
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCuxNiy alloy nanoparticles embedded in a nitrogen–carbon network for efficient conversion of carbon dioxide†
Dongxing
Tan
a,
Jianling
Zhang
 *a,
Xiuyan
Cheng
a,
Xiuniang
Tan
a,
Jinbiao
Shi
a,
Bingxing
Zhang
a,
Buxing
Han
a,
Lirong
Zheng
b and
Jing
Zhang
b
*a,
Xiuyan
Cheng
a,
Xiuniang
Tan
a,
Jinbiao
Shi
a,
Bingxing
Zhang
a,
Buxing
Han
a,
Lirong
Zheng
b and
Jing
Zhang
b
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid, Interface and Chemical Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, School of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, P. R. China. E-mail: zhangjl@iccas.ac.cn
bBeijng Synchrotron Radiation Facility (BSRF), Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 15th March 2019
Abstract
The electrocatalytic conversion of CO2 to CO using non-noble metal catalysts under mild conditions is of great importance. Achieving the combination of high activity, selectivity and current density by developing electrocatalysts with desirable compositions and structures is challenging. Here we prepared for the first time CuxNiy alloy nanoparticles embedded in a nitrogen–carbon network. Such an electrocatalyst not only well overcomes the disadvantages of single Cu and Ni catalysts but has a high CO2 adsorption capacity. Outstandingly, the catalyst can effectively convert CO2 into CO with a maximum faradaic efficiency of 94.5% and current density of 18.8 mA cm−2 at a low applied potential of −0.60 V (versus reversible hydrogen electrode, RHE). Moreover, the catalyst is very stable during long-term electrolysis owing to the stabilization of the nitrogen–carbon network.
Introduction
CO2 is a rich C1 resource and its conversion into chemicals has attracted more and more attention.1–3 Particularly, reducing CO2 into CO, which is a key starting material for the synthesis of important chemicals and commodities, is of great importance.4 Among the transformation approaches, the electrochemical CO2 reduction reaction (CO2 RR) into CO using an electrocatalyst is a promising way.5–9 Compared with precious metal electrocatalysts (Au, Ag and Pd),10–12 it is more desirable to develop alternative earth-abundant metal catalysts for the CO2 RR to CO. However, non-noble metal electrocatalysts often suffer from low activities and selectivities. It has been proposed that the formation of metal alloy or bimetallic catalysts (especially with noble metals) can improve the efficiency of non-precious metals for the CO2 RR, through tuning their binding energy to reaction intermediates and regulating their inherent activity.13–22 However, the performances of these electrocatalysts for the CO2 RR still need improvement in terms of two issues. First, other carbon products (e.g. formic acid, methane, etc.) are inevitably produced in many catalytic systems. Second, the current density is usually low, especially at relatively low overpotentials. It is challenging to develop non-precious metal electrocatalysts with combined high activity, selectivity and current density for the CO2 RR into CO.Here we demonstrate for the first time the formation of Cu/Ni alloy nanoparticles embedded in a three-dimensional (3D) nitrogen–carbon network. Outstandingly, the as-synthesized catalyst can effectively convert CO2 into CO with a maximum faradaic efficiency of 94.5% and current density of 18.8 mA cm−2 at a low applied potential of −0.60 V. Such an electrocatalyst well overcomes the disadvantages of single Cu and Ni catalysts. For example, Cu catalysts usually produce multiple products due to their moderate binding energy to most reaction intermediates.13,23 Ni tends to favor the competitive hydrogen evolution reaction (HER) in aqueous electrolyte.24 Moreover, the Cu/Ni catalyst is very stable during long-term electrolysis owing to the stabilization of the nitrogen–carbon network.
Results and discussion
To get the target product, a Cu/Ni bimetallic complex was first prepared by a hydrothermal method using copper powder (Fig. S1†) and nickel(II) nitrate hexahydrate as precursors. From scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images, the Cu/Ni bimetallic complex has a 3D network structure (Fig. 1a, d and S2†). After calcination of the Cu/Ni bimetallic complex at 900 °C for 1 h, the target product was obtained. It well maintains the network structure of the bimetallic complex, but it is embedded with nanoparticles in the range of 50–300 nm (Fig. 1b and c). The high-resolution transmission electron microscopy (HRTEM) image (Fig. 1e) shows a lattice fringe corresponding to the crystal plane (111) (d = 0.21 nm) of the face centered cubic Cu/Ni bimetallic alloy,25,26 which is consistent with the X-ray diffraction (XRD) pattern (Fig. S3†). Energy dispersive X-ray spectroscopy (EDS) line scans on nanoparticles reveal that Cu and Ni elements are distributed nearly at the same locations (Fig. 1f), further proving the formation of the Cu/Ni alloy. Energy dispersive X-ray elemental (EDX) mapping images show that C, N and O elements are evenly distributed in the whole sample, while Cu and Ni elements aggregate into granular forms, corresponding to the Cu–Ni alloy (Fig. 1g). The Cu and Ni contents were determined to be 37.8 and 33.5 wt% by inductively coupled plasma-mass spectrometry (ICP-MS), respectively. The contents of C and N are 23.2 and 4.2 wt%, respectively, as determined by elemental analysis. The product was denoted as Cu1.0Ni1.0/N–C.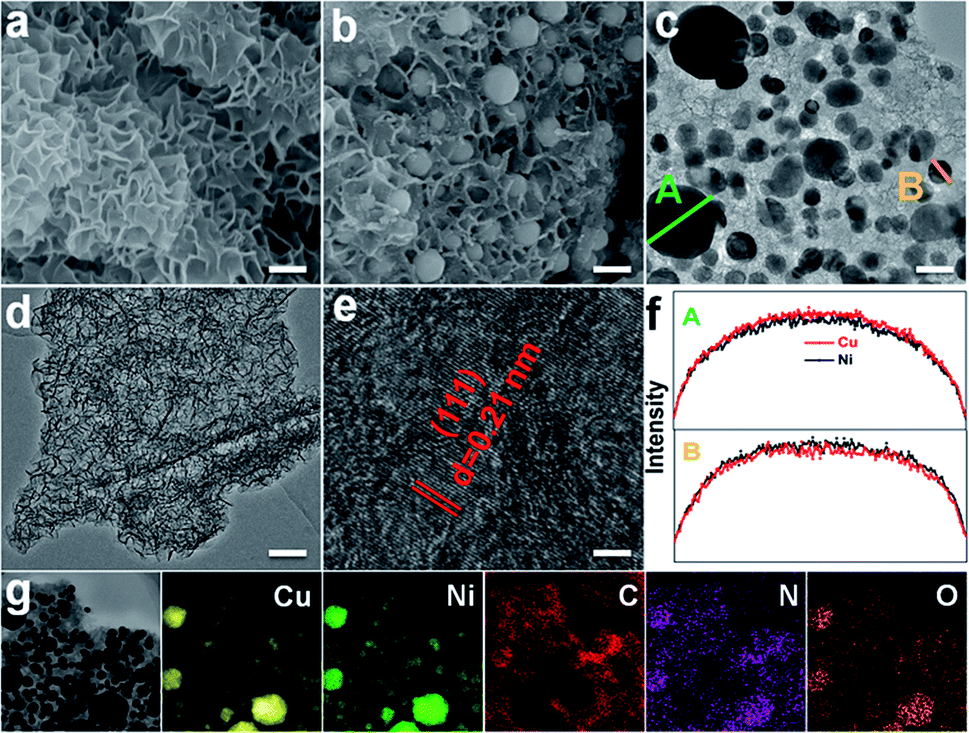 | ||
| Fig. 1 SEM image (a) and TEM image of the Cu/Ni bimetallic complex (d), and SEM image (b), TEM image (c), HRTEM image (e), EDS line scans (f) and EDX mapping of Cu1.0Ni1.0/N–C (g). Scale bars, 300 nm in (a) and (b), 100 nm in (c) and (d), and 2 nm in (e). | ||
The chemical compositions and elemental states of the Cu1.0Ni1.0/N–C catalyst were investigated by X-ray photoelectron spectroscopy (XPS). In the high-resolution Cu 2p spectrum (Fig. 2a), the binding energies at 934.8 eV and 954.9 eV correspond to Cu2+, while those at 932.8 eV and 952.6 eV can be assigned to Cu0 and possibly existing Cu+.27,28 The Auger Cu LMM spectrum shows a peak at a binding energy of 918.5 eV (Fig. S4†), which excludes the presence of Cu+ species.28 In the high-resolution Ni 2p spectrum (Fig. 2b), the binding energies at 852.7 eV, 870.1 eV and 855.9 eV, 873.5 eV correspond to Ni0 and Ni2+, respectively.29,30 The four nitrogen species peaks in the high-resolution N 1s spectrum (Fig. 2c) correspond to pyridinic N (B.E. 398.9 eV), Cu/Ni–N (B.E. 399.5 eV), pyrrolic N (B.E. 400.9 eV) and graphitic N (B.E. 402.1 eV), respectively.9,31 This indicates that the N species on the catalyst surface is mainly in the pyridinic and pyrrolic form, and there is a small amount of the Cu/Ni–N form. The presence of graphitic N species indicates that the 3D network is mainly composed of nitrogen-doped graphene.31 The high-resolution C 1s spectrum (Fig. 2d) shows five peaks at 284.8, 286.1, 287.7, 289.2 and 291.3 eV, which correspond to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N, C–OH, CO and Ph–π* species, respectively.32,33 Furthermore, synchrotron extended X-ray absorption fine structure (XAFS) analysis was performed to further investigate the alloy structure of Cu1.0Ni1.0/N–C. The Cu–Cu bond distance of Cu1.0Ni1.0/N–C shifts to 2.209 Å, which is lower than that of Cu foil (2.240 Å). However, the Ni–Ni bond distance of Cu1.0Ni1.0/N–C is 2.175 Å, which is a higher bond distance than that of Ni foil (2.082 Å). This phenomenon also proves the existence of the Cu/Ni alloy in Cu1.0Ni1.0/N–C (Fig. S5†).34
C, C–N, C–OH, CO and Ph–π* species, respectively.32,33 Furthermore, synchrotron extended X-ray absorption fine structure (XAFS) analysis was performed to further investigate the alloy structure of Cu1.0Ni1.0/N–C. The Cu–Cu bond distance of Cu1.0Ni1.0/N–C shifts to 2.209 Å, which is lower than that of Cu foil (2.240 Å). However, the Ni–Ni bond distance of Cu1.0Ni1.0/N–C is 2.175 Å, which is a higher bond distance than that of Ni foil (2.082 Å). This phenomenon also proves the existence of the Cu/Ni alloy in Cu1.0Ni1.0/N–C (Fig. S5†).34
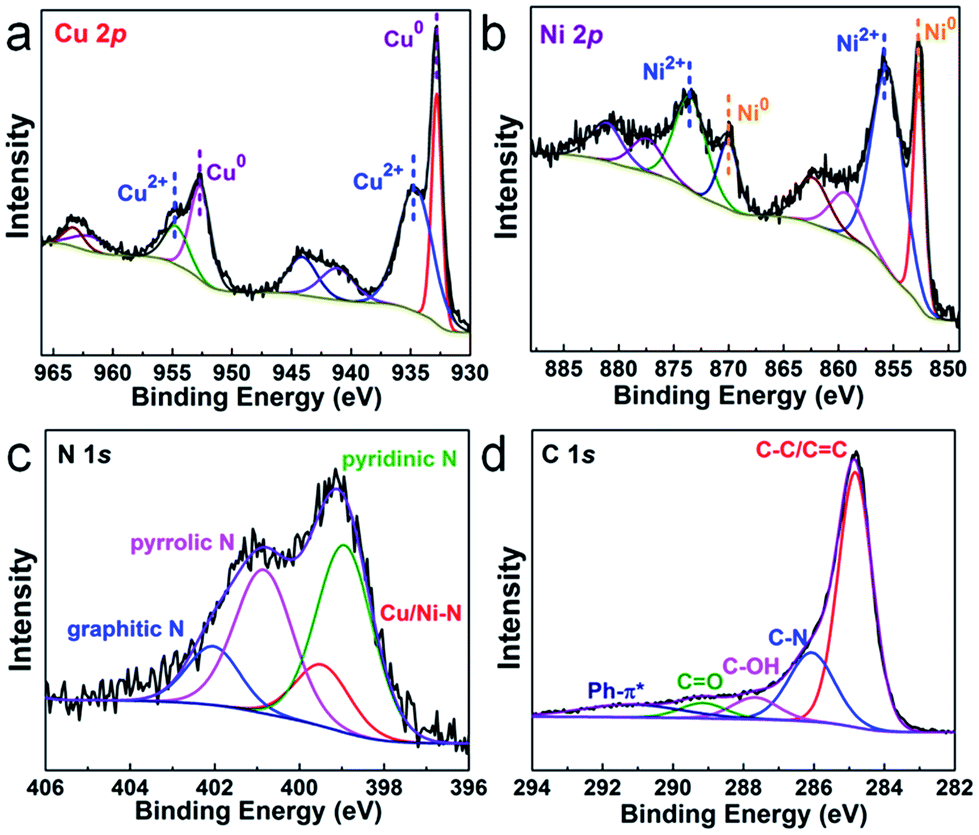 | ||
| Fig. 2 High-resolution XPS spectrum of Cu 2p (a), Ni 2p (b), N 1s (c) and C 1s (d) of Cu1.0Ni1.0/N–C. | ||
A series of CuxNiy/N–C catalysts with different Cu to Ni molar ratios were synthesized and characterized (Fig. S6–S9 and Table S1†), where x/y represents the molar ratio of Cu to Ni. All the catalysts present similar morphologies to Cu1.0Ni1.0/N–C, i.e., the Cu/Ni alloy nanoparticles are embedded in the nitrogen–carbon network. The X-ray diffraction peaks of the CuxNiy/N–C catalysts shift toward a lower diffraction degree with increasing Cu content, implying the mergence of more Cu atoms into the Ni lattice (Fig. S3†).1,35,36 For comparison, Cu/N–C and Ni/N–C catalysts were synthesized using a method similar to that for CuxNiy/N–C synthesis in the absence of nickel(II) nitrate hexahydrate and copper powder, respectively (Fig. S10 and 11†).
The electrocatalytic activities of CuxNiy/N–C for the CO2 RR were studied in a two-compartment cell. To assess the CO2 electroreduction performance of CuxNiy/N–C, linear sweep voltammetry (LSV) measurements were performed in N2 and CO2 saturated 0.5 M KHCO3, respectively (Fig. 3a). For Cu1.0Ni1.0/N–C and Cu4.8Ni1.0/N–C, the increase of current density relates to the HER in the N2 saturated solution, and the combination of the CO2 RR and HER in the CO2 saturated solution.37 The sharp increase of current density in the CO2 saturated solution compared to that in the N2 saturated solution indicates that Cu1.0Ni1.0/N–C and Cu4.8Ni1.0/N–C are more favorable for the CO2 RR than the HER. However, the current density of Cu/N–C in CO2 saturated 0.5 M KHCO3 is close to that in the N2 saturated solution. This implies that the performance of Cu/N–C for CO2 reduction is lower than that of Cu1.0Ni1.0/N–C and Cu4.8Ni1.0/N–C catalysts. Moreover, the current density of Cu1.0Ni1.0/N–C (40.3 mA cm−2) and Cu4.8Ni1.0/N–C (30.1 mA cm−2) at −1.0 V is much higher than that of Cu/N–C (15.5 mA cm−2).
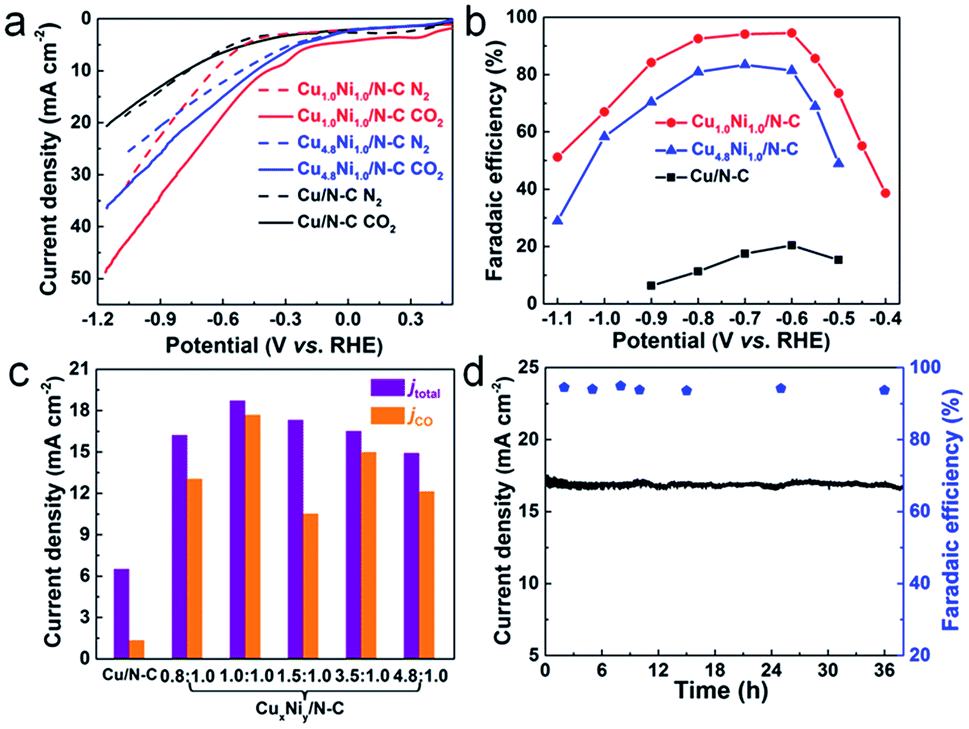 | ||
| Fig. 3 LSV curves acquired in N2-saturated (dotted line) or CO2-saturated (solid line) 0.5 M KHCO3 solution at a scan rate of 50 mV s−1 (a). CO faradaic efficiency at various potentials (b). Total current density and partial current density of different catalysts at an applied potential of −0.60 V vs. RHE (c). Stability of Cu1.0Ni1.0/N–C at a potential of −0.60 V vs. RHE during 38 h (d). Catalyst loading: 0.5 mg cm−2. | ||
The gaseous products and liquid products formed in CO2 reduction were analyzed by gas chromatography (GC) and 1H nuclear magnetic resonance (1H NMR) spectroscopy, respectively. The faradaic efficiencies of CO formation during the CO2 RR at different applied potentials are presented in Fig. 3b. CO and H2 were produced when catalyzed by CuxNiy/N–C and there is no liquid product in the whole electrolysis process. In contrast, H2 is the main catalytic product with a small amount of CO and HCOOH when catalyzed by Cu/N–C, and H2 is the only catalytic product when catalyzed by Ni/N–C (Fig. S12–S14†). Outstandingly, the maximum faradaic efficiency FE(CO) of Cu1.0Ni1.0/N–C is 94.5% at a low potential of −0.60 V, with a high current density of 18.8 mA cm−2. In sharp contrast, the FE(CO) of Cu/N–C is only 20.4% at the same potential, with a small current density of 6.5 mA cm−2. Such a high FE(CO) of Cu1.0Ni1.0/N–C at low potential provides a maximum energy conversion efficiency of 69.2%, which is about 4.6 times that of Cu/N–C (Fig. S15†).38,39 Compared with the reported Cu-based alloys and bimetallic catalysts, the catalytic performance of Cu1.0Ni1.0/N–C is superior owing to the combined high FE(CO) and current density at relatively low potential (Table S2†).18,20,22,27,40–42
Fig. 3c shows the total current density and partial current density of different catalysts at −0.60 V for CO formation. Cu1.0Ni1.0/N–C has a jCO of 17.7 mA cm−2 at −0.60 V, which is about 13.4 times that of the Cu/N–C catalyst (1.32 mA cm−2). Moreover, the partial current density for CO formation increases first and then decreases with applied potential (Fig. S16†), indicating that high applied potentials can accelerate the reaction rate for CO2 reduction.38,43 Attractively, the catalytic performance of Cu1.0Ni1.0/N–C is very stable, because the current density and FE(CO) remain undiminished after 38 h of constant potential electrolysis (Fig. 3d). The reused Cu1.0Ni1.0/N–C catalyst was characterized by different techniques (Fig. S17 and 18†). It is observed that the Cu1.0Ni1.0/N–C catalyst is very stable against long-term electrolysis.
The electrochemical surface area (ECSA), which can be evaluated by comparing the double-layer capacitance (Cdl) of catalysts, is an important factor affecting the catalytic performance.44 From Fig. 4a, the Cdl of Cu1.0Ni1.0/N–C is about 2 times that of Cu/N–C. The large ECSA will provide abundant active sites, and thus can enhance the activity for the CO2 RR.45 The Tafel slopes of Cu1.0Ni1.0/N–C and Cu4.8Ni1.0/N–C are 113.4 and 104.2 mV dec−1, respectively (Fig. 4b), which are close to the theoretical value of 116 mV dec−1. It indicates that the single electron transfer to CO2 to form the surface adsorbed CO2˙− intermediate is the rate-determining step for CuxNiy/N–C.9 The values of CuxNiy/N–C are obviously lower than those of Cu/N–C, implying that the formation of CO by CuxNiy/N–C is more favorable kinetically. Electrochemical impedance spectroscopy (EIS) was used to further explain the reaction kinetics of the CO2 RR. EIS was carried out in 0.5 M KHCO3 solution at an open circuit potential (Fig. 4c), and the equivalent circuit (Fig. S19†) was then used to fit the impedance data. The Nyquist plots confirm that Cu1.0Ni1.0/N–C and Cu4.8Ni1.0/N–C possess smaller charge transfer resistance than Cu/N–C. This indicates a faster electron transfer to CO2 for forming the reduced CO2˙− intermediate in the CO2 RR catalyzed by CuxNiy/N–C, thus leading to greatly enhanced catalytic efficiency for CO2 reduction.46
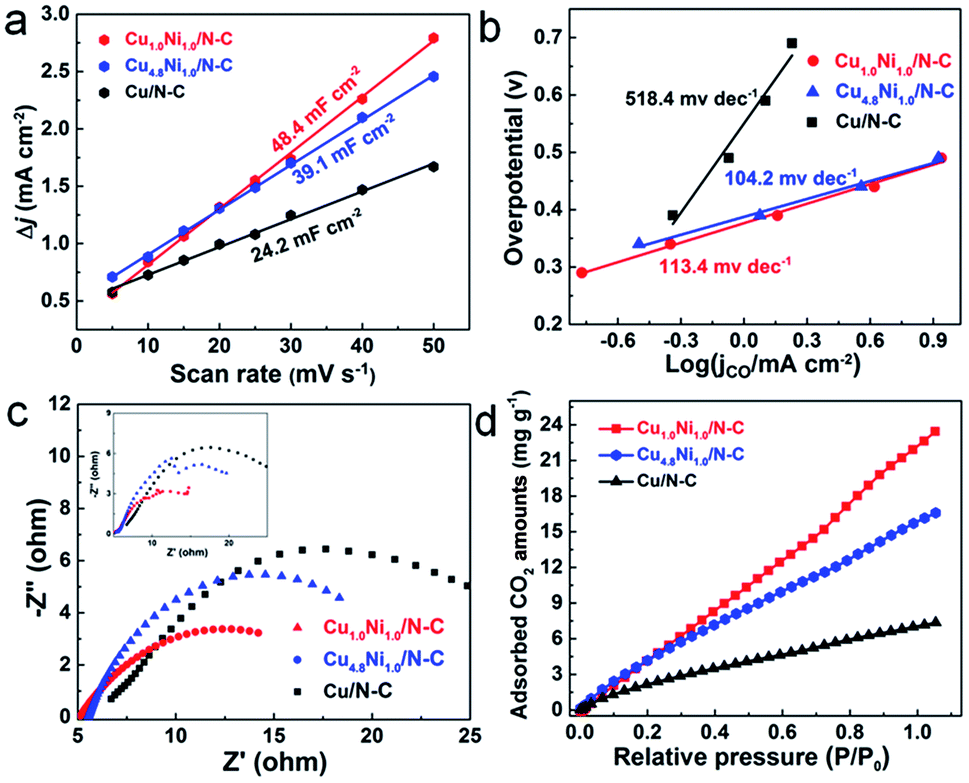 | ||
| Fig. 4 Charging current density differences Δj plotted against scan rates (a). Tafel plots for CO production (b). The experimental (inset) and simulated EIS spectra of the samples (c). CO2 adsorption isotherms of different catalysts at 298 K (d). | ||
The above results reveal that the CuxNiy/N–C catalyst has superior electrocatalytic performance for CO2 reduction to CO, in particular compared with mono-metal catalysts. In comparison with Cu/N–C, the enhanced electrocatalytic activity of CuxNiy/N–C can be attributed to the more stabilized COOH* adsorption and decreased overpotential caused by the CuNi alloy.13,47 Compared with Ni/N–C which produces H2 only, the release of CO from the CuNi alloy surface is more feasible than from the Ni surface, resulting from the reduced binding energy of CO*.48,49 Moreover, it is worth noting that the 3D porous nitrogen–carbon network would improve the CO2 adsorption capacity, contributing to the enhancement of reactivity.50,51 To testify this, the CO2 adsorption isotherms of different catalysts were determined (Fig. 4d). Obviously, the CuxNiy/N–C catalysts present improved CO2 adsorption capacity than Cu/N–C. For example, Cu1.0Ni1.0/N–C reaches a CO2 adsorption capacity of 23.4 mg g−1 at 298 K and 1 atm, which is 3.3 times higher than that of Cu/N–C. Therefore, the large CO2 adsorption capability facilitates the adsorption of a large amount of CO2 on the CuxNiy/N–C catalyst surface, thus promoting its catalytic performance.
Conclusions
In conclusion, we prepared the CuxNiy/N–C catalyst with Cu/Ni alloy nanoparticles embedded in a highly porous nitrogen–carbon network, which has combined advantages for the electrocatalytic conversion of CO2 to CO. For example, it well overcomes the disadvantages of single Cu and Ni electrocatalysts; the highly porous nitrogen–carbon network not only improves the CO2 adsorption ability but stabilizes the Cu/Ni nanoparticles during long-term electrolysis. The CuxNiy/N–C catalyst exhibits a maximum faradaic efficiency of 94.5% and current density of 18.8 mA cm−2 at a relatively low potential of −0.60 V vs. RHE. This study opens up new opportunities for improving the performance of metal electrocatalysts for CO2 reduction under mild conditions, particularly for metals with low intrinsic activity.Experimental
Materials
CO2 (>99.999% purity) was provided by Beijing Analysis Instrument Factory. Copper powder (>98% purity), Nafion N117 membrane and Nafion D-521 dispersion were purchased from Beijing InnoChem Science & Technology Co., Ltd. 2-Methylimidazole, cupric oxide and nickel oxide were purchased from J&K Scientific Co., Ltd. Absolute methanol was provided by Beijing Chemical Works. Nickel nitrate hexahydrate was provided by Sinopharm Chemical Reagent Beijing Co., Ltd.Preparation of CuxNiy/N–C
In a typical synthesis process, appropriate amounts of copper powder, nickel nitrate hexahydrate and 2-methylimidazole were dispersed in 10 mL of anhydrous methanol and stirred at room temperature for 10 min. After this, the solution was transferred into a Teflon lined stainless-steel autoclave, and the autoclave was sealed and heated at 120 °C for 24 h under stirring. After the autoclave cooled to room temperature, the as-prepared sample was washed with methanol and dried in a vacuum oven at 80 °C for 24 h. The solid was annealed in a tube furnace at 900 °C in a N2 atmosphere for 1 h with a heating rate of 2 °C min−1.Characterization
The morphologies of the catalysts were characterized by SEM (HITACHI S-4800). TEM images were taken on JEOL JEM-EM-1011 and JEOL JEM-2100F field-emission high-resolution transmission electron microscopes operated at 200 kV. XRD patterns were recorded on a Rigaku D/max 2400 diffractometer with Cu Kα radiation (λ = 0.15418 nm) with a scanning rate of 4° min−1. XPS was carried out with a multipurpose X-ray photoemission spectrometer (Thermo Scientific ESCALAB 250Xi). The elemental analysis of C, N and O was performed on a FLASH EA1112 elemental analysis instrument. The contents of Cu and Ni elements were determined by ICP-MS. The XAFS experiment was carried out at Beamline 1W1B at BSRF. Data of XAFS were processed using the Athena and Artemis programs of the IFEFFIT package based on FEFF 6. Data were processed with k3-weighting and an Rbkg value of 1.0. Normalized XANES data were obtained directly from the Athena program of the IFEFFIT package.Fabrication of electrodes
The working electrodes were prepared by loading the sample suspension onto carbon paper (CP). Briefly, the as-prepared catalysts and 10 μL Nafion D-521 dispersion were dispersed in absolute ethanol and ultrasonicated for 30 min to form a uniform suspension, and the suspension was loaded on 1 cm × 1 cm CP. The electrode was dried in a vacuum oven at 80 °C for 6 h before electrochemical experiments.A CHI660E electrochemical analyzer (Shanghai Chenhua Instrument co. LTD, China) was used in all the electrochemical experiments. A three-electrode test was carried out in a sealed H-cell which was separated by a Nafion N117 membrane. The reference and counter electrodes are Ag/AgCl and a Pt net (1 × 1 cm), respectively. All experiments were carried out at atmospheric pressure and room temperature. Linear sweep voltammetry (LSV), amperometric i–t curve measurements and electrochemical impedance spectroscopy (EIS) were performed in 0.5 M KHCO3 solution. Before experiments, the electrolyte solution in the working compartment was bubbled for 1 h with N2 (99.99%) and CO2 (99.999%) to form a saturated solution. The pH values of the electrolyte solutions with N2 and CO2 are 8.8 and 7.3, respectively. All potentials reported in this paper are with respect to the reversible hydrogen electrode (RHE), which were converted using eqn (1).
| Potential in the RHE = applied potential vs. Ag/AgCl + 0.21 V + 0.0592 × pH | (1) |
The gaseous product in the gas bag was collected and analyzed using a gas chromatograph (GC, HP 4890D), which was equipped with TCD and FID detectors, using helium as the internal standard. The liquid mixture was analyzed by the 1H-NMR method, and the spectra were recorded on a Bruker Avance III 400 HD spectrometer in deuterium oxide-d2 with TMS as an internal standard.
Electrochemical impedance spectroscopy (EIS)
The EIS measurement was carried out in 0.5 M KHCO3 solution at an open circuit potential (OCP) with an amplitude of 5 mV in the frequency range of 10−2 to 105 Hz.Double-layer capacitance (Cdl)
The Cdl was determined by measuring the capacitive current associated with double-layer charging from the scan-rate dependence of cyclic voltammetric stripping. The scan rates were 5 mV s−1, 10 mV s−1, 15 mV s−1, 20 mV s−1, 25 mV s−1, 30 mV s−1, 40 mV s−1 and 50 mV s−1.Energy efficiency
The energy efficiency of reduction of CO2 to CO was obtained using eqn (2):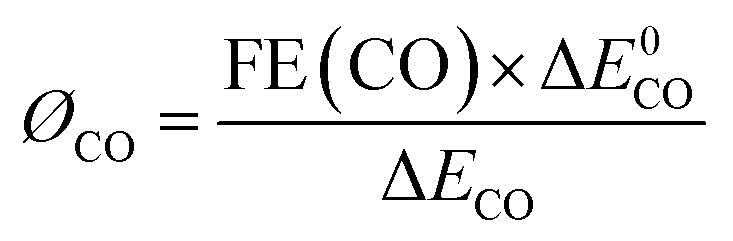 | (2) |
In the equation, ØCO stands for the cathodic energy conversion efficiency of the CO formation; ΔE0CO represents the difference between the standard half reaction potentials for water oxidation (1.23 V vs. RHE) and reduction of CO2 to CO (−0.11 V vs. RHE); ΔECO stands for the difference between the standard water oxidation potential and the applied potential for CO2 reduction.
Conflicts of interest
The authors of this manuscript have no conflicts of interest.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21525316, 21673254), Ministry of Science and Technology of China (2017YFA0403003), Chinese Academy of Sciences (QYZDY-SSW-SLH013), and Beijing Municipal Science & Technology Commission (Z181100004218004).Notes and references
- D. Tan, J. Zhang, J. Shi, S. Li, B. Zhang, X. Tan, F. Zhang, L. Liu, D. Shao and B. Han, ACS Appl. Mater. Interfaces, 2018, 10, 24516–24522 CrossRef CAS PubMed.
- C. Liu, J. Zhang, L. Zheng, J. Zhang, X. Sang, X. Kang, B. Zhang, T. Luo, X. Tan and B. Han, Angew. Chem., Int. Ed., 2016, 55, 11372–11376 CrossRef CAS PubMed.
- T. Luo, J. Zhang, X. Tan, C. Liu, T. Wu, W. Li, X. Sang, B. Han, Z. Li, G. Mo, X. Xing and Z. Wu, Angew. Chem., Int. Ed., 2016, 55, 13533–13537 CrossRef CAS PubMed.
- G. P. Lau, M. Schreier, D. Vasilyev, R. Scopelliti, M. Grätzel and P. J. Dyson, J. Am. Chem. Soc., 2016, 138, 7820–7823 CrossRef CAS PubMed.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, Z. Zhang and B. Han, Angew. Chem., Int. Ed., 2016, 55, 6771–6775 CrossRef CAS PubMed.
- L. He, X. Sun, H. Zhang and F. Shao, Angew. Chem., Int. Ed., 2018, 57, 12453–12457 CrossRef CAS PubMed.
- Y. Cheng, S. Zhao, B. Johannessen, J. P. Veder, M. Saunders, M. R. Rowles, M. Cheng, C. Liu, M. F. Chisholm, R. De Marco, H. M. Cheng, S. Z. Yang and S. P. Jiang, Adv. Mater., 2018, 30, 1706287 CrossRef PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- J. Xu, X. Li, W. Liu, Y. Sun, Z. Ju, T. Yao, C. Wang, H. Ju, J. Zhu, S. Wei and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 9121–9125 CrossRef CAS PubMed.
- W. Zhu, L. Zhang, P. Yang, C. Hu, Z. Luo, X. Chang, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 11544–11548 CrossRef CAS PubMed.
- H. Mistry, Y. W. Choi, A. Bagger, F. Scholten, C. S. Bonifacio, I. Sinev, N. J. Divins, I. Zegkinoglou, H. S. Jeon, K. Kisslinger, E. A. Stach, J. C. Yang, J. Rossmeisl and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2017, 56, 11552–11556 CrossRef.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem, 2018, 4, 1809–1831 CAS.
- M. Ma, H. A. Hansen, M. Valenti, Z. Wang, A. Cao, M. Dong and W. A. Smith, Nano Energy, 2017, 42, 51–57 CrossRef CAS.
- S. Zhu, Q. Wang, X. Qin, M. Gu, R. Tao, B. P. Lee, L. Zhang, Y. Yao, T. Li and M. Shao, Adv. Energy Mater., 2018, 8, 1802238 CrossRef.
- D. Gao, Y. Zhang, Z. Zhou, F. Cai, X. Zhao, W. Huang, Y. Li, J. Zhu, P. Liu, F. Yang, G. Wang and X. Bao, J. Am. Chem. Soc., 2017, 139, 5652–5655 CrossRef CAS PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Norskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS PubMed.
- Z. Yin, D. Gao, S. Yao, B. Zhao, F. Cai, L. Lin, P. Tang, P. Zhai, G. Wang and D. Ma, Nano Energy, 2016, 27, 35–43 CrossRef CAS.
- Z. Chang, S. J. Huo, Z. Wei, J. Fang and H. Wang, J. Phys. Chem. C, 2017, 121, 11368–11379 CrossRef CAS.
- S. Sarfraz, A. T. Garcia-Esparza, A. Jedidi, L. Cavallo and K. Takanabe, ACS Catal., 2016, 6, 2842–2851 CrossRef CAS.
- S. Rasul, D. H. Anjum, A. Jedidi, Y. Minenkov, L. Cavallo and K. Takanabe, Angew. Chem., Int. Ed., 2015, 54, 2146–2150 CrossRef CAS PubMed.
- W. J. Bagger, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef PubMed.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- J. Mondal, P. Saikiran, I. Mondal, A. Shrotri, r. B. Srinivasa and N. Lingaiah, Sustainable Energy Fuels, 2018, 2, 1516–1529 RSC.
- R. Pérez-Hernández, A. Gutiérrez-Martínez, M. E. Espinosa-Pesqueira, M. L. Estanislao and J. Palacios, Catal. Today, 2015, 250, 166–172 CrossRef.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. Kenis, J. Am. Chem. Soc., 2016, 139, 47–50 CrossRef PubMed.
- W. Hong, J. Wang and E. Wang, Nanoscale, 2016, 8, 4927–4932 RSC.
- Z. Wang, M. Jiang, J. Qin, H. Zhou and Z. Ding, Phys. Chem. Chem. Phys., 2015, 17, 16040–16046 RSC.
- R. Zou, K. Xu, T. Wang, G. He, Q. Liu, X. Liu, Z. Zhang and J. Hu, J. Mater. Chem. A, 2013, 1, 8560–8566 RSC.
- P. Sekar, L. Calvillo, C. Tubaro, M. Baron, A. Pokle, F. Carraro, A. Martucci and S. Agnoli, ACS Catal., 2017, 7, 7695–7703 CrossRef CAS.
- S. L. Kuo, W. R. Liu, C. P. Kuo, N. L. Wu and H. C. Wu, J. Power Sources, 2013, 244, 552–556 CrossRef CAS.
- J. Zhang, Z. Xie, W. Li, S. Dong and M. Qu, Carbon, 2014, 74, 153–162 CrossRef CAS.
- E. T. Saw, U. Oemar, X. R. Tan, Y. Du, A. Borgna, K. Hidajat and S. Kawi, J. Catal., 2014, 314, 32–46 CrossRef CAS.
- R. Long, Y. Li, Y. Liu, S. Chen, X. Zheng, C. Gao, C. He, N. Chen, Z. Qi and L. Song, J. Am. Chem. Soc., 2017, 139, 4486–4492 CrossRef CAS PubMed.
- H. Zhang, Y. Shang, J. Zhao and J. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 16635–16643 CrossRef CAS PubMed.
- K. Lv, C. Teng, M. Shi, Y. Yuan, Y. Zhu, J. Wang, Z. Kong, X. Lu and Y. Zhu, Adv. Funct. Mater., 2018, 28, 1802339 CrossRef.
- D. Yang, Q. Zhu, X. Sun, C. Chen, L. Lu, W. Guo, Z. Liu and B. Han, Green Chem., 2018, 20, 3705–3710 RSC.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J. L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- Q. Li, J. Fu, W. Zhu, Z. Chen, B. Shen, L. Wu, Z. Xi, T. Wang, G. Lu and J. Zhu, J. Am. Chem. Soc., 2017, 139, 4290–4293 CrossRef CAS PubMed.
- G. O. Larrazábal, A. J. Martín, S. Mitchell, R. Hauert and J. Pérezramírez, ACS Catal., 2016, 6, 6265–6274 CrossRef.
- W. Zhu, L. Zhang, P. Yang, X. Chang, H. Dong, A. Li, C. Hu, Z. Huang, Z. J. Zhao and J. Gong, Small, 2018, 14, 1703314 CrossRef PubMed.
- S. Liu, H. Yang, X. Huang, L. Liu, W. Cai, J. Gao, X. Li, T. Zhang, Y. Huang and B. Liu, Adv. Funct. Mater., 2018, 28, 1800499 CrossRef.
- Y. Li, H. Su, S. H. Chan and Q. Sun, ACS Catal., 2015, 5, 6658–6664 CrossRef CAS.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu and C. Wu, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- Y. Li, H. Su, S. H. Chan and Q. Sun, ACS Catal., 2015, 5, 6658–6664 CrossRef CAS.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- E. Vesselli, E. Monachino, M. Rizzi, S. Furlan, X. Duan, C. Dri, A. Peronio, C. Africh, P. Lacovig, A. Baldereschi, G. Comelli and M. Peressi, ACS Catal., 2013, 3, 1555–1559 CrossRef CAS.
- W. Zheng, S. Nayak, W. Yuan, Z. Zeng, X. Hong, K. A. Vincent and S. C. Tsang, Chem. Commun., 2016, 52, 13901–13904 RSC.
- H. Wang, J. Jia, P. Song, Q. Wang, D. Li, S. Min, C. Qian, L. Wang, Y. F. Li and C. Ma, Angew. Chem., Int. Ed., 2017, 129, 7955–7960 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and TEM images, XRD patterns, EXAFS spectra, XPS survey spectra, faradaic efficiency, 1H NMR spectra and partial current density details. See DOI: 10.1039/c9sc00174c |
| This journal is © The Royal Society of Chemistry 2019 |