
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Brønsted acid and Pd–PHOX dual-catalysed enantioselective addition of activated C-pronucleophiles to internal dienes†
Sangjune
Park
,
Nathan J.
Adamson
and
Steven J.
Malcolmson
 *
*
Department of Chemistry, Duke University, French Family Science Center, 124 Science Drive, Durham, NC 27708, USA. E-mail: steven.malcolmson@duke.edu
First published on 17th April 2019
Abstract
We describe the development of Pd–PHOX-catalysed enantioselective couplings of internal dienes with malononitrile and other activated C-pronucleophiles. Reactions are dramatically accelerated by the addition of Et3N·HBArF4 as a Brønsted acid co-catalyst, enabling a suite of products bearing a variety of alkyl substituents at the stereogenic carbon to be prepared. A series of mechanism-oriented experiments reveal key aspects of the catalytic cycle and the importance of the non-coordinating BArF4 counterion in not only promoting reactions of internal dienes but also additions of previously unreactive nucleophiles towards terminal dienes.
Introduction
The addition of carbon-based electrophiles to enolates is a hallmark strategy in organic synthesis for the enantioselective formation of C–C bonds. Mannich reactions, aldol and Michael additions1 are among the most common approaches for the construction of small molecules. Enolate alkylations are also widespread but often focused towards controlling stereochemistry at a carbonyl's α-position.2 An important class of enolate alkylation is allylic substitution,3 which enables the assembly of molecules comprised of carbonyls with β-stereogenic centres4 while often concomitantly setting the stereochemistry at the α-position.5,6 Additionally, the products contain pendant unsaturation that might be leveraged for downstream synthesis applications. These enolate alkylations (allylic and non-allylic electrophiles) utilize preactivated electrophiles such as alkyl halides or allylic acetates.The use of olefins as alkylating agents7 offers a number of distinct advantages, not the least of which is the inherent stability of an alkene compared to the lability of a leaving group, facilitating carrying the olefin electrophile through several transformations in a multistep sequence. A handful of researchers have explored this approach in enantioselective catalysis. For example, enol/enolate addition to allenes8 has afforded products with a variety of substituents at the β-stereogenic centre (Scheme 1), in some cases with additional control at the carbonyl's α-position.8a,c Employing allenes as electrophiles has exclusively9 resulted in products bearing a terminal alkene. In a similar process, Rh-bis(phosphine)-catalysed enol additions to internal alkynes delivers analogous products.10‡ Terminal dienes have also emerged as alkyl electrophiles capable of affording products comprised of internal alkenes.11 Recently, our group described the Pd–PHOX-catalysed addition of activated pro-enolates to these dienes,12–14 and Xiao, Zhou and co-workers demonstrated diene reactions in Ni–bis(phosphine)-catalysed additions of enolates formed from simple ketones (Scheme 1).15 However, the products obtained from reactions of terminal dienes bear only a methyl group at the β-stereogenic centre.
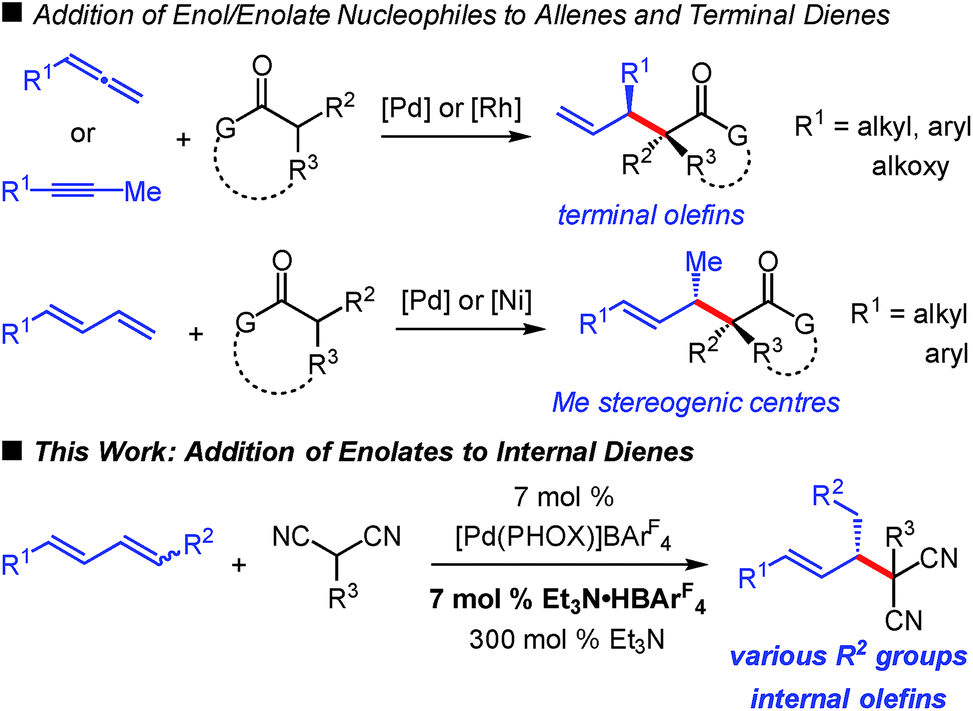 | ||
| Scheme 1 Catalytic enantioselective additions of enols/enolates to olefins. | ||
In comparison, hydrofunctionalisation of internal dienes delivers molecules with both disubstituted olefins and myriad groups at the stereogenic β-position. Still, enantioselective additions to acyclic 1,4-disubstituted dienes are challenging: only indole16 and amine17 couplings have been reported. In this work, we illustrate that malononitrile and related pronucleophiles undergo highly regio- and enantioselective additions to internal dienes (Scheme 1). High reaction efficiency was achievable by the combination of an electron-deficient Pd–PHOX catalyst and Et3N·HBArF4 as Brønsted acid co-catalyst.
Results and discussion
Method development and product functionalisation
We initiated our investigation by examining the addition of malononitrile to diene 1a with Pd–PHOX catalyst Pd-1, the optimal catalyst for internal diene hydroamination,17 which bears the non-coordinating BArF4 counterion (Table 1). We hypothesized that the small size of malononitrile, its low pKa,18 and the relatively high nucleophilicity of its anion19 would make it exceptionally reactive towards alkylation. Indeed, in the addition of malononitrile to 1-phenylbutadiene—a terminal diene—we found it difficult to suppress double alkylation.12 In the absence of any additive, there is no observable reaction with diene 1a (entry 1); however, with 300 mol% Et3N, the desired 2a is generated in >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er as the sole product of the reaction but in only 24% yield (entry 2).
:1 er as the sole product of the reaction but in only 24% yield (entry 2).
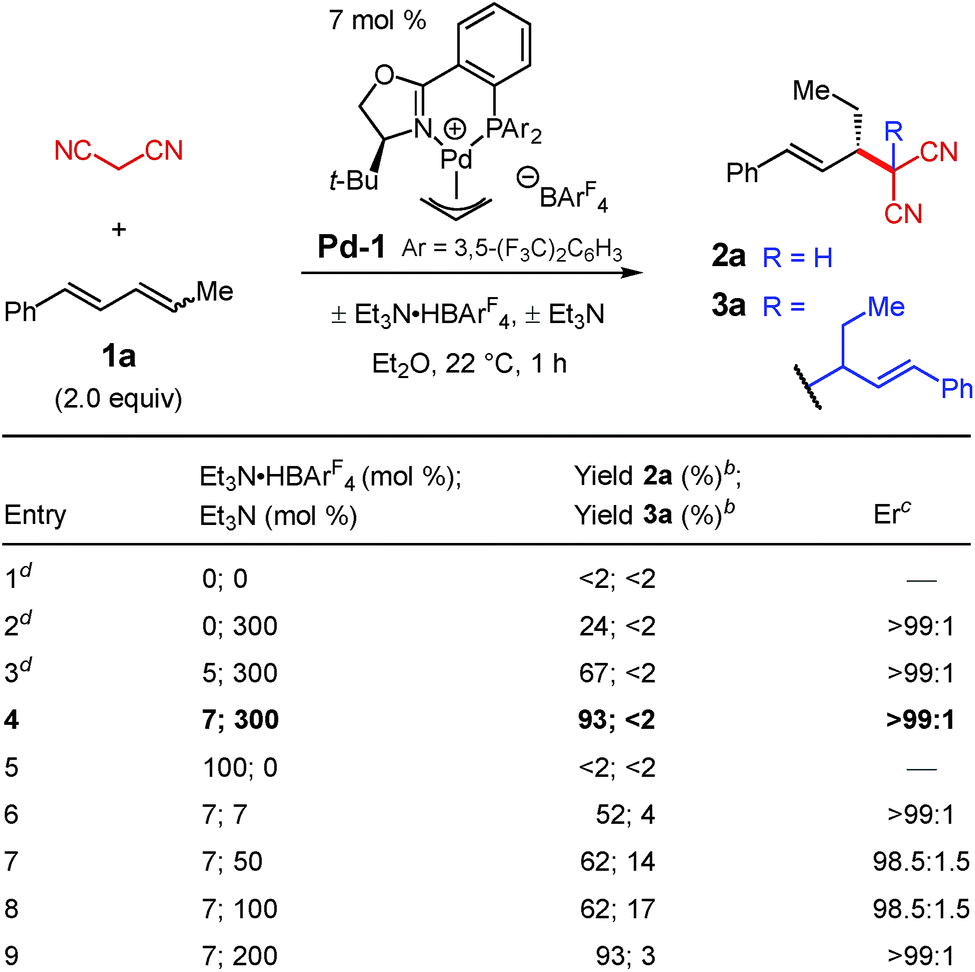
We discovered that the addition of a substoichiometric quantity of Et3N·HBArF4 as a Brønsted acid greatly improves the product yield (entry 3). Notably, the addition of Brønsted acid has been observed as a crucial component in several other transformations involving nucleophilic additions to olefins,20 including reactions of enols.8a–d,10a Further raising the catalyst loading of both Pd-1 and Et3N·HBArF4 to 7 mol%, which proved to be the optimal conditions, allows 2a to be isolated in 93% yield and >99:1 er (entry 4). In the absence of Et3N, even with stoichiometric Et3N·HBArF4 (entry 5), alkylation product 2a is not formed. Lesser quantities of the base, from 7 mol% up to 100 mol%, drastically reduce the yield of 2a while also enabling its conversion to bis-alkylation product 3a (entries 6–8). With 200 mol% Et3N, product 2a is again isolated in 93% yield but is still accompanied by a small quantity of byproduct 3a (entry 9).
Under the optimal conditions, several 1,4-disubstituted dienes couple with malononitrile (Table 2). Electronic perturbations of the diene's aryl group are tolerated, affording 2b–d in 40–66% yield and >95:5 er. Moving the aryl group substituent to the meta or ortho positions generates tolyl products 2e and 2f in higher yields (87–89%) and with excellent enantioselectivity (97.5:2.5 and 96:4 er, respectively). Changing the substituent at the 4-position of the diene enables products with a variety of alkyl groups at the stereogenic carbon to be obtained (2g–k); the reaction is tolerant of both ethereal (2i–j) and carbonyl functionality (2h and 2k). All aryl/alkyl-substituted diene substrates deliver malononitrile products 2a–k as a single regioisomer. Additionally, 1,4-dialkyl-containing dienes also afford malononitrile addition products as a single regioisomer as long as the two alkyl groups are sterically differentiated. Vinylcyclohexane 2l is obtained in 72% yield and 94:6 er. In all cases, enantioselectivity is constant throughout the course of the reaction, indicating that the process is irreversible.

Substituted malononitrile pronucleophiles undergo Pd-1-catalysed addition to diene 1a within 1 h with complete regiocontrol (Table 3). The methyl-, benzyl-, and cinnamyl-substituted pronucleophiles generate products 5a–c in 78–99% yield and >95:5 er. Acetylacetone, both less acidic21 and its anion less nucleophilic19 than malononitrile, leads to diketone 5d in 51% yield and 92.5:7.5 er after 20 h, still as a single regioisomer. Ethyl cyanoacetate addition to diene 1a, however, results in a 60:40 mixture of product regioisomers 5e and 6e (56% yield). Isomer 6e arises from addition of the nucleophile to the phenyl-substituted carbon of the π-allyl–Pd-1 intermediate derived from diene 1a whereas 5e results from nucleophile addition to the ethyl-substituted carbon of the same π-allyl complex. Dimethyl malonate, delivers a similar result (57:43 5f:6f in 49% yield). Despite their lower pKa's21,22 but perhaps partially due to their lower nucleophilicities,19 cyclic pronucleophiles such as Meldrum's acid and dimedone fail to undergo addition to diene 1a under the conditions shown in Table 3.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :
:
The malononitrile adducts of internal dienes are useful building blocks for further synthesis. For example, the malononitrile unit within 2a (Scheme 2) may undergo oxidative methanolysis23via the cyanoketone to deliver α-alkenylester 7a in 71% yield with minimal erosion of enantiopurity. Similarly, oxidation in the presence of pyrrolidine leads to α-alkenylamide 7b in 78% yield (94.5:5.5 er).24
Mechanism studies
A mechanism for Pd–PHOX-catalysed addition of malononitrile to internal dienes is illustrated in Scheme 2.17,25 After initiation of Pd-1 by nucleophilic attack of the malononitrile anion upon its π-allyl ligand, coordination of diene 1a to the Pd(0) species leads to intermediate i. Oxidative protonation at Pd by Et3N·HBArF4 leads to hydrido complex ii, which undergoes migratory insertion, ultimately forming π-allyl–Pd species iii. The equilibration is accelerated and shifted towards complex iii by the addition of exogenous Et3N·HBArF4 as the co-catalyst. At the same time, deprotonation of malononitrile by Et3N leads to the malononitrile anion iv, an equilibrium that favours the neutral molecule. The anion may then attack π-allyl–Pd iii through an outer sphere pathway. The C–C bond-forming event to form v appears to be irreversible and simultaneously regenerates Et3N·HBArF4. Exchange of the olefin within v for diene 1a releases product 2a and regenerates complex i.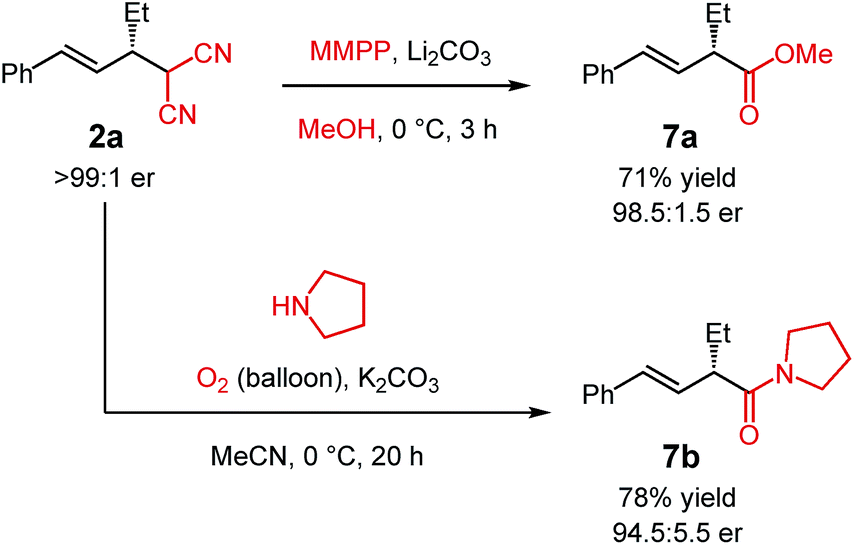 | ||
| Scheme 2 Synthesis of α-alkenyl carbonyls from the malononitrile group. | ||
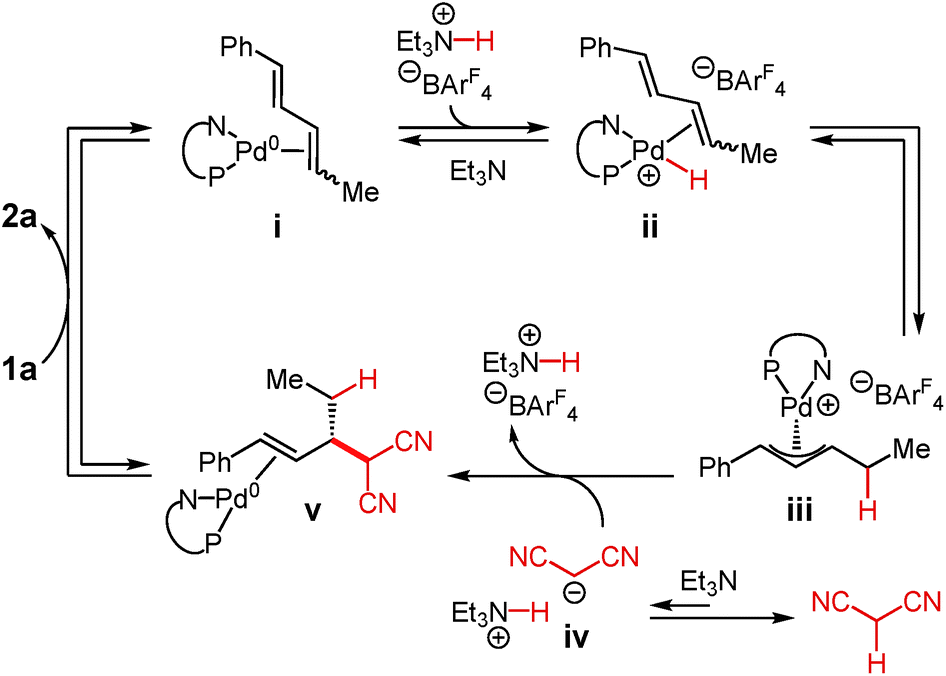 | ||
| Scheme 3 Proposed mechanism of malononitrile addition to internal dienes. | ||
In the context of this mechanistic proposal, we sought to address several facets of the Pd–PHOX-catalysed diene–enolate couplings. These investigations might allow us to glean further information about individual steps of this mechanism and more broadly understand diene couplings to nucleophiles promoted by Pd–PHOX catalysts.
We first set out to investigate the impact of diene stereochemistry upon the reaction. The transformations presented in Tables 1–3 utilize an E,Z/E,E-mixture of diene diastereomers favouring the Z-stereoisomer at the alkyl-substituted alkene. As shown in Scheme 4A, E,Z-1a is significantly more reactive than the E,E-stereoisomer, delivering malononitrile 2a in 90% yield and 99:1 er. Comparatively, E,E-1a (Scheme 4B) leads to only 35% yield of 2a (99:1 er). In both experiments, one equivalent of diene was employed and any left unreacted was completely recovered. From the reaction of E,Z-1a, the recovered diene had almost completely isomerised to the E,E-diastereomer (Scheme 4A). In contrast, the recovered diene from reaction of E,E-1a was still entirely that stereochemistry (Scheme 4B).
 | ||
| Scheme 4 Comparison of reactivity of diene diastereomers; conditions: Pd-1 (7 mol%), Et3N·HBArF4 (7 mol%), Et3N (300 mol%), Et2O, 22 °C, 1 h. | ||
These data also help explain why under the optimal conditions, product 2a does not undergo a second alkylation event despite the presence of excess diene 1a (see Table 1). Of the two equivalents of diene 1a added to the reaction, only ca. 65% (1.3 equiv.) is the more reactive E,Z-stereoisomer. The data also suggest the catalyst is capable of reacting with the diene and isomerising the E,Z- to the E,E-diastereomer without product formation. Therefore, during the course of the reaction, as mono-alkylated product builds up, there is concomitant isomerisation of the diene occurring (likely along with catalyst decomposition), thereby slowing formation of bis-alkylated 3a. With fewer Et3N equivalents, 3a is observed to some degree (Table 1). This might be attributable to the equilibrium (see Scheme 3) between malononitrile and its deprotonated form iv being shifted towards the neutral molecule when less base is available, slowing the rate of formation of 2a. Mono-alkylated 2a that is formed may then engage in subsequent reaction with remaining E,Z-1a prior to its consumption in product formation or its stereoisomerisation. Additionally, although substituted malononitriles, such as those shown in Table 3, are competent reaction partners, compound 2a is significantly more hindered (β-branching) which slows a second alkylation event.
We next further sought to assess the reversibility of the individual elementary steps connecting diene 1a to π-allyl–Pd iii (Scheme 3). To do so, we examined the coupling of a deuterated pronucleophile, (1) to discover the label's position in the anticipated addition product with respect to the site of C–C bond formation and (2) to determine whether deuterium could be incorporated into the diene without product forming. We chose deuterated 2-methylmalononitrile (d-4a) as the pronucleophile (Scheme 5). Incredibly, although the protic version of 4a delivers 5a in 99% yield after 1 h, no coupled product is formed after 20 h with d-4a. This is likely due to the combination of a significant kinetic isotope effect and catalyst decomposition. Diene 1a could be completely recovered from the mixture: a portion of the diene had isomerised from the E,Z-isomer to (E,E)-1a (from 1.8:1 to 1:1.7 E,Z:E,E). A significant percentage of the deuterium label (ca. 43%) was transferred from d-4a to the diene with the label confined solely to the recovered E,E-1a and roughly evenly distributed between C1 and C4.
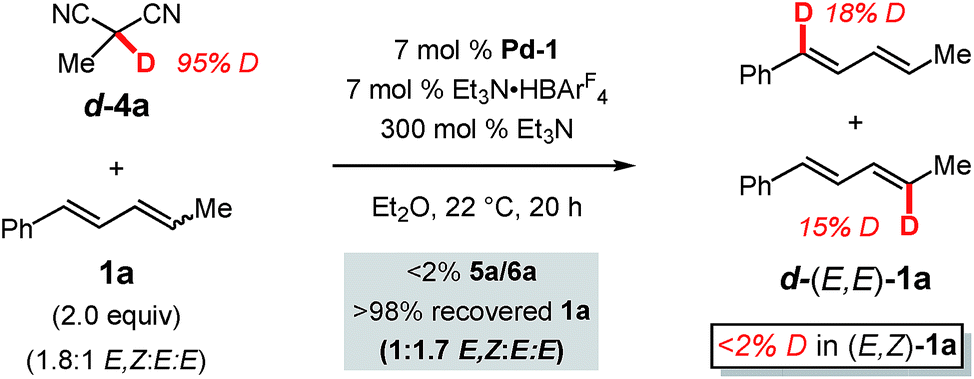 | ||
| Scheme 5 Deuterium labelling studies with deuterated methylmalononitrile | ||
Just as in hydroamination of 1a with Pd-1 (reaction of N-deuterated indoline),17 the kinetic isotope effect observed here is likely at least partially due to an equilibrium isotope effect that exchanges deuterium from d-4a to diene 1a as mediated by Et3N and the Pd catalyst. The lack of label in recovered (E,Z)-1a is owed to its conversion exclusively to d-(E,E)-1a when it reacts with Pd–D: stereochemical isomerisation of the alkene occurs by Pd–D migratory insertion to (E,Z)-1a, bond rotation, and preferential β-hydride elimination (compared to β-deuteride elimination). As mentioned with regard to the reactivity difference of the two diene stereoisomers (vide supra), this process does not occur to incorporate deuterium into (E,Z)-1avia reaction of (E,E)-1a. Finally, the distribution of the label in d-(E,E)-1a indicates that palladium–hydride/deuteride insertion may occur at either olefin of the diene, yet only insertion at the alkyl-substituted olefin leads to product in the reactions presented in Tables 1–3. The results further confirm that E,Z-diene coordination to Pd is reversible.
It is noteworthy that whereas malononitrile and acetylacetone nucleophiles afford a single detectable product regioisomer (Tables 1–3), ethyl cyanoacetate and dimethylmalonate deliver a mixture (5e/6e and 5f/6f, respectively, Table 3). As mentioned earlier, the product isomers arise from attack of the nucleophiles upon two different carbons of the same π-allyl. Differing regioselectivities among nucleophiles is likely due to Curtin–Hammett kinetics in the reaction (Scheme 6) and is the combination of three properties of the nucleophile: (1) the pKa of the pronucleophile (affecting its equilibrium with the active deprotonated nucleophile), (2) the nucleophilicity of the carbanion,19 and (3) the sterics of the nucleophile. As documented previously for Pd–PHOX complexes,3a the π-allyl–Pd species exist as a mixture of the thermodynamically preferred exo-diastereomer and its isomeric endo-complex (Scheme 6). In this transformation, faster attack of the nucleophile upon the exo-isomer at the carbon trans to the phosphine (ethyl-substituted carbon) leads to the observed major enantiomer and regioisomer (2 or 5). Conversely, some nucleophiles undergo competitive addition to the endo-diastereomer, attacking the phenyl-substituted carbon (trans to the phosphine), affording 6. Although ethyl cyanoacetate has a similar pKa (ref. 26) to acetylacetone, generating a certain quantity of active nucleophile, its nucleophilicity is considerably greater (similar to malononitrile19). Likewise, even though dimethyl malonate has an even higher pKa,22 its nucleophilicity is greater still.19 Therefore, it is possible that reactants with greater nucleophilicity lead to similar rates of attack upon the exo- and endo-diastereomers of the π-allyl intermediate. The outlier is malononitrile, whose lower pKa (ref. 18) and high nucleophilicity19 should seemingly lead to an isomeric mixture. The high selectivity for products 2 or 5 with malononitrile (via the exo-isomer) is likely due to the small size of this pronucleophile.
 | ||
| Scheme 6 Curtin–Hammett kinetics likely operative in internal diene–enolate couplings and responsible for the observed regio- and enantioselectivity. | ||
An interesting dichotomy between reactions of terminal dienes, such as 1-phenylbutadiene, and those of internal dienes with activated C-pronucleophiles as catalysed by Pd–PHOX complexes presented itself in this study. In our previous investigations of C-pronucleophile additions to terminal dienes,12 Meldrum's acid proved to be one of the best and most versatile pronucleophiles. Those reactions are promoted by the same PHOX ligand for Pd as in Pd-1 but with a catalyst that bears a tetrafluoroborate counterion in place of BArF4. Additionally, terminal diene reactions do not require Brønsted acid additive. Contrastingly, dimethyl malonate fails to undergo addition to 1-phenylbutadiene under the previously established optimal terminal diene conditions with the BF4-containing catalyst.
We wished to determine whether these differences arise from the BArF4 counterion of Pd-1 or the Et3N·HBArF4 additive. As shown in Scheme 7, dimethyl malonate 4f is able to add to 1-phenylbutadiene in the presence of Pd-1 (BArF4 counterion). Although the yield is slightly higher with the addition of Et3N·HBArF4, both the regioselectivity and enantiopurity of the major product (8) are the same with or without Brønsted acid. Therefore, higher pKa pronucleophiles may react with terminal dienes with a Pd–PHOX catalyst comprised of the non-coordinating BArF4. It is noteworthy that the regiomeric ratio favouring the 4,3-addition product 9 is higher for dimethyl malonate coupling to phenylbutadiene than to internal diene 1a (cf., Table 3, compound 5f).
 | ||
| Scheme 7 Addition of dimethyl malonate to phenylbutadiene with Pd-1. | ||
We finally turned to addressing the lack of reactivity of Meldrum's acid towards internal diene 1a with Pd-1. In fact, not only does C–C bond formation fail to occur in this case, based on the diene recovered from the reaction mixture, the initial 1.8:1 E,Z:E,E ratio of 1a also remains unchanged. The absence of stereochemical isomerisation indicates that catalyst formed from Pd-1 fails to engage the diene at all in the presence of this pronucleophile.
As mentioned, we previously developed the addition of Meldrum's acid to 1-phenylbutadiene with [(η3-C3H5)Pd(PHOX)]BF4 as the catalyst.12 Unfortunately, reactions of internal dienes are completely inhibited by having a BF4 counterion in the reaction medium, preventing our examining Meldrum's acid reaction with internal diene 1a catalysed by [(η3-C3H5)Pd(PHOX)]BF4. However, Meldrum's acid addition to 1-phenylbutadiene occurs smoothly with BArF4-containing Pd-1, delivering a nearly identical result to its BF4 analogue (Scheme 8). These data highlight that the acidic Meldrum's acid is compatible with Pd-1 no matter what its counteranion as long as a more reactive terminal diene is present. This suggests that as the internal diene 1a is more difficult to coordinate to the metal centre, significant catalyst decomposition occurs under the more acidic conditions imposed with Meldrum's acid (lower pKa) compared to other pronucleophiles, thereby impeding the desired reaction pathway.
 | ||
| Scheme 8 Meldrum's acid addition to phenylbutadiene with Pd–PHOX catalysts bearing different counteranions. | ||
Conclusions
We have demonstrated that internal dienes act effectively as alkylating agents for enantioselective intermolecular couplings with in situ-generated enolates under Pd–PHOX catalysis. The atom economic olefin hydrofunctionalisations are accelerated by the addition of Et3N·HBArF4 as a Brønsted acid co-catalyst. Somewhat counterintuitively, the use of superstoichiometric Et3N is needed to generate sufficient quantities of the active nucleophile in order to form the mono-alkylation product selectively (i.e., avoid over-alkylation). Several features of the reaction mechanism have been uncovered through deuterium labelling the pronucleophile and by selectively employing different diastereomerically pure diene stereoisomers in reactions. Notable differences in participating C-pronucleophiles in Pd–PHOX-catalysed reactions of internal versus terminal 1,3-dienes have been unveiled. Further investigation of reaction mechanism and applications to additional methodology development from lessons learned in these studies are ongoing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (CHE-1800012), the ACS Petroleum Research Fund (56575-DNI1), and Duke University. N. J. A. is grateful to the Duke Chemistry Department for a Burroughs-Wellcome Fellowship.Notes and references
- For reviews, see: (a) J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 324 CrossRef; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed; (c) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626 CrossRef CAS PubMed; (d) G. L. Beutner and S. E. Denmark, Angew. Chem., Int. Ed., 2013, 52, 9086 CrossRef CAS PubMed; (e) H. Pellissier, Chem. Rev., 2016, 116, 14868 CrossRef CAS PubMed.
- (a) T. Ooi, T. Miki, M. Shiraishi, M. Takeuchi and K. Maruoka, Angew. Chem., Int. Ed., 2003, 42, 3796 CrossRef CAS; (b) A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2005, 127, 62 CrossRef CAS PubMed; (c) H.-Y. Jang, J.-B. Hong and D. W. C. MacMillan, J. Am. Chem. Soc., 2007, 129, 7004 CrossRef CAS PubMed; (d) S. Hong, J. Lee, M. Kim, Y. Park, C. Park, M.-h. Kim, S.-s. Jew and H.-g. Park, J. Am. Chem. Soc., 2011, 133, 4924 CrossRef CAS PubMed; (e) T. Kano, Y. Hayashi and K. Maruoka, J. Am. Chem. Soc., 2013, 135, 7134 CrossRef CAS PubMed; (f) B. Teng, W. Chen, S. Dong, C. W. Kee, D. A. Gandamana, L. Zong and C.-H. Tan, J. Am. Chem. Soc., 2016, 138, 9935 CrossRef CAS.
- For reviews, see: (a) G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336 CrossRef CAS; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (c) J. T. Mohr and B. M. Stoltz, Chem.–Asian J., 2007, 2, 1476 CrossRef CAS PubMed; (d) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258 CrossRef CAS PubMed; (e) J. C. Hethcox, S. E. Shockley and B. M. Soltz, ACS Catal., 2016, 6, 6207 CrossRef CAS PubMed.
- (a) B. M. Trost and C. B. Lee, J. Am. Chem. Soc., 2001, 123, 3671 CrossRef CAS PubMed; (b) G. Lipowsky, N. Miller and G. Helmchen, Angew. Chem., Int. Ed., 2004, 43, 4595 CrossRef CAS PubMed; (c) D. J. Weix and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 7720 CrossRef CAS PubMed; (d) B. Mao, Y. Ji, M. Fañanás-Mastral, G. Caroli, A. Meetsma and B. L. Feringa, Angew. Chem., Int. Ed., 2012, 51, 3168 CrossRef CAS PubMed; (e) W. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 15249 CrossRef CAS PubMed; (f) M. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 2014, 53, 8691 CrossRef CAS PubMed; (g) J. C. Hethcox, S. E. Shockley and B. M. Stoltz, Angew. Chem., Int. Ed., 2018, 57, 1 CrossRef PubMed; (h) S.-B. Tang, X. Zhang, H.-F. Tu and S.-L. You, J. Am. Chem. Soc., 2018, 140, 7737 CrossRef CAS PubMed; (i) T. W. Butcher and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 13125 CrossRef CAS PubMed.
- (a) B. M. Trost and C. Lee, J. Am. Chem. Soc., 2001, 123, 12191 CrossRef CAS; (b) W.-H. Zheng, B.-H. Zheng, Y. Zhang and X.-L. Hou, J. Am. Chem. Soc., 2007, 129, 7718 CrossRef CAS PubMed; (c) W. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2068 CrossRef CAS PubMed; (d) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 Search PubMed; (e) W.-B. Liu, C. M. Reeves and B. M. Stoltz, J. Am. Chem. Soc., 2013, 135, 17298 CrossRef CAS PubMed; (f) W. Chen, M. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 15825 CrossRef CAS PubMed; (g) K. Ikeda, T. Futamura, T. Hanakawa, M. Minakawa and M. Kawatsura, Org. Biomol. Chem., 2016, 14, 3501 RSC; (h) X. Jiang, W. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 5819 CrossRef CAS PubMed; (i) K. Balaraman and C. Wolf, Angew. Chem., Int. Ed., 2017, 56, 1390 CrossRef CAS PubMed; (j) J. Liu, Z. Han, X. Wang, F. Meng, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2017, 56, 5050 CrossRef CAS PubMed; (k) P. Starkov, J. T. Moore, D. C. Duquette, B. M. Stoltz and I. Marek, J. Am. Chem. Soc., 2017, 139, 9615 CrossRef CAS PubMed.
- For examples where only an α-stereogenic centre is set, see: (a) B. M. Trost and G. M. Schroeder, J. Am. Chem. Soc., 1999, 121, 6759 CrossRef CAS; (b) J. T. Mohr, T. Nishimata, D. C. Behenna and B. M. Stoltz, J. Am. Chem. Soc., 2006, 128, 11348 CrossRef CAS PubMed; (c) B. M. Trost, R. Koller and B. Schäffner, Angew. Chem., Int. Ed., 2012, 51, 8290 CrossRef CAS PubMed; (d) T. B. Wright and P. A. Evans, J. Am. Chem. Soc., 2016, 138, 15303 CrossRef CAS PubMed; (e) A. Ngamnithiporn, C. Jette, S. Bachman, S. C. Virgil and B. M. Stoltz, Chem. Sci., 2018, 9, 2547 RSC.
- For reviews, see: (a) F. Dénès, A. Pérez-Luna and F. Chemla, Chem. Rev., 2010, 110, 2366 CrossRef; (b) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333 CrossRef CAS.
- (a) B. M. Trost, C. Jäkel and B. Plietker, J. Am. Chem. Soc., 2003, 125, 4438 CrossRef CAS PubMed; (b) B. M. Trost, A. B. C. Simas, B. Plietker, C. Jäkel and J. Xie, Chem.–Eur. J., 2005, 11, 7075 CrossRef CAS PubMed; (c) B. M. Trost, J. Xie and J. D. Sieber, J. Am. Chem. Soc., 2011, 133, 20611 CrossRef CAS PubMed; (d) T. M. Beck and B. Breit, Angew. Chem., Int. Ed., 2017, 56, 1903 CrossRef CAS PubMed . For an example that leads to products with only an α-stereogenic centre, see: ; (e) H. Zhou, Y. Wang, L. Zhang, M. Cai and S. Luo, J. Am. Chem. Soc., 2017, 139, 3631 CrossRef CAS PubMed.
- For a non-enantioselective exception, see: B. M. Trost and V. J. Gerusz, J. Am. Chem. Soc., 1995, 117, 5156 CrossRef CAS.
- For an enantioselective intermolecular example, see: (a) F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1029 CrossRef CAS PubMedFor non-enantioselective intermolecular examples, see: (b) F. A. Cruz, Z. Chen, S. I. Kurtoic and V. M. Dong, Chem. Commun., 2016, 52, 5836 RSC; (c) C. Li, C. P. Grugel and B. Breit, Chem. Commun., 2016, 52, 5840 RSC.
- For reviews regarding enantioselective reactions of dienes, see: (a) Y. Xiong, Y. Sun and G. Zhang, Tetrahedron Lett., 2018, 59, 347 CrossRef CAS; (b) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026 CrossRef CAS.
- N. J. Adamson, K. C. E. Wilbur and S. J. Malcolmson, J. Am. Chem. Soc., 2018, 140, 2761 CrossRef CAS PubMed.
- For an example that leads to products with only an α-stereogenic centre, see: Y.-L. Su, L.-L. Li, X.-L. Zhou, Z.-Y. Dai, P.-S. Wang and L.-Z. Gong, Org. Lett., 2018, 20, 2403 CrossRef CAS PubMed.
- For intermolecular non-enantioselective enol/enolate additions to dienes, see: (a) K. Takahashi, A. Miyake and G. Hata, Bull. Chem. Soc. Jpn., 1972, 45, 1183 CrossRef CAS; (b) B. M. Trost and L. Zhi, Tetrahedron Lett., 1992, 33, 1831 CrossRef CAS; (c) A. Leitner, J. Larsen, C. Steffens and J. F. Hartwig, J. Org. Chem., 2004, 69, 7552 CrossRef CAS PubMed; (d) M. J. Goldfogel and S. J. Meek, Chem. Sci., 2016, 7, 4079 RSC; (e) M. J. Goldfogel, C. C. Roberts, R. S. Manan and S. J. Meek, Org. Lett., 2017, 19, 90 CrossRef CAS.
- L. Cheng, M.-M. Li, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, J. Am. Chem. Soc., 2018, 140, 11627 CrossRef CAS.
- J. S. Marcum, C. C. Roberts, R. S. Manan, T. N. Cervarich and S. J. Meek, J. Am. Chem. Soc., 2017, 139, 15580 CrossRef CAS.
- S. Park and S. J. Malcolmson, ACS Catal., 2018, 8, 8468 CrossRef CAS.
- W. S. Matthews, J. E. Bares, J. E. Bartmess, F. G. Bordwell, F. J. Cornforth, G. E. Drucker, Z. Margolin, R. J. McCallum, G. J. McCollum and N. R. Vanier, J. Am. Chem. Soc., 1975, 97, 7006 CrossRef CAS.
- R. Lucius, R. Loos and H. Mayr, Angew. Chem., Int. Ed., 2002, 41, 91 CrossRef CAS.
- (a) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2000, 122, 9546 CrossRef CAS; (b) O. Löber, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 4366 CrossRef; (c) J. Pawlas, Y. Nakao, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 3669 CrossRef CAS PubMed; (d) M. Utsunomiya and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 2702 CrossRef CAS; (e) A. B. Pritzius and B. Breit, Angew. Chem., Int. Ed., 2015, 54, 15818 CrossRef CAS PubMed; (f) K. Xu, Y.-H. Wang, V. Khakyzadeh and B. Breit, Chem. Sci., 2016, 7, 3313 RSC; (g) X.-H. Yang and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1774 CrossRef CAS PubMed; (h) X.-H. Yang, A. Lu and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 14049 CrossRef CAS PubMed; (i) C. P. Grugel and B. Breit, Org. Lett., 2018, 20, 1066 CrossRef CAS PubMed; (j) S.-Z. Nie, R. T. Davison and V. M. Dong, J. Am. Chem. Soc., 2018, 140, 16450 CrossRef CAS PubMed.
- W. N. Olmstead and F. G. Bordwell, J. Org. Chem., 1980, 45, 3299 CrossRef CAS.
- E. M. Arnett, S. G. Maroldo, S. L. Schilling and J. A. Harrelson, J. Am. Chem. Soc., 1984, 106, 6759 CrossRef CAS.
- S. Förster, O. Tverskoy and G. Helmchen, Synlett, 2008, 2803 Search PubMed.
- B. H. Oh, I. Nakamura and Y. Yamamoto, Angew. Chem., Int. Ed., 2016, 55, 9060 CrossRef PubMed.
- A. M. Johns, M. Utsunomiya, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 1828–1839 CrossRef CAS PubMed.
- F. G. Bordwell and H. E. Fried, J. Org. Chem., 1981, 46, 4327 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00633h |
| ‡ Reactions of alkynes proceed through the identical metal–π-allyl intermediate as those of allenes. |
| This journal is © The Royal Society of Chemistry 2019 |