DOI:
10.1039/C8SC04160A
(Edge Article)
Chem. Sci., 2019,
10, 4476-4485
Superoxide dismutase transcellular shuttle constructed from dendritic MOF and charge reversible protein derivatives†
Received
19th September 2018
, Accepted 7th March 2019
First published on 11th March 2019
Abstract
The development of molecular biology has led to the identification of protein-based therapeutics as an intriguing approach for the treatment of a wide range of diseases. To manufacture transcellular protein delivery shuttles, we attempted charge reversal chemistry on native proteins [e.g., superoxide dismutase (SOD): an enzyme capable of scavenging detrimental reactive oxygen species] by the selective conversion of the positively charged amino residues of native SOD to conjugated negatively charged citraconic moieties, eliciting overall negatively charged polyelectrolytes for the subsequent electrostatic self-assembly with cationic metal–organic framework (MOF) derivatives into protein delivery systems. Please note that the charge conversion was reversible, restoring the original amino groups in intracellular acidic endosome compartments (pH 5), which afforded facile charge reversible functions to reclaim the active SOD in the cell interior. In particular, the strategic manufacture of dendritic MOF supramolecular architectures as transcellular shuttles for the aforementioned charge-reversible SOD derivatives is noteworthy. The MOF was surface-functionalized with several polycationic segments, thus contributing to the hyper-charged architecture for the easy accommodation of the negatively charged SOD derivatives. Consequently, the SOD derivatives managed to internalize into cells by hitchhiking via endocytosis of the positively charged MOF. Once they resided in the acidic endosomes, the charge reversal of the SOD derivatives could occur smoothly and result in reduced interactions between the charged-reversed SOD and MOF, leading to the release of active SOD. Simultaneously, the dendritic MOF due to protein release presented a highly positive-charged architecture to provoke endosome membrane disruption, consequently spurring the translocation of SOD to the cytosol for the execution of its enzymatic activities. Herein, the intracellular ROS level of the activated macrophages was validated to be markedly suppressed by our proposed transcellular SOD shuttles, thereby indicating their wide availability to diverse functional proteins for biomedical applications.
1. Introduction
Eukaryotic cells contain a myriad of proteins that account for fundamental structural components and critical cellular functions, where a staggering number of diseases have been validated to be a consequence of the irregular levels of functional proteins. Hence, protein-based therapeutics have been perceived as one of the most fundamental therapeutic modalities. Unlike nucleic acid-based therapeutics (incessant concerns due to the potentials of nucleic acid-based therapeutics in altering the genomics of the affected cells), the protein therapeutic approach, envisaged to transport therapeutic proteins into cells with the aim of restoring their biological functionalities, is postulated to be a straightforward and safe approach in disease treatments.1–3 However, despite the appealing characteristics of protein therapeutics, intracellular transportation of native proteins remains an onerous task with reference to the poor cellular internalization activity of the native proteins and their inability in circumventing the subcellular barriers (e.g., degradation in lysosomes) to arrive at the targeted intracellular sites. Although the protein-recombinant technique (a variety of functional domains schemed into original proteins) has been postulated to tackle the aforementioned extracellular and intracellular barriers,4–6 the complicated protein-customized structural design and the potential recombinant-incurred immunogenicity have encouraged alternative facile and versatile pathways to promote the wide availability of protein therapeutics. Accordingly, the manufacture of delivery vehicles is deemed to be a viable approach to explore the intracellular transportation of cell-impermeable therapeutic payloads, e.g., nucleic acids (plasmid DNA, messenger RNA, small interference RNA, etc.).7–10 Negatively charged nucleic acids could be facilely complexed with positively charged species through electrostatic reactions to formulate nanoscaled nucleic acid delivery systems.11,12 Accordingly, the charge-neutralized nucleic acids possessing a condensed nanostructure imparted favorable affinity to the negatively charged cell membrane and activated cell internalization behaviors.13 Unfortunately, impeded by the intrinsic molecular characters of proteins, the self-assembly of the proteins into delivery structures was difficult to accomplish. Please note that the fundamental thermodynamic drive forces for the supramolecular self-assembly (e.g., electrostatic interactions or hydrophobic interactions) are hardly beneficial for the construction of protein supramolecular assemblies due to the unmethodical positive charge from the amine groups and the negative charge from the carboxyl groups in native proteins.14–16 To date, the development of protein delivery systems has focused on vesicles, i.e., either liposomes or polymersomes, whose internal cavities can serve as protein reservoirs. Nonetheless, protein encapsulation in vesicles has been reported to exclusively rely on the process of non-specific physical entrapment; thus, it is impossible to anticipate high protein encapsulation efficiency (documented to be approximately 5% at maximum).17,18 This obstacle forced us to revert to the self-assembly pathway to acquire high protein-loading efficiency. Considering the unmethodical charges of protein payloads, we aimed to develop a chemical strategy to synchronize the charges of proteins via the conversion of the positively charged amino groups of the native proteins to negatively charged carboxyl groups, consequently accounting for overall negatively charged polyelectrolytes. Please note that the aforementioned charge conversion was designed to allow facile reversal to the original amino groups in the intracellular acidic endosome compartments. To this end, the resulting protein derivatives obtained using the above chemical strategy were utilized for the manufacture of delivery nanostructures with cationic species analogous to negatively charged nucleic acids. Nevertheless, the charge density of the protein derivatives was significantly low (considering that the R groups in a variety of amino acids are non-charged) as compared to that of the anionic phosphate residues in every nucleotide of nucleic acids. Hence, the assembled colloidal structures with conventional cationic species (such as polyethyleneimine, cationic lipids or chitosan) were conjectured to provide insufficient structural stability in the biological milieu. In the present study, we attempted to construct a highly charged dendritic polycationic metal–organic framework (MOF), characterized by several dendritic polymeric cationic segments propagated from the ligands of UiO66-Zr-MOF, as a nanoscaled protein transcellular shuttle template to accommodate negatively charged protein derivatives (Scheme 1). It should be noted that our previous study validated the improved colloidal stabilities of self-assembled dendritic MOF and nucleic acids.19 Therefore, the proposed dendritic MOF is postulated to steadily accommodate the protein cargos in the physiological milieu and permit endocytosis inside cells. After residing in acidic endosomes, the charge reversal of the protein derivatives could smoothly occur and lead to reduced interactions between the charged-reversed protein and MOF as well as the consequent release of proteins. Simultaneously, the dendritic MOF due to protein release presented a highly positive-charged architecture to exert endosome membrane disruption, inducing the translocation of the released proteins to the cytosol. In this research, Cu, Zn superoxide dismutase 1 (SOD-1) was employed as a model protein to test the feasibility of our strategies for intracellular protein transportation. SOD-1 is a critical enzyme capable of scavenging reactive oxygen species and consequently, it has potentials in the treatment of a variety of irregular inflammatory-relevant diseases via the inhibition of ROS production in macrophages.20,21 The ROS production from macrophages plays a central role in irritating inflammatory diseases, including arthritis, sepsis and acute liver failure.22,23 Unfortunately, the clinical administration of native SOD-1 has been validated to be ineffective, which is due to the poor membrane permeabilities of the native SOD-1. In this research, we were motivated to construct a robust SOD-1 delivery vehicle and provide a worthy methodology for the development of protein delivery systems.
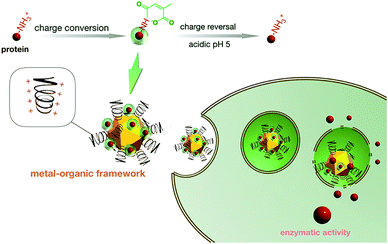 |
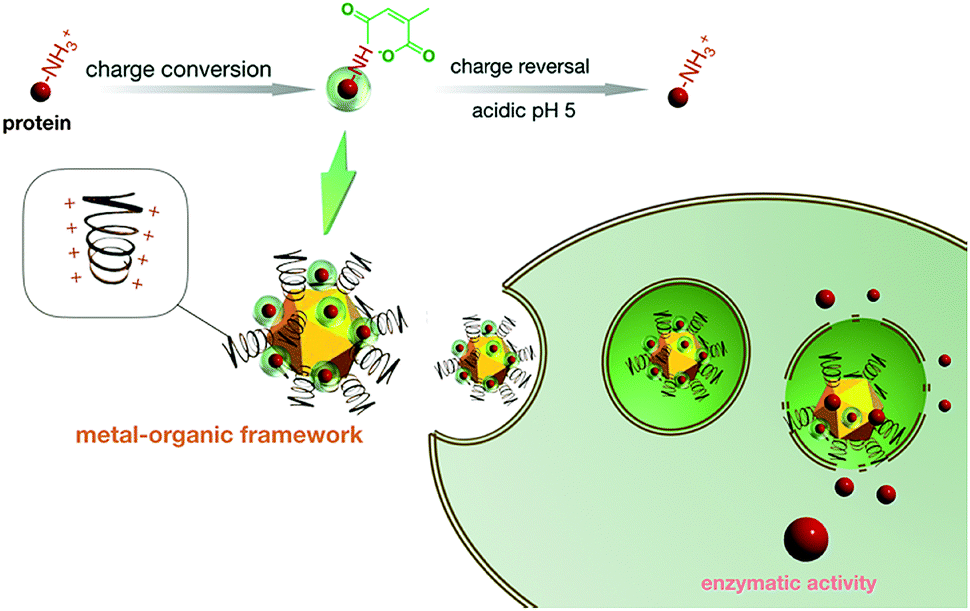
| | Scheme 1 Schematic illustration of the manufacture of the protein transcellular shuttle from acid-responsive charge reversible protein derivatives and hyper-charged dendritic MOF catiomers. Herein, the amino groups of SOD-1 were conjugated with acidic labile carboxyl moieties to create anionic polyelectrolytes, which were subsequently loaded into a hyper-charged dendritic MOF architecture through electrostatic interactions. The resulting SOD-1 derivative could readily shuttle inside cells by hitchhiking via cell endocytosis of cationic MOF. After residing in the subcellular compartment of acidic endosomes post endocytosis, the hyper-charged MOF is envisioned to provoke potent destabilization activities to the entrapped endosome as a result of the charge reversal of the SOD-1 derivatives and the ensuing SOD-1 departure from the MOF, ultimately accounting for the release of the charge reversed native SOD-1 in the cytosol to execute enzymatic functions. | |
2. Materials and methods
2.1 Materials
All chemicals were purchased from Tansoole, Titan Co. Ltd. (Shanghai, China). Alexa Fluor 647 for labelling SOD-1 (derivatives) was purchased from Invitrogen Molecular Probes (Eugene, OR) and used according to the manufacturer's protocol. Cy3 and Cy5 were purchased from Thermo Fisher Scientific (Waltham, MA). Dulbecco's modified Eagle's medium (DMEM) and Dulbecco's phosphate-buffered saline (DPBS) were purchased from Sigma-Aldrich Co. (Madison, WI). Fetal bovine serum (FBS) was purchased from GIBCO. Penicillin and streptomycin were purchased from Beyotime Institute of Biotechnology Co. (Jiangsu, China). Ethidium bromide (EtBr) and Cu, Zn-SOD 1 from bovine liver erythrocytes were purchased from Sigma Aldrich China (Shanghai, China). Human umbilical vein endothelial cells (HUVECs) were purchased from Syndean Biotech. Co. (Dalian, China). The culture EGM-2 Bullet Kit medium was purchased from Lonza (Logan, MA). Human recombinant TNF-α was provided by Hygeia Medi-Tech. Co. Ltd (Ningbo, China). YOYO™-1 iodide, CellLight™ early endosomes-GFP, BacMam 2.0 and CellLight™ late endosomes-GFP, BacMam 2.0 were purchased from ThermoFisher Scientific (Waltham, MA).
2.2 Charge conversion of SOD-1
The purchased SOD-1 was dissolved at a concentration of 1 mg mL−1 in NaHCO3 buffer (0.1 M, pH 9.0), followed by titration with varied concentrations of citraconic anhydride and incubation for reaction under stirring at 4 °C for 5 h. The unreacted citraconic anhydride was removed using a Microcon centrifugal MWCO 3 kDa filter unit (Sigma-Aldrich, St. Louis, MI) three times in 10 mM NaHCO3 buffer (pH 7.4). The degree of charge conversion was estimated according to a fluorescamine method.24 In brief, the yielded SOD-1 derivatives were dissolved in PBS buffer (0.1 mL, 0.5 mg mL−1, pH 7.4) and mixed with fluorescamine solution (10 μL, 5 mg mL−1 in DMF). Following 10 min reaction at room temperature, the fluorescence emission was recorded using an ND-3300 fluorospectrometer (Nanodrop, Wilmington, DE). The charge-conversion degree of the yielded SOD-1 derivatives was expressed as the percentage of the converted amino groups to the total available amino groups (determined by fluorescamine measurement for the native SOD-1). Note that a small fraction of Alexa Fluor 680 NHS Ester (succinimidyl ester, Thermo Fisher Scientific, Waltham, MA)-conjugated SOD-1 enzyme (2.5%) was included in the original SOD-1 solution for the quantification of SOD-1 and its concentration based on the absorbance measurement at 680 nm. Note that the preliminary results were validated by the absence of the absorbance of SOD-1 and the strong absorbance of the Alexa Fluor 680-conjugated SOD-1 enzyme at 680 nm.
2.3 Charge reversal of SOD-1 derivatives upon acidic treatment
The charge reversal behaviour of the yielded SOD-1 derivatives upon acidic treatment was determined by incubating the SOD-1 derivatives in acidic 10 mM phosphate buffer (pH 5.5, 150 mM NaCl) at 37 °C or in neutral phosphate buffer (10 mM, pH 7.4, and 150 mM NaCl). At a pre-defined period post incubation, the reaction solution was supplemented with an equal volume of phosphate buffer (50 mM, pH 7.4) to neutralize the acidic reaction solutions. The recovered amine groups were quantified according to the aforementioned fluorescamine assay.
2.4 Recovered enzymatic activities of SOD-1 derivatives upon acidic treatment
The recovered activities of the SOD-1 derivatives were estimated using a superoxide dismutase colorimetric activity kit according to the manufacture's protocol. In brief, the DetectX® superoxide dismutase activity kit was employed to quantitatively measure the SOD-1 activity for a variety of samples. Varied concentrations of the native SOD-1 solution were utilized to generate a standard curve for the assay. The activities of all samples were calculated according to the standard curve. Furthermore, the substrate was added, followed by xanthine oxidase reagent and incubated at room temperature for 20 minutes. The xanthine oxidase generated superoxide in the presence of oxygen, which converted the colourless substrate in the detection reagent into a yellow-coloured product. The absorption of the coloured product was measured at 450 nm. An increase in the levels of SOD-1 in the samples caused a decrease in superoxide concentration and a reduction in yellow product. The results were expressed in terms of SOD-1 activity of SOD-1 derivatives relative to the native SOD-1.
2.5 Preparation of polyionic complexes
The polymer of PGMA(EA) or MOF-PGMA(EA) and SOD-60 were separately dissolved in 10 mM PBS buffer (pH 7.4). Polyionic complexes were created by instantly mixing equal volumes of the aforementioned polycationic solution and SOD-60 solution at an N/C ratio of 0.9. The N/C ratio is defined as the residual molar ratio of amine (N) groups of PGMA(EA) or MOF-PGMA(EA) to the total carboxyl groups (C) groups of SOD-60.
2.6 DLS measurement
The hydrodynamic diameters together with the polydispersity index (PDI) of the SOD derivative-included polymeric complexes were determined by DLS measurements with a Zetasizer Nanoseries instrument (Malvern Instruments Ltd., UK). Each measurement was conducted three times at 25 °C. The rate of decay in the photon correlation function was analyzed according to a cumulant method, and the corresponding diameter was calculated using the Stokes–Einstein equation.
2.7
ζ-Potential measurements
The ζ-potentials of the polyionic complexes were also measured by Nano ZS (ZEN3600, Malvern Instruments, Ltd., UK). The ζ-potential was determined via laser-doppler electrophoresis using the Zetasizer Nanoseries (Malvern Instruments Ltd., UK). From the obtained electrophoretic mobility, the ζ-potential was calculated using the Smoluchowski equation: ζ = 4πην/ε, where η is the electrophoretic mobility, ν is the viscosity of the solvent, and ε is the dielectric constant of the solvent. The results are expressed as the average of three experiments.
2.8 Cellular uptake
Human umbilical endothelial cells (HUVECs, Lonza Walkersville, Walkersville, MD) were cultured onto 6 well culture plates (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well) with 2 mL EGM-2 Bullet Kit medium (Lonza, Logan, MA) with 10% fetal bovine serum supplemented with 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (GIBCO) in a humidified atmosphere supplemented with 5% CO2 at 37 °C. After 24 h incubation, the medium was replaced with a fresh medium. The cells were treated with the polyionic complex solutions (1 μg of Alexa Fluro 647-labeled SOD per well), followed by another 24 h incubation. The medium was discarded, and the cells were washed three times with PBS to remove extracellular fluorescence. The cells were detached by trypsin–EDTA treatment and harvested from the cell culture plate as a suspension in 1 mL ice-cold PBS. The cellular uptake efficiency was measured using a BD LSR II flow cytometer equipped with the FACS-Diva software (BD Biosciences). The obtained data were expressed as the mean fluorescence intensity from three independent samples (n = 3).
000 cells per well) with 2 mL EGM-2 Bullet Kit medium (Lonza, Logan, MA) with 10% fetal bovine serum supplemented with 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (GIBCO) in a humidified atmosphere supplemented with 5% CO2 at 37 °C. After 24 h incubation, the medium was replaced with a fresh medium. The cells were treated with the polyionic complex solutions (1 μg of Alexa Fluro 647-labeled SOD per well), followed by another 24 h incubation. The medium was discarded, and the cells were washed three times with PBS to remove extracellular fluorescence. The cells were detached by trypsin–EDTA treatment and harvested from the cell culture plate as a suspension in 1 mL ice-cold PBS. The cellular uptake efficiency was measured using a BD LSR II flow cytometer equipped with the FACS-Diva software (BD Biosciences). The obtained data were expressed as the mean fluorescence intensity from three independent samples (n = 3).
2.9 Endosome escape
The endosome entrapment ratio was determined by evaluating the colocalization degree of SOD-1 (derivatives) and endosomes via confocal laser scanning microscopy (CLSM). In brief, the Alexa Fluor 647-labeled SOD-60 derivative was used to prepare a polyionic complex. HUVECs were cultured in a 35 mm glass-bottom dish in an EGM-2 Bullet Kit medium (Lonza) with 10% fetal bovine serum supplemented with 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (GIBCO). The medium was replaced with fresh medium, followed by the addition of 75 mL of polyionic complex solution (20 μg SOD-60 per mL) into each cell culture dish. After 24 h of incubation, the medium was removed, and the cells were rinsed thrice with PBS prior to imaging. The intracellular distribution of polyionic complexes was observed by CLSM after staining acidic late endosomes and lysosomes with LysoTracker Green (Molecular Probes, Eugene, OR). CLSM observation was performed using LSM 510 (Carl Zeiss, Germany) at excitation wavelengths of 488 nm (Ar laser) and 633 nm (He–Ne laser) for LysoTracker Green (green) and Alexa Fluor 647 (red), respectively. The colocalization ratio was calculated as previously described according to the following formula:
| Colocalization ratio = the pixel number of yellow pixels/the sum-up pixel numbers of yellow and red pixels |
Here, yellow corresponds to SOD trapped in the late endosome/lysosomes and red corresponds to SOD in the cytosol.
2.10
In situ observation of endosome membrane destabilization
HUVECs were cultured in a 35 mm glass-bottom dish in EGM-2 Bullet Kit medium with 10% fetal bovine serum supplemented with 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. The medium was replaced with fresh medium, followed by the addition of 30 μL of CellLight™ early endosome GFP to selectively label the early endosome membrane for 48 h incubation. Aiming to visualize the intracellular distribution, the proposed constructs of MOF-PGMA(EA)/SOD-60 were added to HUVECs for 1 h incubation on top of an ice bath. The medium was removed, and the cells were rinsed 3 times with PBS (with the aim of synchronizing the cell internalization process), followed by another 2 h incubation at 37 °C. The cell nuclei were further stained with Hoechst 33342 (Dojindo Laboratories, Kumamoto, Japan). CLSM observation was performed using LSM 780 (Carl Zeiss, Oberkochen, Germany) at excitation wavelengths of 488 nm (Ar laser) for GFP, 640 nm for MOF-Cy5 and 405 nm (diode laser) for Hoechst 33342. Following a similar procedure, the late endosome membrane was stained with CellLight™ late endosome GFP, and 8 h post incubation was conducted to gain insights into the reaction between MOF-PGMA(EA)/SOD-60 (MOF was stained by Cy5) and late endosome (stained by CellLight™ late endosome GFP).
2.11 Colocalization of SOD-1 (derivatives) and MOF-PGMA(EA)
HUVECs were cultured in a 35 mm glass-bottom dish in EGM-2 Bullet Kit medium (Lonza) with 10% fetal bovine serum supplemented with 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (GIBCO). The medium was replaced with fresh medium, followed by the addition of 30 μL of MOF-PGMA(EA)/SOD-60 solution. Aiming to visualize the intracellular distribution of MOF-PGMA(EA) and SOD-1 derivatives, the proposed constructs were added to HUVECs for 1 h incubation on top of an ice bath. The medium was removed and the cells were rinsed 3 times with PBS (with the aim of synchronizing the cell internalization process), followed by another 2 h or 8 h incubation at 37 °C. The cell nuclei were further stained with Hoechst 33342 (Dojindo Laboratories, Kumamoto, Japan). The Cy3 and Cy5 signals were detected simultaneously using 561 and 640 nm excitation lasers and band-pass emission filters of 576/40 and 700/75 nm, respectively.
2.12 Cytoplasm membrane destabilization activity
The membrane destabilizing ability was assessed by measuring intracellular membrane-impermeable YOYO™-1 iodide from the cells contacting with diverse samples. In brief, HUVECs (5000 cells per well) were plated on 96 well plates and incubated overnight in 100 μL of MCDB131 containing 10% FBS and 10 ng mL−1 b-FGF. The cell culture medium was changed to 100 μL of diverse samples [including catiomer solutions of PGMA(EA) and MOF-PGMA(EA) or complex solution of MOF-PGMA(EA)/SOD-60] in 20 mM HEPES buffer (pH 7.4) or 20 mM MES buffer (pH 6.0 or 5.0) with 150 mM NaCl together with extra component of YOYO™-1 iodide (1 mM). After incubation at 37 °C for 30 min, the fluorescence intensity of YOYO™-1 iodide in each nucleus (stained by 2 mg mL−1 Hoechst 33342) was quantitatively evaluated using a fluorescence microscope equipped with an image-analysis software (IN Cell Analyzer 1000: GE Healthcare UK Ltd., Buckinghamshire, England).
2.13 SOD-1 (derivatives) release profiles
The release of SOD-1 (derivatives) from MOF-PGMA(EA) was estimated based on a fluorescence resonance energy transfer (FRET) method. In brief, SOD-60 and MOF-PGMA(EA) were labelled by Cy3 and Cy5, respectively, which were utilized to prepare a polyionic complex at pH 7.4 (5 mM PBS buffer). Furthermore, the prepared polymeric solution was mixed with an equal volume of MES buffer (50 mM, pH 5) for incubation under stirring at 37 °C. The FRET intensity (excitation of Cy3: 548 nm, emission of Cy5: 680) was recorded in a time-dependent manner. To probe the intracellular release profiles of SOD (derivatives), the polyionic complex of MOF-PGMA(EA)/SOD-60 was incubated with HUVECs for 2 h. HUVECs were washed with PBS three times and immersed in a fresh culture medium. The time-dependent FRET signals were determined using EnVision Multilabel Plate Readers (PerkinElmer, Waltham, MA) (excitation of Cy3: 548 nm, emission of Cy5: 680–720 nm).
2.14 Intracellular biological activities
The capacities of diverse SOD-1 formulations in scavenging superoxide in macrophages were investigated in a culture of TIB-186 macrophages. In brief, TIB-186 macrophages (105 cells per well, 96 well plate) were incubated with diverse SOD-1 formulations (containing constant SOD-1: 1 μg) for 2 h. The cells were washed 3 times and then stimulated with 0.2 μg mL−1 phorbol myristate acetate (PMA) for 30 min. The superoxide production from these macrophages was measured using a cytochrome c-based assay.25
3. Results and discussion
3.1 Synthesis and characterizations of dendritic MOF derivatives and charge reversible SOD-1 derivatives
The MOF derivatives were synthesized according to Scheme 1. In brief, the amino-functionalized MOF kernel of UiO-66 was achieved by the reaction of 2-amino-benzendicarboxylic acid and ZrCl4. The introduced amino moieties were further replaced by a bromide terminus and employed for the atom transfer radical polymerization of the monomers of glycidyl methacrylate to yield external polymeric segments. Ultimately, the obtained polymeric segments were transferred for an aminolysis reaction to afford hyper-charged supramolecular MOF-PGMA(EA) (Fig. S1†). XRD measurements confirmed the kernel MOF formation (Fig. S2†), and 1H-NMR measurements confirmed the successful propagation of the PGMA(EA) segments from the MOF kernel (Fig. S3†). For comparison, a linear poly[(glycidyl methacrylate)-(ethanolamine)] PGMA(EA) was synthesized as a control. The linear PGMA(EA) and supramolecular MOF-PGMA(EA) catiomers (chemical structures provided in Fig. 1a and the chemical descriptions provided in Table S1†) were proposed to electrostatically complex with the charge-reversible SOD-1 derivative to formulate a protein delivery system.
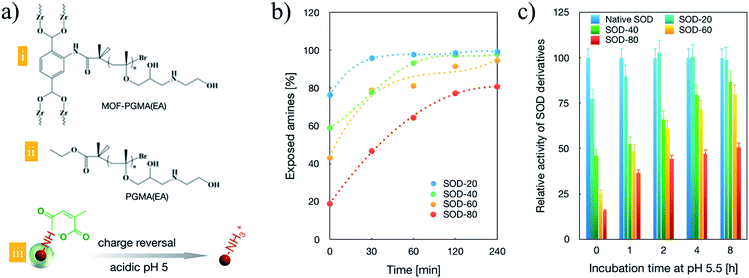 |
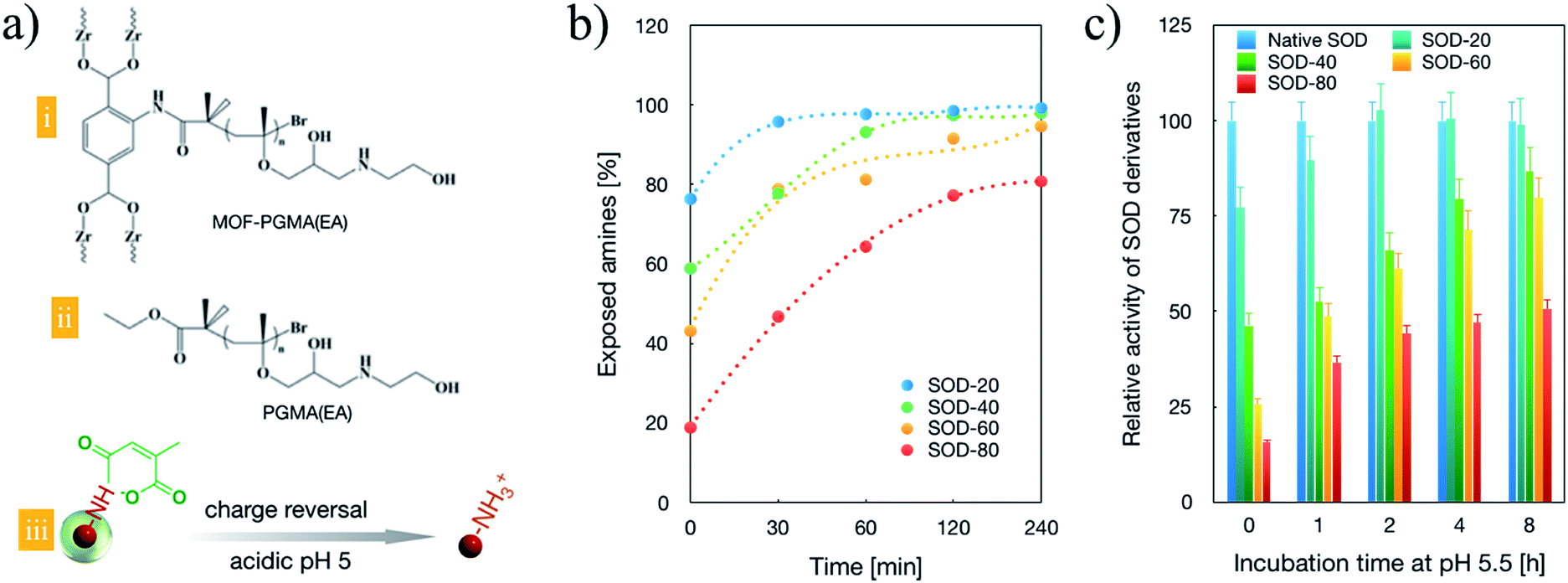
| | Fig. 1 Chemical structures and characterizations of the constituent components of SOD transcellular shuttles. The chemical structure of MOF-PGMA(EA). (a) Chemical structures of the dendritic MOF-PGMA(EA) (i) and linear PGMA(EA) (ii) and the molecular mechanism of charge reversal of SOD-1 derivatives upon acidic treatment (iii). (b) Acid (pH 5.5)-stimulated charge-reversal kinetics of SOD-1 derivatives. (c) SOD-1 activities of a class of SOD-1 derivatives upon incubation at pH 5 for varied periods. | |
Furthermore, the quantitative conversion of the amino groups in SOD-1 to carboxyl groups was accomplished by its reaction with cis-aconitic anhydride to yield cis-aconitic amide (Scheme 1). The resulting cis-aconitic amide was fairly stable at the physiological pH of 7.4, and the primary amino groups could be readily regenerated via the acidic hydrolysis of cis-aconitic amide to detach cis-aconitic anhydride under acidic incubation (e.g., endosome entrapment, the pH of which has been documented to approach 5 prior to the transformation into digestive lysosomes, Fig. 1a). Herein, a class of SOD-1 derivatives with varied charge conversion ratios (defined as the percentages of the converted amino groups to the total amino groups in SOD-1) was prepared. The detailed chemical descriptions of the SOD-1 derivatives are summarized in Table 1.
Table 1 Numbers of the residual amines and the converted carboxyl groups and percentages of the converted amines of native SOD-1 and SOD-1 derivatives at pH 7.4
| Sample |
Number of residual amines |
Converted amines (%) |
Created carboxyl |
|
The numbers in the sample names represent the percentage of charge-converted amino groups in the SOD-1 derivatives.
|
| Native SOD |
15 |
0 |
0 |
| SOD-20a |
11.4 |
23.8 |
7.1 |
| SOD-40a |
8.8 |
41.2 |
12.4 |
| SOD-60a |
6.5 |
56.9 |
17.1 |
| SOD-80a |
2.8 |
81.2 |
24.4 |
To validate the feasibility of the charge reversal capacities of the synthesized SOD derivatives, the exposed amino groups were quantified based on a fluorescamine assay. As shown in Fig. 1b, prior to acidic treatment (pH 5.5), approximately 20%, 40%, 60% and 80% of the residual amino groups were confirmed for SOD-20, SOD-40, SOD-60 and SOD-80, respectively, approving the facile quantitative modulation of amino groups of native SOD-1 with carboxyl groups (Fig. 1b, time 0). Progressive charge reversion to amino groups was observed for the class of SOD-1 derivatives under extended incubation in endosome-representative milieu (pH 5.5). Complete charge reversal was achieved for SOD-20, SOD-40 and SOD-60 upon 240 min acidic incubation (Fig. 1b), suggesting their potential for the construction of transcellular delivery vehicles. Given that the pioneering research on the chemical modification of SOD to SOD-1 validated that SOD-1 derivatives substantially inhibited its enzymatic activities,26–29 it is imperative for SOD-1 derivatives to detach the chemical modifications at the targeted subcellular compartment. Herein, to test the feasibilities of reclaiming the enzymatic activity upon acidic treatment, a class of SOD-1 derivatives post acidic incubation was transferred for the estimation of the enzymatic activity. Herein, the SOD activity was evaluated using SOD-1 activity assay kits (Cayman Chemicals Inc., Ann Arbor, Michigan) according to the manufacturer's protocol, where native SOD-1 was utilized as the 100% standard reference. As shown in Fig. 1c, a progressive increase in SOD activities is observed with extended incubation in endosome-representative milieu (pH 5.5), which is in accordance with the charge reversal behaviours of the cis-aconitic amide-functionalized SOD-1 derivatives (Fig. 1a). Notably, the enzymatic activities of SOD-1 could be fairly preserved with cis-aconitic amide modification, with over enzymatic 75% activities for SOD-20, SOD-40 and SOD-60 upon 480 min acidic incubation, confirming our strategical harnessing of cis-aconitic amide-based chemistry for the facile restoration of the protein activities in response to an acidic gradient. However, SOD-80 was observed to exhibit merely 50% enzymatic activity relative to native SOD-1, which can possibly be attributed to its difficulties in retrieving the original structure of SOD-1 as a consequence of exceeding cis-aconitic amide modification to SOD-1. To this end, in the quest to reclaim high biological activities and in the pursuit of a large number of carboxyl groups for subsequent electrostatic complexation with cationic MOF-PGMA(EA), SOD-60 was selected for the subsequent experiments.
3.2 Dendritic MOF and charge reversible SOD-1 derivatives for facilitated cellular uptake and endosome escape
SOD-60 was utilized for complexation with dendritic MOF-PGMA(EA) for the manufacture of protein delivery vehicles. In comparison, the linear PGMA(EA) polycation was employed as a control. The formation of MOF-PGMA(EA)/SOD-60 or PGMA(EA)/SOD-60 [prepared at an N/C ratio of 0.9, defined as the equal molar quantities of amino groups from the cationic MOF-PGMA(EA) or PGMA(EA) and the carboxyl groups from SOD-60] was characterized by DLS measurements. As shown in Fig. 2a, both possess nanoscaled structures with comparable sizes and unimodal polydispersity (PDI < 0.1), and the hydrodynamic diameters of MOF-PGMA(EA)/SOD-60 and PGMA(EA)/SOD-60 were determined to be 36.8 nm and 35.5 nm, respectively. Moreover, the zeta-potential measurement confirmed the comparable positive net charge of MOF-PGMA(EA)/SOD-60 (+19.8 mV) and PGMA(EA)/SOD-60 (+17.4 mV).
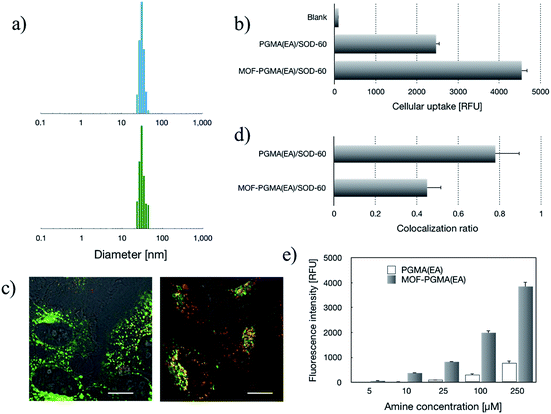 |
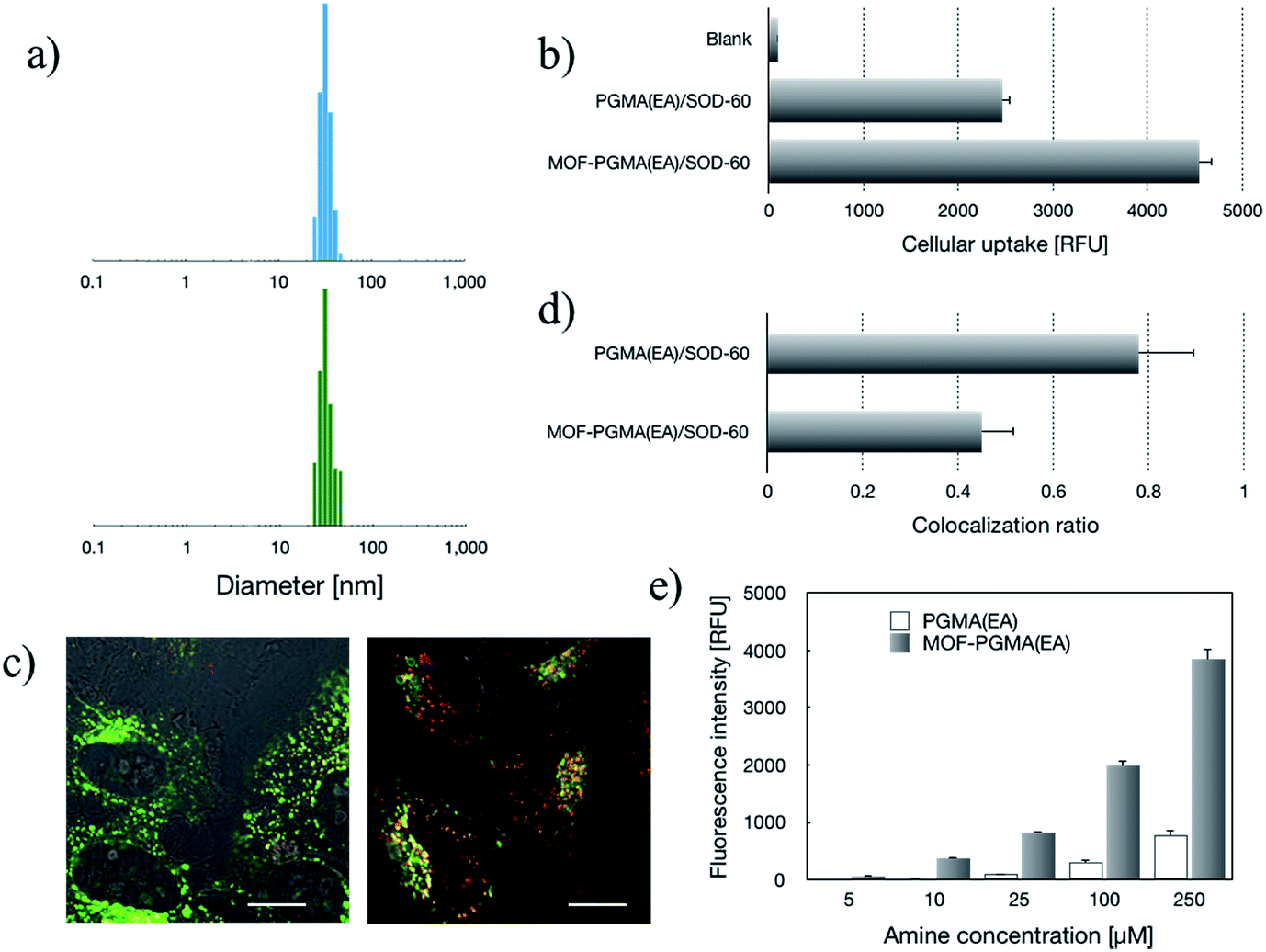
| | Fig. 2 Structural characterizations and in vitro evaluation of the SOD-60 delivery systems from cationic linear PGMA(EA) and dendritic MOF-PGMA(EA). (a) DLS measurements for MOF-PGMA(EA)/SOD-60 (upper) and PGMA(EA)/SOD-60 (lower). (b) Cellular uptake efficiency. (c) Intracellular distributions of the SOD-60 delivery systems from cationic linear PGMA(EA) (left) and dendritic MOF-PGMA(EA) (right), wherein SOD (derivatives) was stained red and endosomes were stained green; scale bar: 10 μm. The quantified colocalization ratio of SOD (derivatives) and endosomes is summarized as bar graph in (d). (e) Cytoplasm membrane destabilization activities at pH 5. | |
The relatively positive surface charge of the SOD-60-based formulations is conjectured to be favorable for cellular internalization. Herein, SOD-60 labelled with Alexa Fluor 488 was used to prepare MOF-PGMA(EA)/SOD-60 and PGMA(EA)/SOD-60. The cellular uptake activities of SOD-60 were visualized via fluorescence microscopy for cells incubated with native SOD and MOF-PGMA(EA)/SOD-60. Unlike the negligible cellular uptake for native SOD (Fig. S5†), abundant intracellular SOD could be observed for MOF-PGMA(EA)/SOD-60. This observation suggested the utility of cationic MOF-PGMA(EA) in the neutralization of SOD-60 to attain a relatively positive surface charge for favorable affinity to the negatively charged cellular membrane and subsequent cell internalization activities. The quantitative measurement of the intracellular MOF derivatives was conducted via flow cytometry. In accordance with the fluorescence microscopy measurements, significantly higher cellular uptake efficiencies were confirmed for MOF-PGMA(EA)/SOD-60 and PGMA(EA)/SOD-60 (Fig. 2b). Close examinations indicated a relatively higher intracellular level of MOF-PGMA(EA)/SOD-60 than that of PGMA(EA)/SOD-60 (Fig. 2b). A plausible reason for the higher cellular uptake of MOF-PGMA(EA)/SOD-60 could be the improved colloidal stabilities of MOF-PGMA(EA)/SOD-60 relative to that of PGMA(EA)/SOD-60. Thus, it was capable of withstanding premature dissociation in the biological milieu, thereby permitting a larger number of MOF-PGMA(EA)/SOD-60 nanoparticles to be internalized in the cells. The markedly high cellular uptake efficiency from the constructed MOF-PGMA(EA)/SOD-60 complex as compared to that of commercial liposome-based protein transfection reagents (Xfect™ protein transfection reagent, Lipofectamine™ 3000 and Proteinfectin™) was notable, where approximately one-order of magnitude higher intracellular SOD-1 using our proposed strategy was confirmed as compared to the commercial reagents (Fig. S6†). A plausible reason for this contrast is the low protein encapsulation efficiency by the aforementioned liposome formulations.
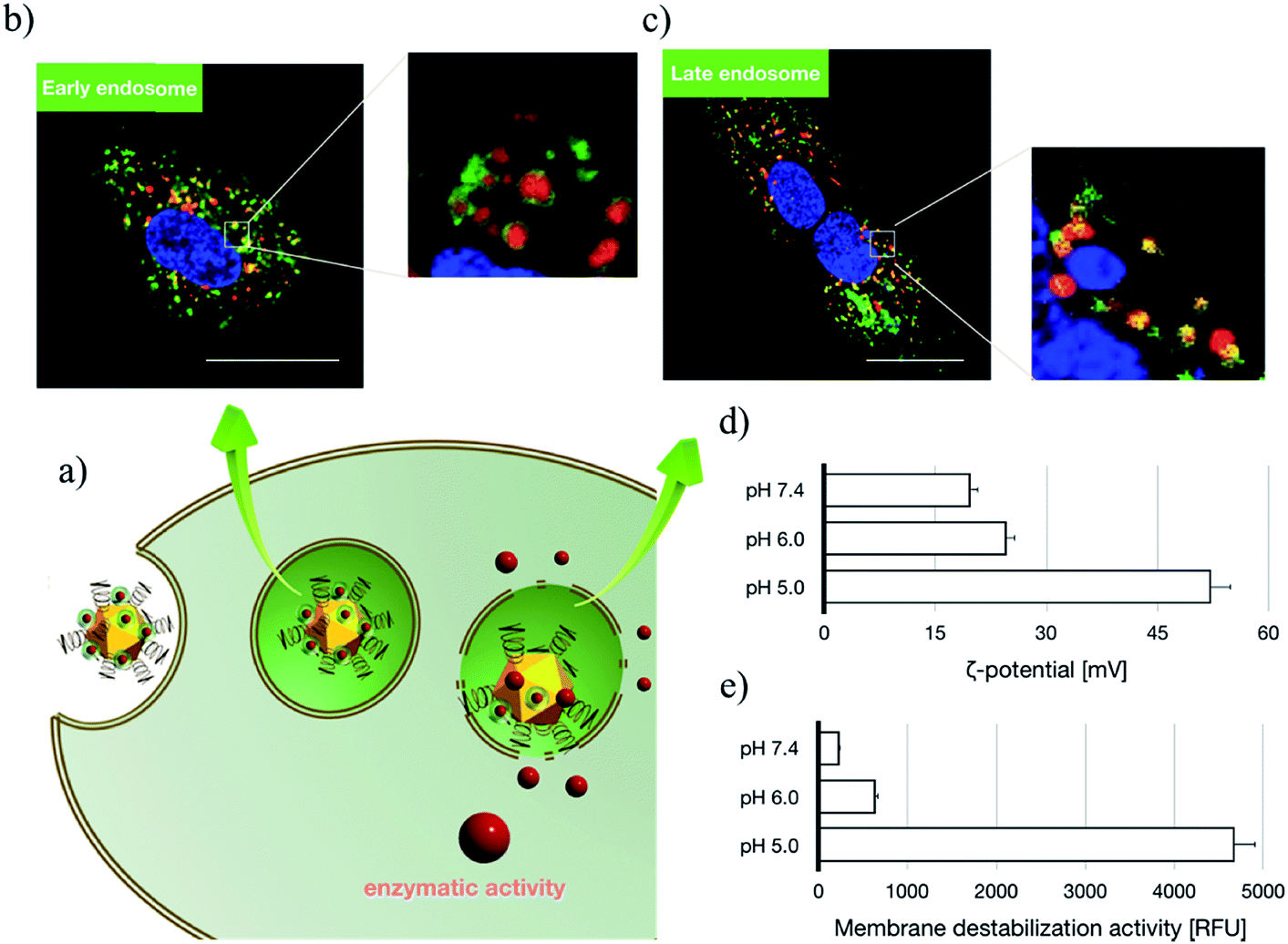
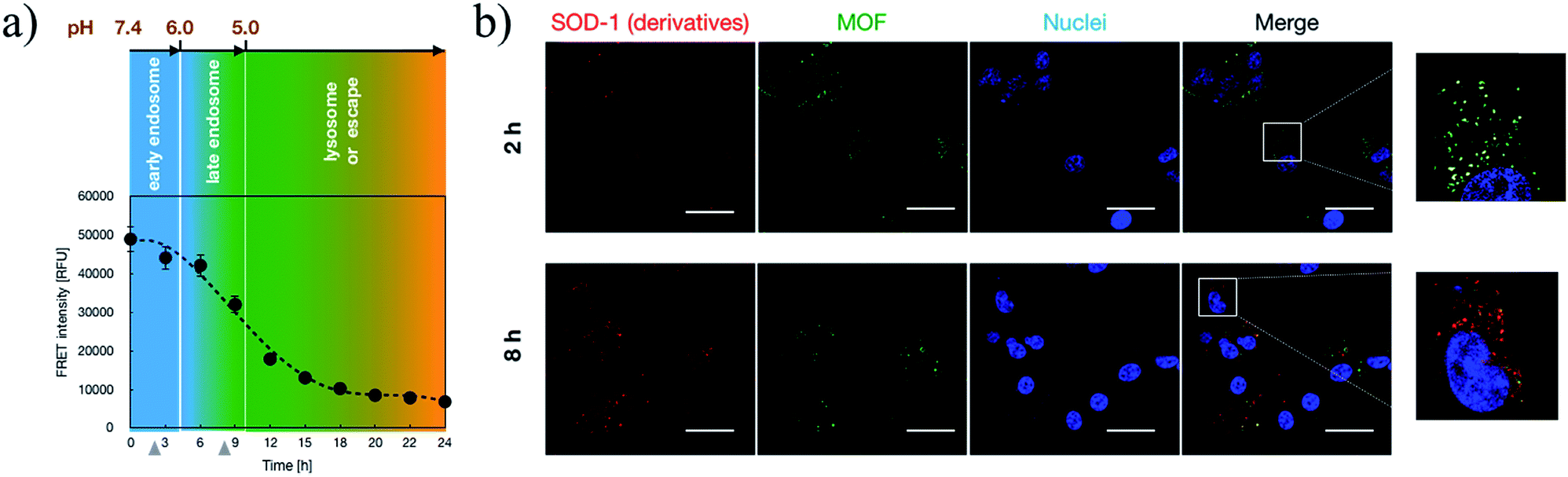
Subsequent to cellular internalization, the internalized species were entrapped in the acidic endosomes and subjected to degradation in the ensuing digestive lysosomes. Thus, the delivery vehicles afforded adequate facilities to retrieve the therapeutic payloads from endosome entrapment. The cationic species retrieved the entrapped payloads from the endosomes via electrostatic destabilization of the negatively charged plasma membrane.30,31 Thus, we compared the membrane destabilization potencies of MOF-PGMA(EA) and PGMA(EA) in the representative acidic endosome pH (pH 5). The membrane destabilizing activities were assessed by measuring the intracellular membrane-impermeable YOYO™-1 iodide from the cells in contact with MOF-PGMA(EA) or PGMA(EA). Markedly stronger destabilization to induce the loss of cell membrane integrity was observed for MOF-PGMA(EA) as compared to that for PGMA(EA) (Fig. 2e). This result supported our strategic arrangement of abundant and dense amino groups on the surface of the MOF architecture to achieve explosive interactions and destabilization reactions to the endosome membrane. To explore evidence for the potential electrostatic destabilization of MOF-PGMA(EA) to the anionic endosome membrane, we obtained high-resolution CLSM measurements to characterize the structures of the endosome membrane in the presence of MOF-PGMA(EA)/SOD-60, where the early endosome membrane and late endosome membrane were stained by CellLight™ early endosomes-GFP and late endosome GFP, respectively. According to the preliminary experiments, we identified that the proposed SOD-1 delivery constructs localized in the early endosomes up to 3 h, followed by acidic late endosomes for approximately 10 h. Accordingly, we attempted to explore the potential endosome escape capacity of our proposed pH-responsive constructs in pH-neutral early endosomes (2 h post transfection) and pH-acidic late endosomes (8 h post transfection) by high-resolution CLSM. As shown in Fig. 3b, the proposed constructs (where the MOF derivatives were stained red) are fairly entrapped in the pH-neutral early endosomes, as evidenced by the observation that the proposed MOF-PGMA(EA)/SOD-60 constructs are entirely encompassed by the early endosome membrane (green) in the magnified inset of Fig. 3b. On the contrary, the proposed constructs appeared to exert marked disruption to the late endosome membrane (Fig. 3c). A plausible reason for this contrast is the pH-responsive character of our proposed constructs. The charge reversal activity of SOD-1 derivatives gradually resulted in a positive charge from their original negative charge (Fig. 1b), thereby leading to the release of the SOD-1 derivative from the positively charged MOF and elevated charge density of MOF (Fig. 3d). This accounted for the electrostatic reaction with the negatively charged endosome membrane. Considering the significantly higher membrane destabilizing potency of MOF-PGMA(EA) than that of PGMA(EA), it is reasonable to anticipate the intensive destabilization potency from MOF-PGMA(EA) to the negatively charged endosome membrane (Fig. 3e), ultimately accounting for its easy escape from digestive late endosome/lysosome entrapment. The endosome escape activities of MOF-PGMA(EA)/SOD-60 and PGMA(EA)/SOD-60 were studied by CLSM observations (Fig. 2c). Their endosome escape capacities were studied by quantifying the colocalization degrees of SOD-60 (red) and late endosomes/lysosomes (green); yellow corresponds to SOD-60 trapped in endosomes, while red corresponds to SOD-1 (derivatives) released into the cytosol. Thus, a lower colocalization degree represents a higher endosome escape capacity. Consistent with the cell membrane destabilizing activities, significantly lower colocalization of SOD (derivatives) and endosome was calculated for the MOF-PGMA(EA)/SOD-60 sample than that for PGMA(EA)/SOD-60 (Fig. 2d). The reduced entrapment in endosomes due to MF-PGMA(EA) indicates the increased possibility of SOD (derivatives) evading degradation in the subcellular digestive endosomes/lysosomes.
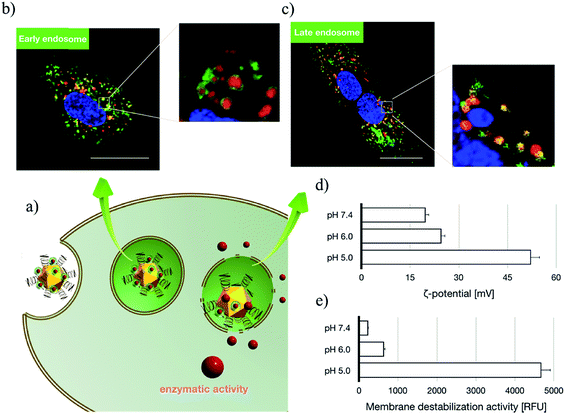 |
| | Fig. 3 Insights into endosome escape activities of the proposed transcellular SOD-1 delivery constructs. (a) Schematic illustration of the intracellular trafficking behaviours of the proposed constructs; (b) intracellular distribution of the proposed constructs at 2 h post transfection by CLSM, where MOFs were stained red, early endosomes were stained green and nuclei were stained blue. Scale bar: 20 μm. (c) Intracellular distribution of the proposed constructs at 8 h post transfection by CLSM, where MOFs were stained red, late endosomes were stained green and nuclei were stained blue. Scale bar: 20 μm. (d) ζ potential of the proposed transcellular SOD-1 delivery constructs upon 30 min incubation at pH 7.4, 6.0 and 5.0. (e) YoYo Pro-1 assay for the estimation of the membrane destabilization of the proposed constructs upon 30 min incubation at pH 7.4, 6.0 and 5.0. | |
3.3 Charge reversal of SOD derivatives for facilitated SOD release and recovered enzymatic activities
Upon acidic endosome incubation, the release of SOD-1 derivatives was speculated to occur as a result of the charge reversal of SOD derivatives. To test our speculations, the release of SOD (derivatives) apart from that of MOF-PGMA(EA) was probed by referring to a FRET method. In principle, the FRET partners Cy3 and Cy5 were used to separately label SOD-60 and MOF-PGMA(EA), respectively. The decrease in FRET intensity corresponding to the spatial separation of the SOD-1 derivatives and MOF-PGMA(EA) could represent the release of SOD-1 derivatives from MOF-PGMA(EA). In agreement with our speculation, a consistent decline in FRET intensity was observed for MOF-PGMA(EA)/SOD-60 under acidic incubation (pH 5) (Fig. S7†). This decline was also observed in the cell experiments; particularly, a progressive drop was noticed at 6–15 h post transfection (Fig. 4a), which was speculated to correspond to the acidification process of the endosomes. Indeed, subsequent CLSM observations confirmed our speculations. As shown in Fig. 4b, when trapped in the relatively pH-neutral early endosomes (2 h), where charge reversal activity can hardly occur, the electrostatically assembled MOF-PGMA(EA) and SOD-1 derivative complex remains, as evidenced by the colocalization of SOD-1 derivatives (red) and MOF-PGMA(EA) (green). In contrast, with the easy charge reversal behaviours of the SOD-1 derivatives together with the association of positively charged MOF-PGMA(EA) and endosome-membrane, the release of MOF-PGMA(EA) and SOD-1 derivatives could be clearly observed, as evidenced by low colocalization. Most likely, upon acidification in late endosomes at a pH of approximately 5 (8 h post incubation) with SOD-60, the negative charge of the SOD-1 derivatives was reduced to reclaim a positive charge as a result of the charge reversal of the amino groups (Fig. 1b), consequently yielding diminished interactive potency (even repulsive force) of SOD-1 (derivatives) to cationic MOF-PGMA(EA).
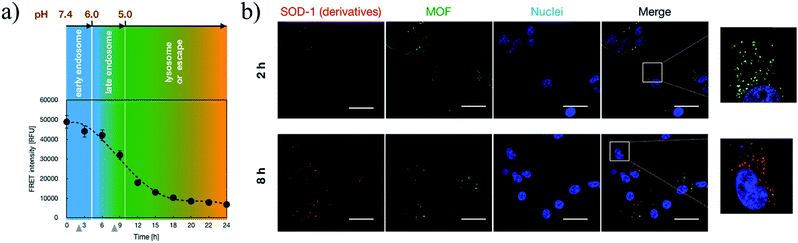 |
| | Fig. 4 Insights into the intracellular release of SOD-1 derivatives from the proposed transcellular SOD-1 delivery constructs. (a) Intracellular FRET intensities of MOF-PGMA(EA)/SOD-60 in HUVECs as a function of incubation time, where MOF-PGMA(EA) and SOD-60 were labelled by Cy3 and Cy5, respectively. (b) Intracellular distributions of MOF (green) and SOD-1 (red) upon 2 h and 8 h incubation in HUVECs. Scale bar: 20 μm. | |
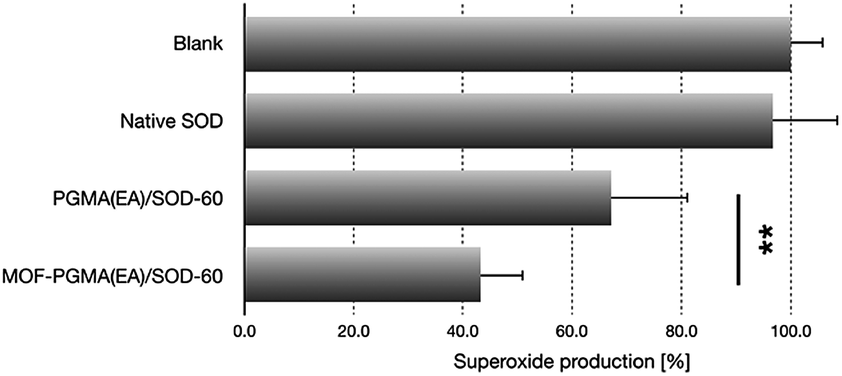
Furthermore, the released SOD-1 in the cytosol exhibited ROS scavenging activities. The ability of SOD-1 to scavenge superoxide was investigated in myristate acetate-activated TIB-186 macrophages. Herein, the suppression of the superoxide levels in TIB-186 macrophages was measured for native SOD-1, PGMA(EA)/SOD-60 and MOF-PGMA(EA)/SOD-60 based on a cytochrome c-based assay. As shown in Fig. 5, native SOD-1 elicits very limited inhibition to superoxide production, whereas distinctive reduction in superoxide production is determined for both MOF-PGMA(EA)/SOD-60 and PGMA(EA)/SOD-60. In particular, an appreciable 56% suppression in the level of superoxide was confirmed for MOF-PGMA(EA)/SOD-60 in superoxide production as compared to 33% superoxide suppression for PGMA(EA)/SOD-60, verifying the utility of our strategically engineered MOF for improved colloidal stability for transcellular protein delivery to the cytosol and the ensuing enzymatic activities.
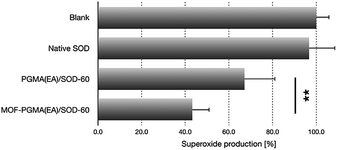 |
| | Fig. 5 Inhibitory efficacy to superoxide production in macrophages from diverse SOD formulations (**p < 0.01, Student's t test). | |
4. Conclusions
We exploited charge-reversal chemistry in therapeutic protein payloads and a dendritic polycationic MOF architecture as the protein delivery template. In particular, the strategic manufacture of a dendritic MOF template whose hyper-charged architecture not only amplified the interactive potency to hypo-charged protein derivatives for a stable delivery construct but also intensified disruptive potencies to the acidic endosome membrane for appreciable endosome escape was noteworthy. Moreover, the charge reversible function of the protein derivatives allowed facile attachment to the MOF template in the extracellular milieu and detachment from the MOF template in the intracellular milieu, consequently releasing protein payload post charge-reversal to regain the native form, exerting excellent enzymatic activities. Therefore, the proposed hyper-charged MOF-catiomer and charge reversible functionalization of therapeutic proteins manifested a robust platform for the promotion of protein therapeutics, which should be highlighted as important chemistry in the development of protein delivery constructs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by National Natural Science Foundation of China (No. 21878041, U1608222) and the funding support from the Fundamental Research Funds for the Central Universities [No. DUT17RC(3)059].
Notes and references
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21 CrossRef CAS PubMed.
- S. D. Putney and P. A. Burke, Nat. Biotechnol., 1998, 16, 153 CrossRef CAS PubMed.
- A. S. De Groot and D. W. Scott, Trends Immunol., 2007, 28, 482 CrossRef CAS PubMed.
- A. Dinca, W. M. Chien and M. T. Chin, Int. J. Mol. Sci., 2016, 17, 263 CrossRef PubMed.
- A. Bolhassani, B. S. Jafarzade and G. Mardani, Peptides, 2017, 87, 50 CrossRef CAS PubMed.
- G. Walsh, Drug Discovery Today, 2010, 15, 773 CrossRef CAS PubMed.
- E. Mastrobattista, M. A. van der Aa, W. E. Hennink and D. J. A. Crommelin, Nat. Rev. Drug Discovery, 2006, 5, 115 CrossRef PubMed.
- M. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259 CrossRef CAS PubMed.
- K. Osada, J. R. Christie and K. Kataoka, J. R. Soc., Interface, 2009, 6, S325 CrossRef CAS PubMed.
- K. Itaka and K. Kataoka, Eur. J. Pharm. Biopharm., 2009, 71, 475 CrossRef CAS PubMed.
- Y. Li, J. Li, B. Chen, Q. Chen, G. Zhang and Z. Ge, Biomacromolecules, 2014, 15, 2914 CrossRef CAS PubMed.
- K. Miyata, M. Oba, M. Nakanishi and K. Kataoka, J. Am. Chem. Soc., 2008, 130, 16287 CrossRef CAS PubMed.
- Y. Li, H. Tian, J. Ding, X. Dong, J. Chen and X. Chen, Polym. Chem., 2014, 5, 3598 RSC.
- Y. Yin and D. Talapin, Chem. Soc. Rev., 2013, 42, 2484 RSC.
- G. M. Whitesides and B. A. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS PubMed.
- C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608 CrossRef CAS PubMed.
- K. Y. Lee and S. H. Yuk, Prog. Polym. Sci., 2007, 32, 669 CrossRef CAS.
- B. Chatin, M. Mével, J. Devallière, L. Dallet, T. Haudebourg, P. Peuziat, T. Colombani, M. Berchel, O. Lambert, A. Edelman and B. Pitard, Mol. Ther.--Nucleic Acids, 2015, 4, 244 CrossRef PubMed.
- S. Dong, Q. Chen, W. Li, Z. Jiang, J. Ma and H. Gao, J. Mater. Chem. B, 2017, 5, 8322 RSC.
- V. L. Kinnula and J. D. Crapo, Am. J. Respir. Crit. Care Med., 2003, 167, 1600 CrossRef PubMed.
- G. N. Landis and J. Tower, Mech. Ageing Dev., 2005, 126, 365 CrossRef CAS PubMed.
- C. A. Hitchon and H. S. El-Gabalawy, Arthritis Res. Ther., 2004, 6, 265 CrossRef PubMed.
- H. Kamata, S. Honda, S. Maeda, L. Chang, H. Hirata and M. Karin, Cell, 2005, 120, 649 CrossRef CAS PubMed.
- A. Kim, Y. Miura, T. Ishii, O. F. Mutaf, N. Nishiyama, H. Cabral and K. Kataoka, Biomacromolecules, 2016, 17, 446 CrossRef CAS PubMed.
- K. Z. Voter, J. C. Whitin, A. Torres, P. E. Morrow, C. Cox, Y. Tsai, M. J. Utell and M. W. Frampton, Inhalation Toxicol., 2001, 13, 465 CrossRef CAS.
- C. L. Fisher, D. E. Cabelli, J. A. Tainer, R. A. Hallewell and E. D. Getzoff, Proteins, 1994, 19, 24 CrossRef CAS PubMed.
- E. D. Getzoff, J. A. Tainer, P. K. Weiner, P. A. Kollman and J. S. Richardson, Nature, 1983, 306, 287 CrossRef CAS PubMed.
- D. P. Malinowski and I. Fridovich, Biochemistry, 1979, 18, 5909 CrossRef CAS PubMed.
- S. Rasouli, A. Abdolvahabi, C. M. Croom, D. L. Plewman, Y. Shi, J. I. Ayers and B. F. Shaw, J. Biol. Chem., 2017, 292, 19366 CrossRef CAS PubMed.
- Q. Chen, K. Osada, T. Ishii, M. Oba, S. Uchida, T. A. Tockary, T. endo, Z. Ge, H. Kinoh, M. R. Kano, K. Itaka and K. Kataoka, Biomaterials, 2012, 33, 4722 CrossRef CAS PubMed.
- Q. Chen, K. Osada, Z. Ge, S. Uchida, T. A. Tockary, A. Dirisala, A. Matsui, K. Toh, K. M. Takeda, X. Liu, T. Nomoto, T. Ishii, M. Oba, Y. Matsumoto and K. Kataoka, Biomaterials, 2017, 113, 253 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Amino acids sequence of SOD; chemical structures of linear PGMA(EA) and dendritic MOF-PGMA(EA); synthetic procedure of MOF-PGMA(EA); 1H-NMR, XRD and GPC spectrum of MOF-PGMA(EA); fluorescence microscopy measurement for cellular uptake of SOD and MOF-PGMA(EA)/SOD-60 in HUVECs; cell uptake efficiency and cell viability of diverse commercial liposome-based protein transfection reagents. See DOI: 10.1039/c8sc04160a |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Zhen
Li
e,
Hongyan
Cui
b,
Shuseng
Lin
b,
Jingyi
Zhu
b and
Qixian
Chen
ab,
Zhen
Li
e,
Hongyan
Cui
b,
Shuseng
Lin
b,
Jingyi
Zhu
b and
Qixian
Chen