
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthetic reconstitution of deoxysugar phosphoramidate metalloprotease inhibitors using an N–P-bond-forming kinase†
Alexandra
Baulig
ab,
Irina
Helmle
a,
Marius
Bader
ab,
Felix
Wolf
ab,
Andreas
Kulik
 c,
Arwa
Al-Dilaimi
d,
Daniel
Wibberg
d,
Jörn
Kalinowski
d,
Harald
Gross
ab and
Leonard
Kaysser
*ab
c,
Arwa
Al-Dilaimi
d,
Daniel
Wibberg
d,
Jörn
Kalinowski
d,
Harald
Gross
ab and
Leonard
Kaysser
*ab
aDepartment of Pharmaceutical Biology, Pharmaceutical Institute, University of Tübingen, 72076 Tübingen, Germany. E-mail: leonard.kaysser@uni-tuebingen.de
bGerman Centre for Infection Research (DZIF), partner site Tübingen, 72076 Tübingen, Germany
cInterfaculty Institute for Microbiology and Infection Medicine Tübingen (IMIT), Microbiology/Biotechnology, University of Tübingen, 72076 Tübingen, Germany
dCenter for Biotechnology (CeBiTec), Bielefeld University, 33615 Bielefeld, Germany
First published on 21st March 2019
Abstract
Phosphoramidon is a potent metalloprotease inhibitor and a widespread tool in cell biology research. It contains a dipeptide backbone that is uniquely linked to a 6-deoxysugar via a phosphoramidate bridge. Herein, we report the identification of a gene cluster for the formation of phosphoramidon and its detailed characterization. In vitro reconstitution of the biosynthesis established TalE as a phosphoramidate-forming kinase and TalC as the glycosyltransferase which installs the L-rhamnose moiety by phosphoester linkage.
Introduction
Phosphoramidate and phosphonamidate moieties are common motifs in medicinal chemistry. Nucleoside phosphoramidates are developed as antiviral prodrugs with great success.1 In protease inhibitors they serve as peptidomimetics replacing the carbonyl carbon in the amide bond.2 They can be seen as transition-state analogues of the enzyme reaction and coordinate to the active site metal ion in metalloproteases.3 In natural products, however, N–P bonds are comparably rare and little is known about their biosynthesis.4 Phosphoramidon (Fig. 1; 1) and its stereoisomer talopeptin (2) have been isolated from Streptomyces culture broths and are potent inhibitors of metalloendopeptidases including thermolysin, Pseudomonas elastase LasB, neutral endopeptidase and endothelin-converting enzyme.5–7 Phosphoramidon was extensively investigated in animal models and pilot clinical studies for its effects on various diseases.8–10 Today, this compound is still a widely used pharmacological tool in cell biological research. The inhibitory activity of 1 and 2 has been attributed to the metal-chelating properties of a phosphoramidate moiety which uniquely links a deoxysugar to the N-terminus of a L-Leu–L-Trp dipeptide backbone.11 The deoxysugar consists of L-rhamnose in the case of 1 and 6-deoxy-L-talose in the case of 2. The two molecules belong to a small group of structurally diverse natural products with N–P bonds (Fig. 1). The biosynthesis of such compounds has drawn a lot of interest and the biosynthetic gene clusters (BGCs) were identified for microcin C7 (3) in Escherichia coli,12,13 agrocin 84 in Agrobacterium radiobacter,14 fosfazinomycin in Streptomyces sp. WM6372![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 and phaseolotoxin in Pseudomonas syringae pv. phaseolicola.16 However, the enzymatic mechanism for the formation of the N–P bond has so far only been established for 3. Here, the adenylating enzyme MccB catalyzes the formation of the phosphoramidate linkage of a precursor peptide and AMP via the generation of a succinimide intermediate and the consumption of two molecules of ATP.17 Given the structural difference to 3 and the absence of an adenylyl group we speculated about a distinct biosynthetic strategy to form the phosphoramidate bridge in 1 and 2.
15 and phaseolotoxin in Pseudomonas syringae pv. phaseolicola.16 However, the enzymatic mechanism for the formation of the N–P bond has so far only been established for 3. Here, the adenylating enzyme MccB catalyzes the formation of the phosphoramidate linkage of a precursor peptide and AMP via the generation of a succinimide intermediate and the consumption of two molecules of ATP.17 Given the structural difference to 3 and the absence of an adenylyl group we speculated about a distinct biosynthetic strategy to form the phosphoramidate bridge in 1 and 2.
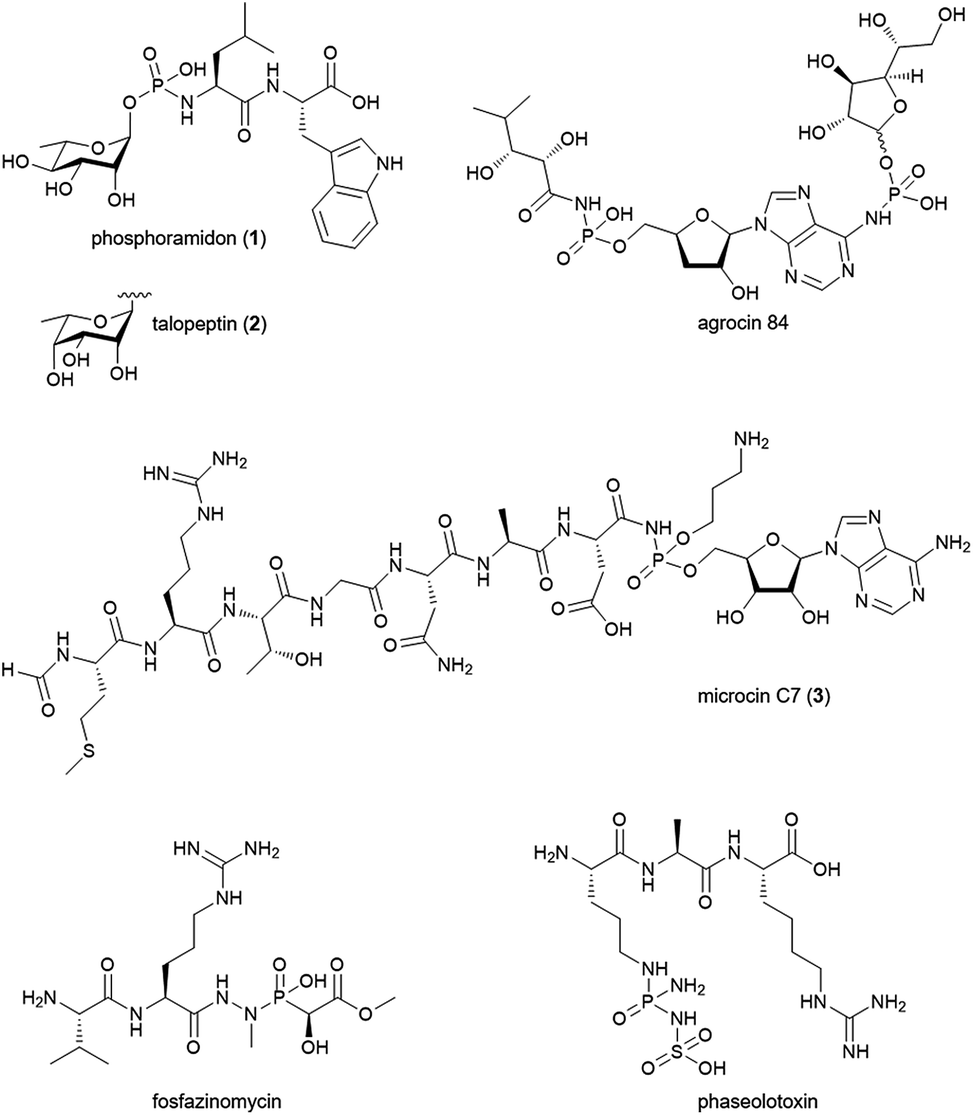 | ||
| Fig. 1 Selected natural products with phosphoramidate or phosphonamidate moieties. | ||
Results and discussion
Identification of the talopeptin biosynthetic gene cluster
To explore the genetic basis for the assembly of 1 and 2 we sequenced the genome of Streptomyces mozunensis MK-23, the original talopeptin producer. This approach resulted in a high quality draft genome with eight scaffolds and 7708 predicted genes. Unexpectedly, analysis of the draft genome for the presence of non-ribosomal peptide synthetase (NRPS) genes did not result in the identification of a reasonable candidate pathway. We thus used Cpz31, a rhamnosyltransferase from the caprazamycin gene cluster,18 as reference to scan the genome sequence on the assumption, that a homologous enzyme could be involved in the installation of the deoxysugar in 2. A BLAST comparison revealed one homolog, TalC, with 45% sequence identity to Cpz31. The respective gene was co-located with genes for a putative ATP-grasp enzyme (TalD) and a putative PEP synthase/pyruvate, phosphate dikinase (TalE; Fig. 2A). Adjacent to this small operon-like structure were putative genes encoding for transport and regulatory functions. Based on these findings a three-reaction pathway to 2 (and 1) would be conceivable (Fig. 2B). TalD would form a dipeptide (4) from L-Leu and L-Trp. This could serve as a precursor for TalE to install an N-terminal phosphoramidate generating 5. The subsequent transfer of the deoxysugar could be catalyzed by TalC to afford 2.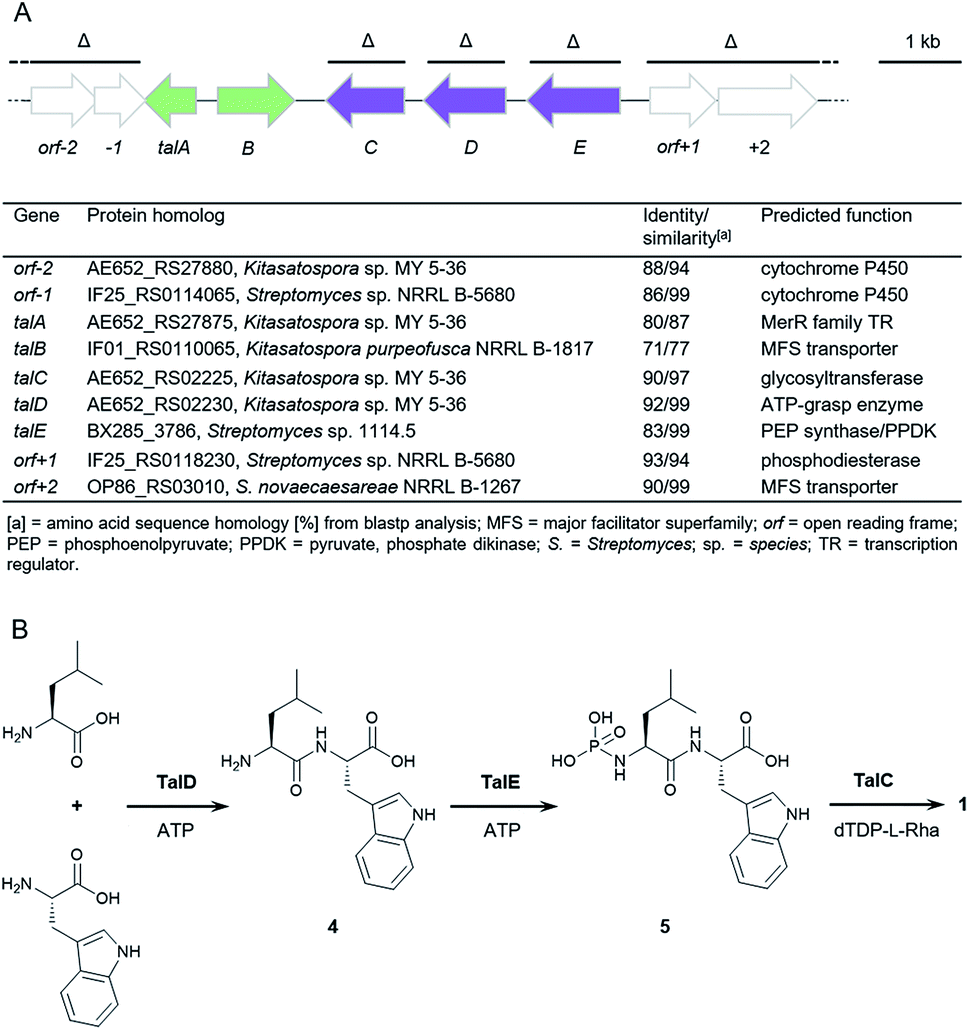 | ||
| Fig. 2 Biosynthesis of 1 and 2. (A) Organization of the talose BGC from S. mozunensis MK-23 and gene cluster table. Genes deleted in the course of this study are indicated (Δ). Purple arrows: biosynthetic genes. Green arrows: transport and regulation. (B) Proposed pathway for the biosynthesis of 1. | ||
Heterologous expression and gene deletion studies
To confirm the identity of the putative tal gene cluster, we transferred the pathway on an integrative pCC1FOS derivative into Streptomyces sp. MK730-62F2 for heterologous expression. This strain is known to provide activated L-rhamnose for the endogenous production of caprazamycins and the respective deoxysugar biosynthesis genes have recently been identified.18,19 To our satisfaction, LC-MS analysis of culture extracts from the heterologous mutant showed the production of a single new peak with a mass, retention time and UV-spectrum identical to a commercial standard of 1 (Fig. 3, S1, S4 and S5†) but with a 0.2 min shift in retention time to 2. This mass peak was absent in the culture extracts of the host without the tal cluster. This finding confirmed that we have found a BGC for the production of 1 and 2, likely comprising the talCDE operon. It appears that the deoxysugar transferase TalC is flexible towards the stereoconfiguration of the hydroxy group at C-4. To unambiguously determine the organization and the boundaries of the tal cluster we individually deleted the genes talC, talD and talE as well as the bordering regions orf-7 to orf-1 and orf+1 to orf+12 and expressed the mutant clusters heterologously. LC-MS analysis of culture extracts from the mutant strains showed that the biosynthesis of 1 was not affected by the Δorf-7–orf-1 and the Δorf+1–orf+12 deletions. In contrast, the ΔtalC, ΔtalD and ΔtalE mutations led to an apparent loss of production (Fig. 3). This strongly indicates that talA, a MerR-like regulatory gene, and talB, encoding a putative MFS transporter, form the BGC for 1 and 2 together with the talCDE operon. Notably, trace amounts of 1 could still be detected in cultures of the ΔtalD mutant by MS analysis. This might be explained by the supply of 4 from standard protein degradation processes.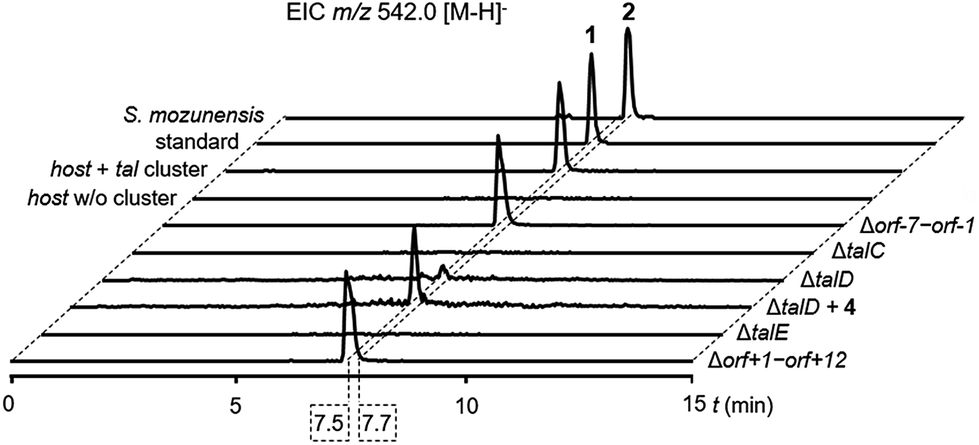 | ||
| Fig. 3 LC-MS analysis of culture extracts for the production of 1 in Streptomyces sp. MK730-62F2/talMB01 and 2 in S. mozunensis MK-23. Extracted ion chromatograms (EICs). | ||
ATP-grasp enzymes such as TalD activate carboxylic acid substrates as acylphosphates under consumption of ATP for subsequent condensation reactions. They have received increasing attention as discrete amide ligases for their ability to form peptide bonds in natural product biosynthesis.20 Our results suggest that the peptide backbone of 1 and 2 is assembled by ATP-grasp biochemistry in analogy to other small-molecule protease inhibitors e.g. the ketomemicins but in contrast to the class of hydroxamic acid-containing metalloprotease inhibitors which derive from NRPS systems.21–23 To unambiguously assign the role of TalD in the biosynthesis of 1 and 2 we supplemented cultures of the ΔtalD mutant with a synthetic 4 precursor. LC-MS analysis of the culture extracts showed that the addition of the dipeptide largely restored the production of 1 in the mutant (Fig. 3). This result confirms the function of TalD as a dipeptide synthetase in the formation of 1 and 2.
Identification of TalE as an N–P-bond-forming kinase
Next, we wanted to explore the biosynthetic mechanism that installs the unique deoxysugar phosphoramidate moiety onto 4. Therefore, we cloned the remaining two biosynthetic genes talC and talE for protein production in E. coli. The putative kinase His-TalE could be produced in yields of 2.71 mg L−1, whereas His-TalC could not be obtained as soluble protein under various conditions (Fig. S10 and S11†). TalE has homology to PEP synthases and pyruvate, phosphate dikinases (PPDKs). PPDKs catalyze the 2-step conversion of pyruvate, ATP and phosphate to phosphoenolpyruvate, AMP and PPi in plants and the reversible reaction bacteria.24 They use a catalytic histidine residue to generate a phosphohistidine intermediate with a high-energy N–P bond that serves as a phosphate shuttle between the two reaction centers of the enzyme.25 Phosphohistidine intermediates can be found in various kinases.26 Some of those enzymes e.g. Hpr or nucleoside diphosphate kinases, were shown to transfer the phosphoryl group onto histidine residues of other proteins to regulate their enzymatic activity.27We thus wondered if TalE might catalyze a similar reaction using the terminal amine of a dipeptide as the acceptor substrate instead of a proteinogenic histidine. To test this hypothesis we incubated purified His-TalE with ATP and synthetic 4 for 2 hours at 30 °C. LC-MS analysis of the reaction mixture showed the presence of an additional mass peak 5 at retention time 8.4 min with m/z 396.1 [M – H]− (Fig. 4 and S7†). The production of this new compound was dependent on the concentration of TalE (Fig. S9†) and absent when one of the assay components was omitted (Fig. 4). HR-MS measurements of 5 indicated a molecular formula of C17H23N3O6P (m/zmeas for [M − H]− 396.13400 vs. m/zcalcd for [M − H]− 396.13300, Δ = +2.53 ppm; Fig. S2 and S3†) and thus a possible phosphoramidate as the TalE product. To unequivocally determine the structure of 5 we purified the compound for comparison of the spectral properties of the product with those of its precursor 4. Both compounds were subjected to a series of 1H/13C/15N-based 1D/2D NMR standard experiments (Fig. 4 and S12–S27†). Subsequent analysis of all obtained spectra confirmed the presence of the Leu–Trp skeleton in 4 and 5. The 1H and 13C NMR spectrum of 5 was highly similar to the corresponding spectra of 4, with the exception of the resonances of Leu-α and Leu-β whose proton resonances shifted upfield, while the corresponding carbon resonances shifted downfield (Tables S1 and S2†), suggesting already that the phosphorylation had occurred at the N-terminal leucine residue of 4. This hypothesis was further corroborated by the interpretation of a 1H decoupled 31P NMR spectrum, which revealed that 5 contained, in contrast to 4 a phosphorus resonance, whose shift value δP −0.25 was indicative for a phosphoramidate group (Fig. S28 and S29†). Final evidence for the existence of a N–P bond in 5 was provided by a cross-correlation between Leu-α and P, observed in the 1H–31P HMBC NMR spectrum of 5 (Fig. 4, S30†). A complementary MS/MS analysis of 5 of the [M + H]+ ion at m/z 398 produced b- and y-series fragment ions that were also in full agreement with the proposed structure (Fig. S8†).
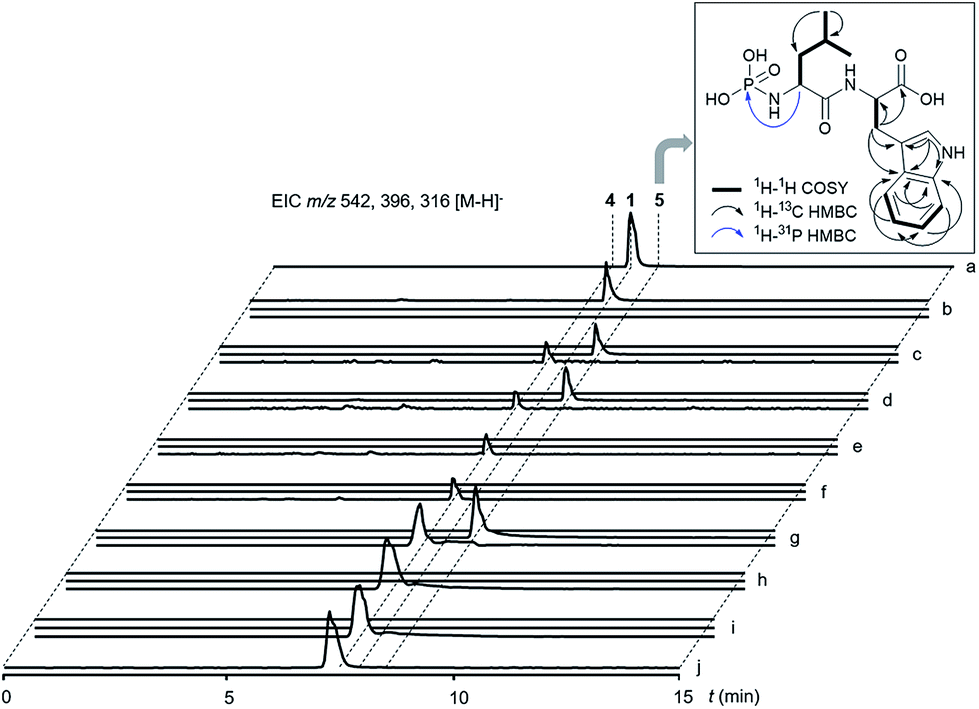 | ||
| Fig. 4 LC-MS analysis of enzyme reactions. Extracted ion chromatograms (EICs) for m/z 542 [M − H]− (1), m/z 396 [M − H]− (5) and m/z 316 [M − H]− (4). (a) Commercial standard of 1. (b) Enzymatic assay with TalC (protein extract of ΔtalE mutant), dTDP-L-rhamnose, TalE, 4 and ATP showed conversion of 4 to 1. (c) Control assay of (b) without TalC using a protein extract of the ΔtalC mutant. (d) Control assay of (b) without dTDP-L-rhamnose. (e) Control assay: protein extract of ΔtalC mutant with 4. (f) Control assay: protein extract of ΔtalE mutant with 4. (g) Enzymatic assay of TalE with 4 and ATP showed conversion of 4 to 5. (h) Control assay of (g) without TalE. (i) Control assay of (g) without ATP. (j) Control assay of (g) without 4. Box: observed NMR correlations of 5. | ||
These results confirm that TalE catalyzes the second step in the biosynthesis of 1 and 2 using the TalD product 4 and ATP as substrates to form 5. To the best of our knowledge, TalE represents the first enzyme in natural product biosynthesis that generates a small molecule phosphoramidate by simple phosphorylation of an amine. Very recently, Raushel and co-workers reported on a so far unprecedented enzyme-catalyzed phosphorylation of an amide to generate γ-phospho-L-glutamine.28,29 The respective enzyme, Cj1418, is involved in O-methyl phosphoramidate modification of capsular polysaccharides in Campylobacter jejuni. Notably, Cj1418 shows conserved protein domains typical for PEP synthases and PPDKs but has no significant sequence similarity to TalE (<20% on AA level). It appears that members of this family of kinase enzymes have evolved separately in bacteria to synthesize small-molecule phosphoramidates in the primary and secondary metabolism. It is tempting to speculate that putative PPDK homologs encoded in the BGCs for agrocin 84 and phaseolotoxin might have an analogous role in the respective pathways.14,16
In vitro biosynthesis of phosphoramidon
We next aimed to establish 5 as a genuine intermediate in the biosynthesis of 1. Therefore, we added dTDP-L-rhamnose and a TalC-containing protein extract to the TalE assay. After incubation for 16 h at 30 °C we analyzed the reaction mixture by LC-MS and identified a single product peak matching 1 in retention time, UV spectrum, mass and MS/MS fragmentation (Fig. 4, S1, S6 and 7†). 1 was not produced when either 5, dTDP-L-rhamnose or TalC were absent in the assay. These results show that TalC uses 5 as an acceptor substrate to catalyze the transfer of a deoxysugar to the phosphoryl group. In general, glycosylation is a common theme in natural product biosynthesis altering target specificity and pharmacological properties of a parent scaffold.30 Particularly, 6-deoxyhexoses are prevalent features in bacterial secondary metabolites. The substrate promiscuity of the respective glycosyltransferases has been extensively used to engineer the structure of bioactive small-molecules.31 In most cases these enzymes build a direct O-glycosidic link to the aglycon although the formation of thioglycosides, N-glycosides and C-glycosides is also known.32–34 Attachment of sugars in natural product biosynthesis via phosphoester linkage, however, is very rare and has only been functionally investigated for moenomycins. Here, Walker and co-workers showed that MoeGT1 transfers galacturonic acid from UDP-GalUA to a farnesyl phosphoglycerate precursor.35 The enzymatic installation of an L-rhamnose moiety to the phosporamidate of 5 thus represents a so far undescribed reaction in natural product biochemistry and adds TalE as a unique biocatalyst to the family of 6-deoxysugar transferases.Conclusions
In conclusion, we have identified the gene cluster for the biosynthesis of phosphoramidate metalloprotease inhibitors. Chemical complementation and in vitro reconstitution revealed a three-reaction pathway to form 1 and 2, which involves exceptional biochemistry. An ATP-grasp amide ligase, a phosphoryl transferase and a deoxysugar transferase work together in the condensation of two amino acids and the subsequent linkage of a 6-deoxyhexose to the resulting dipeptide via a phosporamidate bridge. A preliminary search in public genome databases found a number of homologous orphan pathways containing a talCDE-like putative operon (Fig. S31†). Given the possibility that the encoded ATP-grasp enzymes and glycosyltransferases may express a different substrate tolerance than TalD and TalC, respectively, the identified gene clusters could direct the synthesis of new metalloprotease inhibitors. Additionally, both enzymes have potential as flexible biocatalysts for in vivo and in vitro engineering of bioactive small-molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank Hamada Saad for helpful advises, Dr Dorothee Wistuba for HR-MS support and Anika Winkler for expert assistance during DNA sequencing. This work was generously supported by the DFG (KA 3071/4-1 and GRK 1708). A. K. was supported by grants from the SFB 766.Notes and references
- Y. Mehellou, J. Balzarini and C. McGuigan, ChemMedChem, 2009, 4, 1779–1791 CrossRef CAS PubMed.
- J. Vagner, H. Qu and V. J. Hruby, Curr. Opin. Chem. Biol., 2008, 12, 292–296 CrossRef CAS PubMed.
- D. E. Tronrud, A. F. Monzingo and B. W. Matthews, Eur. J. Biochem., 1986, 157, 261–268 CrossRef CAS PubMed.
- A. J. Waldman, T. L. Ng, P. Wang and E. P. Balskus, Chem. Rev., 2017, 117, 5784–5863 CrossRef CAS PubMed.
- S. Umezawa, K. Tatsuta, O. Izawa, T. Tsuchiya and H. Umezawa, Tetrahedron Lett., 1972, 13, 97–100 CrossRef.
- H. Suda, T. Aoyagi, T. Takeuchi and H. Umezawa, J. Antibiot., 1973, 26, 621–623 CrossRef CAS PubMed.
- S. Murao, M. Katsura, K.-i. Fukuhara and K. Oda, Agric. Biol. Chem., 1980, 44, 701–703 CAS.
- N. Crimi, R. Polosa, G. Pulvirenti, S. Magri, G. Santonocito, G. Prosperini, C. Mastruzzo and A. Mistretta, Thorax, 1995, 50, 505–510 CrossRef CAS PubMed.
- H. H. Kramer, K. Schmidt, S. Leis, M. Schmelz, C. Sommer and F. Birklein, Exp. Neurol., 2005, 195, 179–184 CrossRef CAS PubMed.
- M. P. Love, W. G. Haynes, G. A. Gray, D. J. Webb and J. J. McMurray, Circulation, 1996, 94, 2131–2137 CrossRef CAS PubMed.
- L. H. Weaver, W. R. Kester and B. W. Matthews, J. Mol. Biol., 1977, 114, 119–132 CrossRef CAS PubMed.
- M. A. Novoa, L. Diaz-Guerra, J. L. San Millan and F. Moreno, J. Bacteriol., 1986, 168, 1384–1391 CrossRef CAS PubMed.
- J. E. Gonzalez-Pastor, J. L. San Millan, M. A. Castilla and F. Moreno, J. Bacteriol., 1995, 177, 7131–7140 CrossRef CAS PubMed.
- J. G. Kim, B. K. Park, S. U. Kim, D. Choi, B. H. Nahm, J. S. Moon, J. S. Reader, S. K. Farrand and I. Hwang, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8846–8851 CrossRef CAS PubMed.
- J. T. Gao, K. S. Ju, X. M. Yu, J. E. Velasquez, S. Mukherjee, J. Lee, C. M. Zhao, B. S. Evans, J. R. Doroghazi, W. W. Metcalf and W. A. van der Donk, Angew. Chem., Int. Ed., 2014, 53, 1334–1337 CrossRef CAS PubMed.
- S. Aguilera, K. Lopez-Lopez, Y. Nieto, R. Garciduenas-Pina, G. Hernandez-Guzman, J. L. Hernandez-Flores, J. Murillo and A. Alvarez-Morales, J. Bacteriol., 2007, 189, 2834–2843 CrossRef CAS PubMed.
- R. F. Roush, E. M. Nolan, F. Lohr and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 3603–3609 CrossRef CAS PubMed.
- L. Kaysser, L. Lutsch, S. Siebenberg, E. Wemakor, B. Kammerer and B. Gust, J. Biol. Chem., 2009, 284, 14987–14996 CrossRef CAS PubMed.
- L. Kaysser, E. Wemakor, S. Siebenberg, J. A. Salas, J. K. Sohng, B. Kammerer and B. Gust, Appl. Environ. Microbiol., 2010, 76, 4008–4018 CrossRef CAS PubMed.
- Y. Ogasawara and T. Dairi, Chem.–Eur. J., 2017, 23, 10714–10724 CrossRef CAS PubMed.
- J. Kawata, T. Naoe, Y. Ogasawara and T. Dairi, Angew. Chem., Int. Ed., 2017, 56, 2026–2029 CrossRef CAS PubMed.
- F. Wolf, J. S. Bauer, T. M. Bendel, A. Kulik, J. Kalinowski, H. Gross and L. Kaysser, Angew. Chem., Int. Ed., 2017, 56, 6665–6668 CrossRef CAS PubMed.
- F. Leipoldt, J. Santos-Aberturas, D. P. Stegmann, F. Wolf, A. Kulik, R. Lacret, D. Popadic, D. Keinhorster, N. Kirchner, P. Bekiesch, H. Gross, A. W. Truman and L. Kaysser, Nat. Commun., 2017, 8, 1965 CrossRef PubMed.
- H. J. Evans and H. G. Wood, Proc. Natl. Acad. Sci. U. S. A., 1968, 61, 1448–1453 CrossRef CAS.
- O. Herzberg, C. C. Chen, G. Kapadia, M. McGuire, L. J. Carroll, S. J. Noh and D. Dunaway-Mariano, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2652–2657 CrossRef CAS.
- S. Cheek, H. Zhang and N. V. Grishin, J. Mol. Biol., 2002, 320, 855–881 CrossRef CAS PubMed.
- S. R. Fuhs and T. Hunter, Curr. Opin. Cell Biol., 2017, 45, 8–16 CrossRef CAS PubMed.
- Z. W. Taylor, H. A. Brown, T. Narindoshvili, C. Q. Wenzel, C. M. Szymanski, H. M. Holden and F. M. Raushel, J. Am. Chem. Soc., 2017, 139, 9463–9466 CrossRef CAS PubMed.
- Z. W. Taylor, A. R. Chamberlain and F. M. Raushel, Biochemistry, 2018, 57, 5447–5455 CrossRef CAS PubMed.
- S. I. Elshahawi, K. A. Shaaban, M. K. Kharel and J. S. Thorson, Chem. Soc. Rev., 2015, 44, 7591–7697 RSC.
- R. W. Gantt, P. Peltier-Pain and J. S. Thorson, Nat. Prod. Rep., 2011, 28, 1811–1853 RSC.
- C. Durr, D. Hoffmeister, S. E. Wohlert, K. Ichinose, M. Weber, U. Von Mulert, J. S. Thorson and A. Bechthold, Angew. Chem., Int. Ed., 2004, 43, 2962–2965 CrossRef PubMed.
- T. J. Oman, J. M. Boettcher, H. Wang, X. N. Okalibe and W. A. van der Donk, Nat. Chem. Biol., 2011, 7, 78–80 CrossRef CAS PubMed.
- A. P. Salas, L. Zhu, C. Sanchez, A. F. Brana, J. Rohr, C. Mendez and J. A. Salas, Mol. Microbiol., 2005, 58, 17–27 CrossRef CAS PubMed.
- B. Ostash, E. H. Doud, C. Lin, I. Ostash, D. L. Perlstein, S. Fuse, M. Wolpert, D. Kahne and S. Walker, Biochemistry, 2009, 48, 8830–8841 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00641a |
| This journal is © The Royal Society of Chemistry 2019 |