via the following equation to model the ORR.20,32,33