
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of α-heterosubstituted ketones through sulfur mediated difunctionalization of internal alkynes†
Zhong
Zhang
 ,
Yuzheng
Luo
,
Hongguang
Du
*,
Jiaxi
Xu
* and
Pingfan
Li
*
,
Yuzheng
Luo
,
Hongguang
Du
*,
Jiaxi
Xu
* and
Pingfan
Li
*
State Key Laboratory of Chemical Resource Engineering, Department of Organic Chemistry, Faculty of Science, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: dhg@mail.buct.edu.cn; jxxu@mail.buct.edu.cn; lipf@mail.buct.edu.cn
First published on 16th April 2019
Abstract
Synthesis of α-heterosubstituted ketones was achieved through sulfur mediated difunctionalization of internal alkynes in one pot. The reaction design involves: phenyl substituted internal alkyne attacking triflic anhydride activated diphenyl sulfoxide to give a sulfonium vinyl triflate intermediate, hydrolysis to give an α-sulfonium ketone, and then substitution with various nucleophiles. This method provides a unified route to access α-amino ketones, α-acyloxy ketones, α-thio ketones, α-halo ketones, α-hydroxy ketones, and related heterocyclic structures, in a rapid fashion.
Introduction
Ketones bearing α-heteroatom substituents constitute a class of compounds with significant synthetic interest.1 Besides being important building blocks for more complex target structures, many drugs and biologically active molecules contain α-amino or α-hydroxy ketone moieties. For example, they are key substructures of bupropion2 and brephedron,3 which are used in the clinical treatment of psychological disorders.The most widely used method for synthesizing these compounds include: electrophilic α-halogenation/α-oxidation/α-amination of ketones or corresponding enolates,4 as well as nucleophilic substitution of α-bromo ketones with oxygen, chalcogen, or nitrogen nucleophiles.5 Oxidative difunctionalization of alkenes is another attractive route to give α-heterosubstituted ketones.6 Recently, difunctionalization of alkynes also emerged as an alternative route to access such structural motifs.7 Murakami's group8 developed a copper/rhodium catalyzed two-step reaction sequence for converting terminal alkynes to α-amino ketones via triazole intermediates (Scheme 1a). Liang's group9 reported the synthesis of α-succinimide/phthalimide substituted ketones though bromohydration of alkynes and subsequent substitution (Scheme 1b). Hou et al. reported the conversion of terminal alkynes to α-acetoxy ketones by using PhI(OAc)2 as an oxidant (Scheme 1c).10 Zhang et al. developed a gold catalyzed reaction for converting terminal alkynes to α-acyloxy ketones (Scheme 1d).11
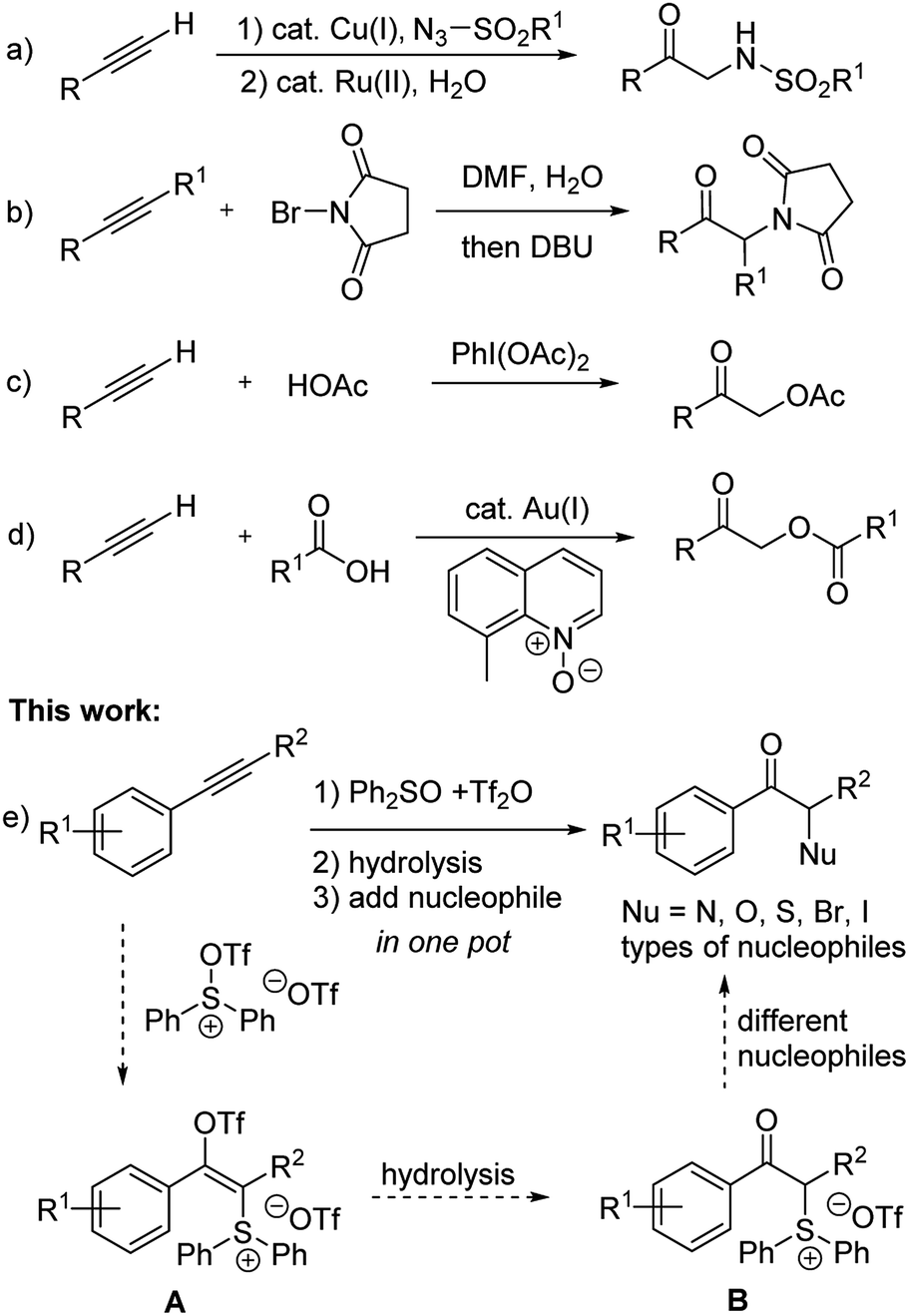 | ||
| Scheme 1 Synthesis of α-heterosubstituted ketones through difunctionalization of alkynes. | ||
On the other hand, to the best of our knowledge, a general one-pot method for converting alkynes to different types of α-heterosubstituted ketone products has not been reported.12 It should be noted that the nitroso aldol reaction of ketones or aldehydes did provide a way to introduce either nitrogen or oxygen atom at the α-position of carbonyls under complementary organocatalyzed or acid catalyzed conditions.4a,13 But extra steps were usually required to cleave the resulting N–O bond of such nitroso aldol products. MacMillan's group14 also developed an efficient CuBr2 catalyzed coupling of α-carbonyls with dialkyl amines. However, extension of such a catalytic system to other nucleophiles has not been reported. Recently, Hartwig's group reported a general solution for enantioselective α-functionalization of ketones via an iridium catalyzed reaction of allylic carbonates containing silyl enol ethers as masked ketones.15 Herein, we describe a one-pot, transition-metal-free method for the synthesis of α-heterosubstituted ketones through sulfur mediated difunctionalization of internal alkynes (Scheme 1e).
In connection with our recent interest in sulfur mediated C–H functionalization of alkenes16 and alkynes,17 we are interested in the reactivity of sulfonium vinyl triflate intermediate A generated through alkyne attack of triflic anhydride activated diphenyl sulfoxide (see Scheme 1e).17a,18 We reasoned that if hydrolysis of A could give α-sulfonium ketone B, subsequent substitution with various nucleophiles in one pot would then afford different types of α-heterosubstituted ketones in a rapid and unified fashion.
Results and discussion
We chose 1-phenyl-1-butyne (1a) and aniline (2a) as our model substrates to find the optimal reaction conditions for the synthesis of α-amino ketone 3aa (Table 1). For the first step, i.e. alkyne (1a) attack of triflic anhydride activated diphenyl sulfoxide, similar conditions were adopted to our previous studies on a sulfur mediated propargylic C–H alkylation reaction of alkynes.17a We soon realized that the key to success of this one-pot reaction design was the optimal aqueous hydrolysis and buffer conditions for the conversion of sulfonium vinyl triflate intermediate A to α-sulfonium ketone B. Hydrolysis of A seems to proceed best with basic aqueous sodium hydroxide, presumably through an addition/elimination pathway. However, with an excessive amount of sodium hydroxide, α-sulfonium ketone B would be converted to the corresponding carbonyl stabilized sulfur ylide C, which could not be used as the electrophile in our subsequent nucleophilic substitution reaction with aniline (2a). And thus, we have to acidify the reaction mixture appropriately to suppress the deprotonation of B, but not to acidify it too much as to protonate our nucleophilic amine base 2a. Eventually, we found that the use of 3 equivalents of sodium hydroxide and 20 equivalents of water, followed by addition of 2 equivalents of sodium dihydrogen phosphate and 1.5 eq. (2a), gave 72% yield of product 3aa (Table 1, entry 1). The results of deviation from the optimized reaction conditions are shown in Table 1 (entries 2–7).19 First, the amounts of sodium hydroxide and sodium dihydrogen phosphate were varied (entries 2–4). Then, tetramethylammonium hydroxide pentahydrate was used as the base (entry 5), and triflic acid (TfOH) or p-toluenesulfonic acid (PTSA) was used as the acid in step 2 (entries 6–7). None of these deviations afforded better yields than our standard conditions.|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield/%b |
| a Standard conditions A: a solution of 1a (0.4 mmol) and Ph2SO (0.48 mmol) in 2 mL of CH2Cl2 was treated with Tf2O (0.48 mmol) at −78 °C, warmed up to 0 °C, NaOH (1.2 mmol) and H2O (8 mmol) were added, and stirred at 40 °C for 12 h. Then, NaH2PO4 (0.8 mmol) and 2a (0.6 mmol) were added to the reaction mixture and stirred at 40 °C for 12 h before work-up and purification. b Isolated yield after column chromatography. | ||
| 1 | None | 72 |
| 2 | Using 5 eq. of NaOH, 4 eq. of NaH2PO4 in step 2 | 50 |
| 3 | Using 1.5 eq. of NaH2PO4 in step 2 | 62 |
| 4 | Using 2.5 eq. of NaH2PO4 in step 2 | 71 |
| 5 | Using 3 eq. of Me4NOH·5H2O instead of NaOH and H2O in step 2 | 49 |
| 6 | Using TfOH instead of NaH2PO4 in step 2 | 63 |
| 7 | Using PTSA instead of NaH2PO4 in step 2 | 63 |
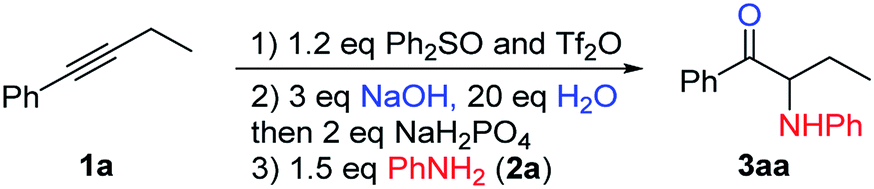
With the optimized reaction conditions in hand, the internal alkyne substrate scope was examined (Table 2). For phenyl substituted alkynes 1b, 1f, 1g, and 1h, corresponding α-amino ketone products 3ba, 3fa, 3ga, and 3ha were obtained in good yields. Olefin containing product 3da was obtained in 52% yield, showcasing that the reactivity of phenyl substituted internal alkynes toward triflic anhydride activated sulfoxide reagents was higher than that of the terminal alkene moiety. Phenyl alkyne substrates bearing alkyl or halide (–F, –Cl, and –Br) substituents all afforded products (3ca, 3la, 3ja, 3ka, 3na, 3oa, 3pa and 3qa) in good yields. When an electron-withdrawing acyl group was attached to the phenyl alkyne, a lower yield for product 3ma was observed, and we recovered a significant amount of alkyne substrate 1m. The yield of product 3ra decreased to 21% when using alkyne 1r bearing a strongly electron-withdrawing cyano group. For methoxy substituted phenyl alkynes 1e and 1i, their reactions with triflic anhydride activated sulfoxide reagents could proceed completely, but it seems that the hydrolysis step was slower, which led to decreased yields for products 3ea and 3ia. For the same reason, substrate 1s gave the product 3sa in a lower yield of 52%. Using terminal alkynes as substrates could not afford this type of product, but would give alkynyl diphenyl sulfonium salts instead.18h When dialkyl or alkyl silyl substituted alkynes were used, propargylic C–H arylation type products were obtained as previously reported by Procter et al. under similar reaction conditions.18j
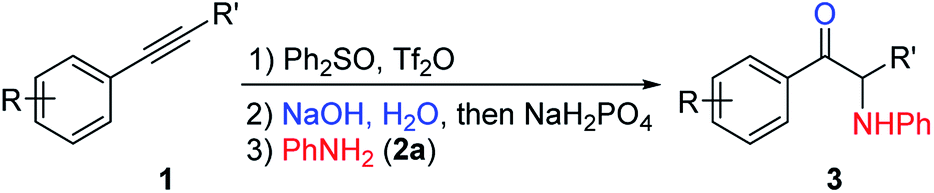
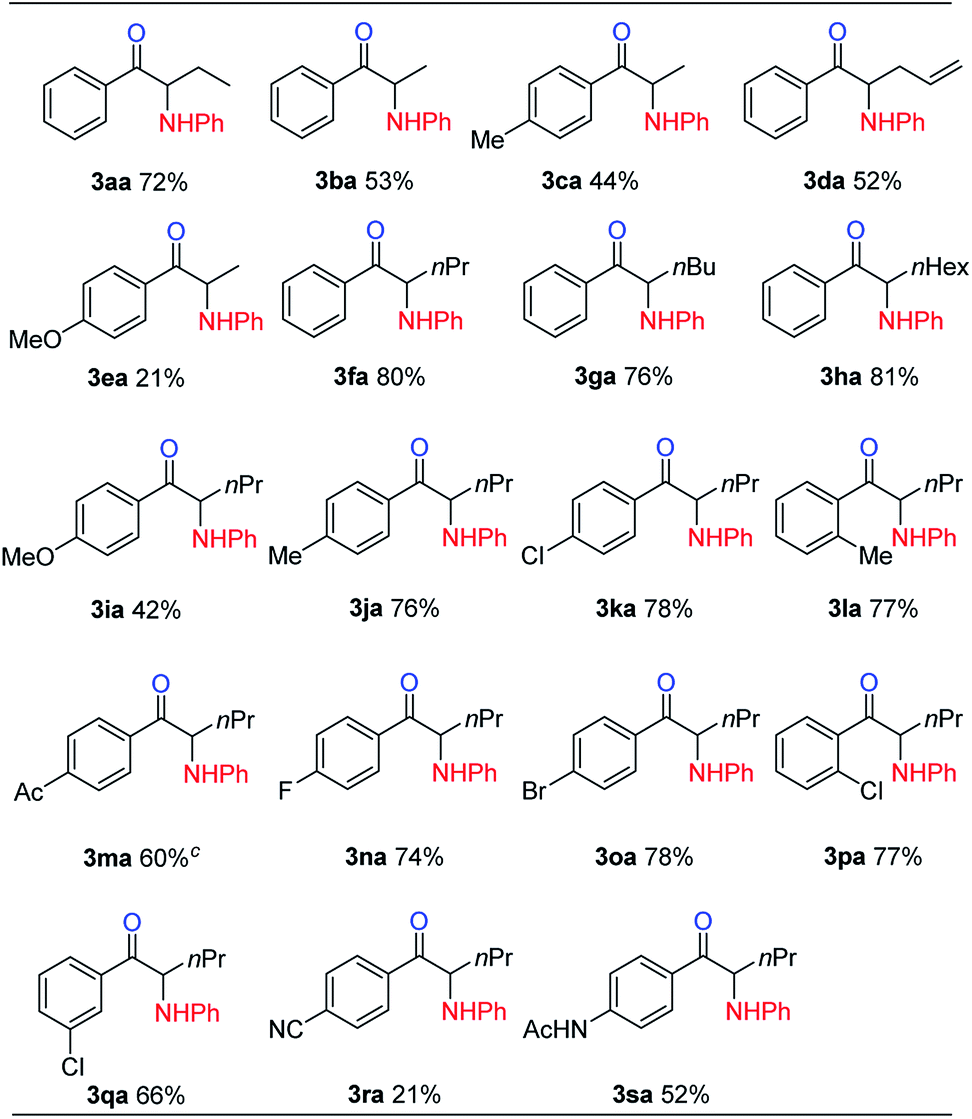
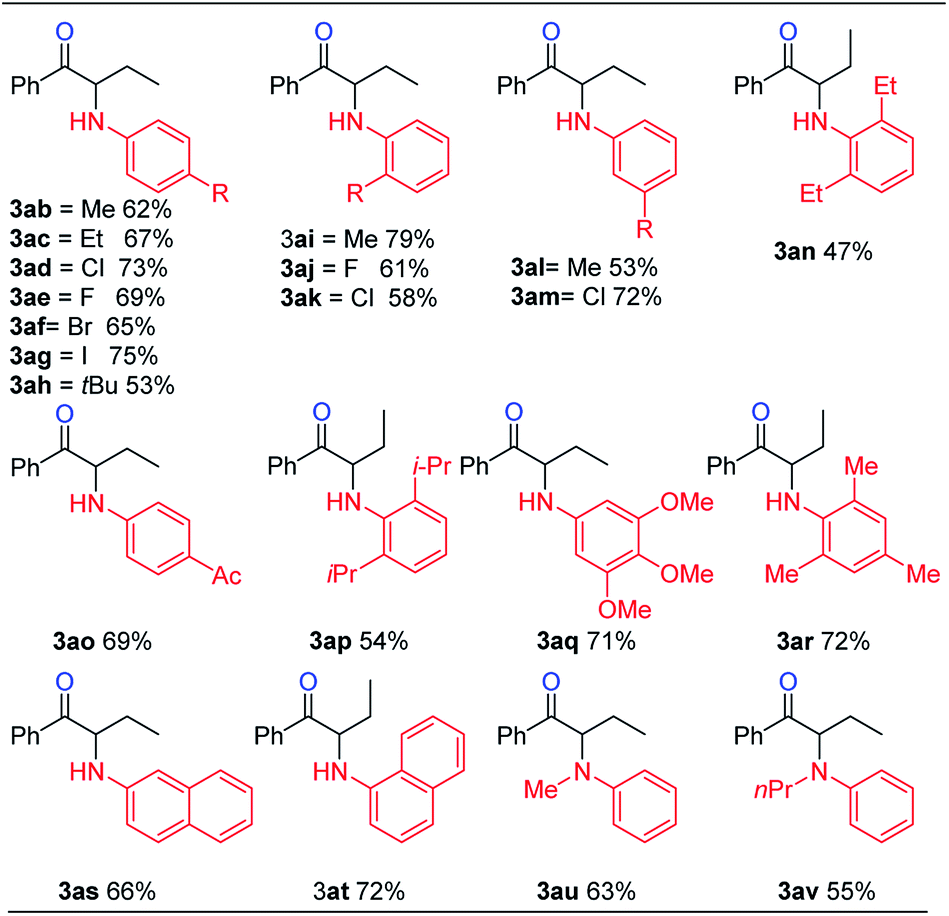
We then examined the electronic and steric effects of various arylamine nucleophiles on the reaction with alkyne 1a (Table 3). To our delight, amines having both electron-donating and electron-withdrawing groups, as well as having increased steric hindrance at the ortho positions, all reacted smoothly to afford the desired products (from 3ab to 3av) in good to moderate yields. Even with sterically very hindered 2,6-diisopropylphenylamine 2p, the desired product 3ap was still obtained in 54% yield. Secondary amines 2u and 2v could also afford the corresponding α-amino ketones 3au and 3av in 63% and 55% yields, respectively.
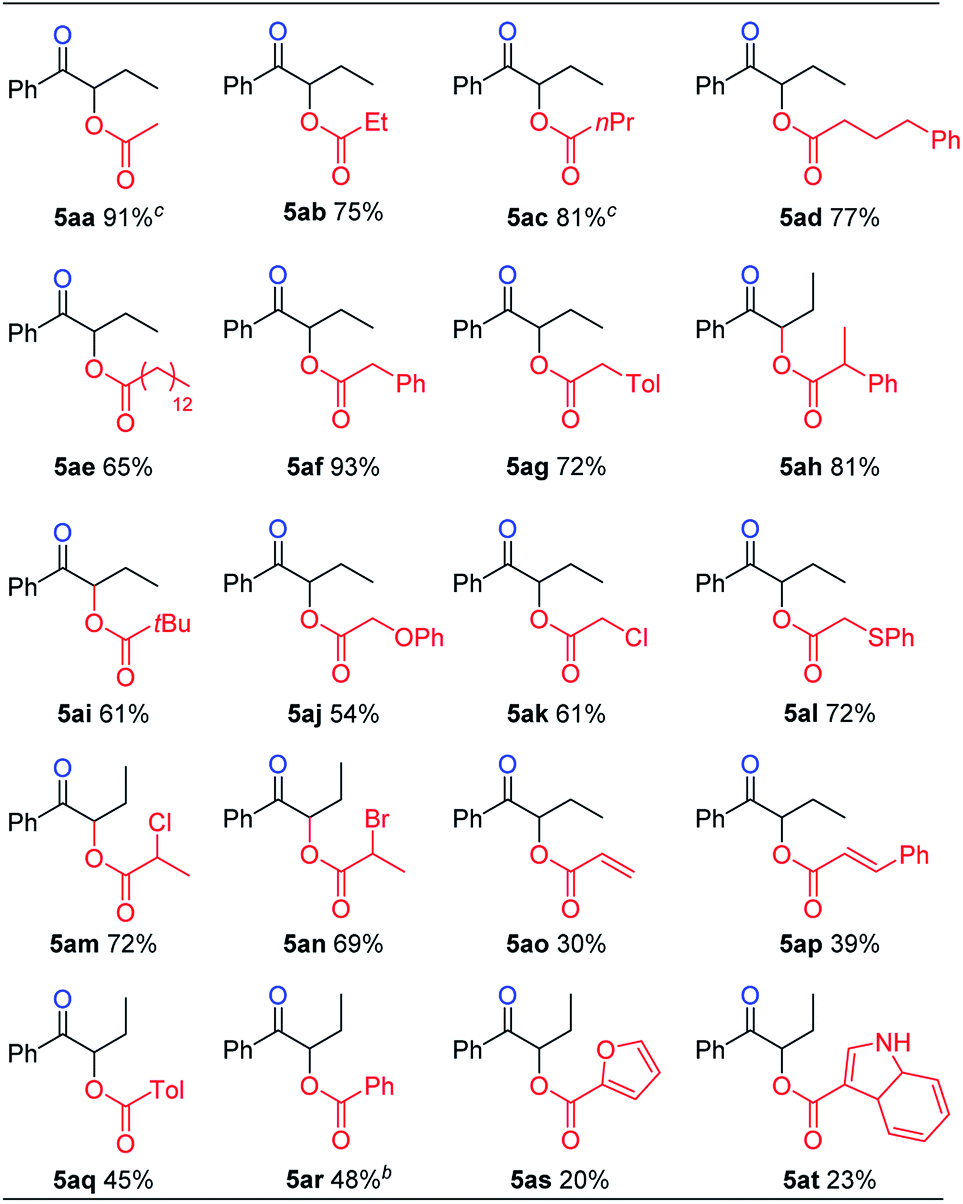
We then turned our attention to the preparation of α-acyloxy ketones. It is reasoned that carboxylic acid substrates could act not only as nucleophiles, but also as acidic additives to replace the use of sodium dihydrogen phosphate to protonate the initially formed sulfur ylide during the hydrolysis step, and give the desired α-sulfonium ketone intermediate. Under further optimized reaction conditions, various carboxylic acids reacted nicely to give α-acyloxy ketone products in good yields (Table 4). In general, saturated carboxylic acids gave higher yields (5aa–5an), while alkenyl, aryl and hetero-aryl substituted carboxylic acids gave lower yields (5ao–5at).
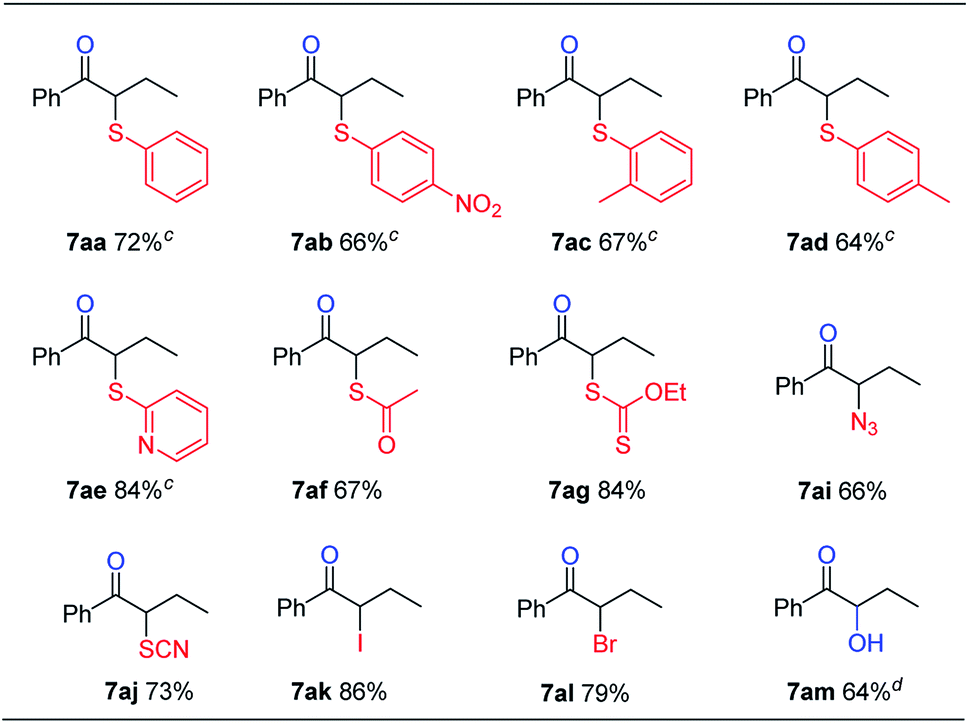
This reaction could also be applied with other nucleophiles (Table 5). A number of thiophenols gave good yields (7aa–7ad). Other sulfur, nitrogen, or halogen based nucleophiles, such as 2-mercaptopyridine (6e), potassium thioacetate (6f), potassium O-ethyl carbonodithioate (6g), sodium azide (6i), potassium thiocyanate (6j), potassium iodide (6k) and potassium bromide (6l), all gave the desired products (7ae–7al) in good yields. Besides, α-hydroxyl ketone 7am could also be obtained in 64% yield in the same fashion by using tetrabutylammonium hydroxide (6m) as both the base and nucleophile. Compared with the previous results obtained with sodium hydroxide or tetramethylammonium hydroxide (Table 1, entry 5), the better phase-transfer nature of 6m seems to be the key factor for the successful formation of 7am.
| a Standard conditions A: (1) 1a (0.4 mmol), Ph2SO (0.48 mmol), and Tf2O (0.48 mmol); (2) NaOH (1.2 mmol), H2O (8 mmol), and NaH2PO4 (0.8 mmol); (3) 6 (0.6 mmol). b Isolated yield. c The reaction was performed without adding NaH2PO4 in step 2. d Skipped step 2 and added aqueous nBu4NOH solution as the nucleophile in step 3. |
|---|
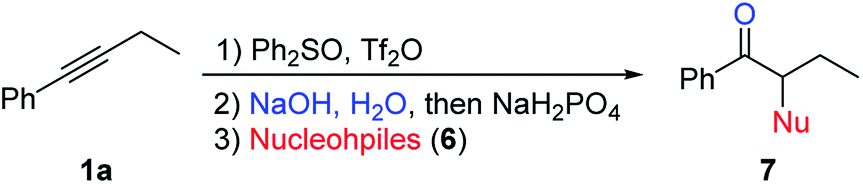
|
|
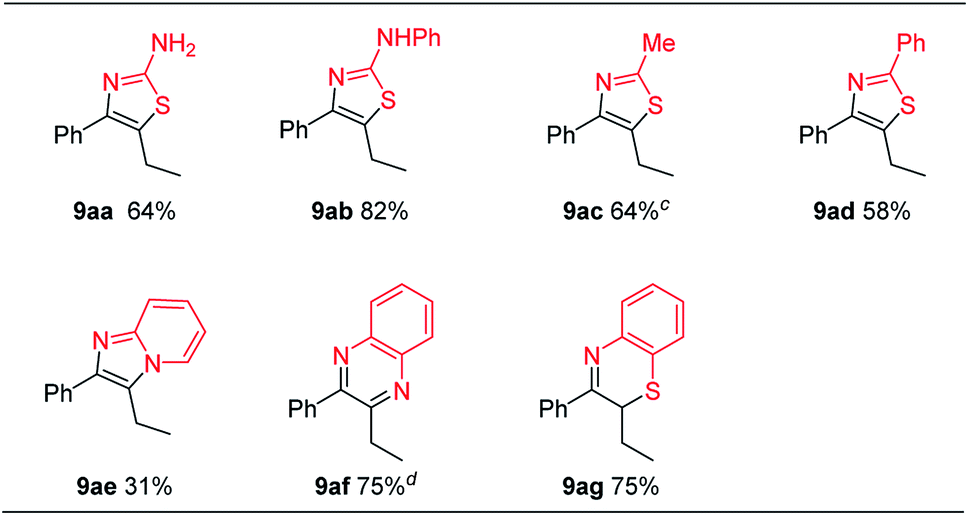
By using appropriate ambident nucleophiles, a number of interesting heterocyclic structures can be prepared, since the initially formed α-hetero substituted ketones could undergo intramolecular condensation reactions (Table 6).20 When thiourea 8a and phenylthiourea 8b were used as nucleophiles, 2-aminothiazoles 9aa and 9ab were afforded in 64% and 82% yields, respectively. Similarly, when thioamides 8c and 8d were used, thiazole products 9ac and 9ad were obtained in 64% and 58% yields, respectively. 2-Aminopyridine 8e gave 3-ethyl-2-phenylimidazo[1,2-a]pyridine 9ae in 31% yield. When ortho-diaminobenzene 8f was used as the nucleophile, the initially formed product was easily oxidized and hard to purify. So we added 3 equivalents of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in one pot after step 3 to give quinoxaline 9af in 75% yield. Similarly, 2-aminothiophenol 8g gave product 9ag in 75% yield.
Our proposed reaction pathway is supported by the isolation and reactivity study of potential reaction intermediates (Scheme 2). When alkyne 1a was treated with triflic anhydride activated diphenyl sulfoxide, we could isolate the sulfonium vinyl triflate intermediate A in 93% yield, which could afford the product 5aa after reacting with potassium acetate and water (Scheme 2a). The attempted hydrolysis of intermediate A with water alone did not occur in this case before the addition of basic potassium acetate. While hydrolysis of intermediate A did occur with sodium hydroxide and water, sulfonium salt B and sulfur ylide C were isolated in 42% and 46% yields, respectively (Scheme 2b). With the weaker base cesium carbonate instead of the stronger base sodium hydroxide, α-sulfonium ketone intermediate B was obtained in 41% yield in addition to the 55% yield of sulfonium vinyl triflate A (Scheme 2c).21
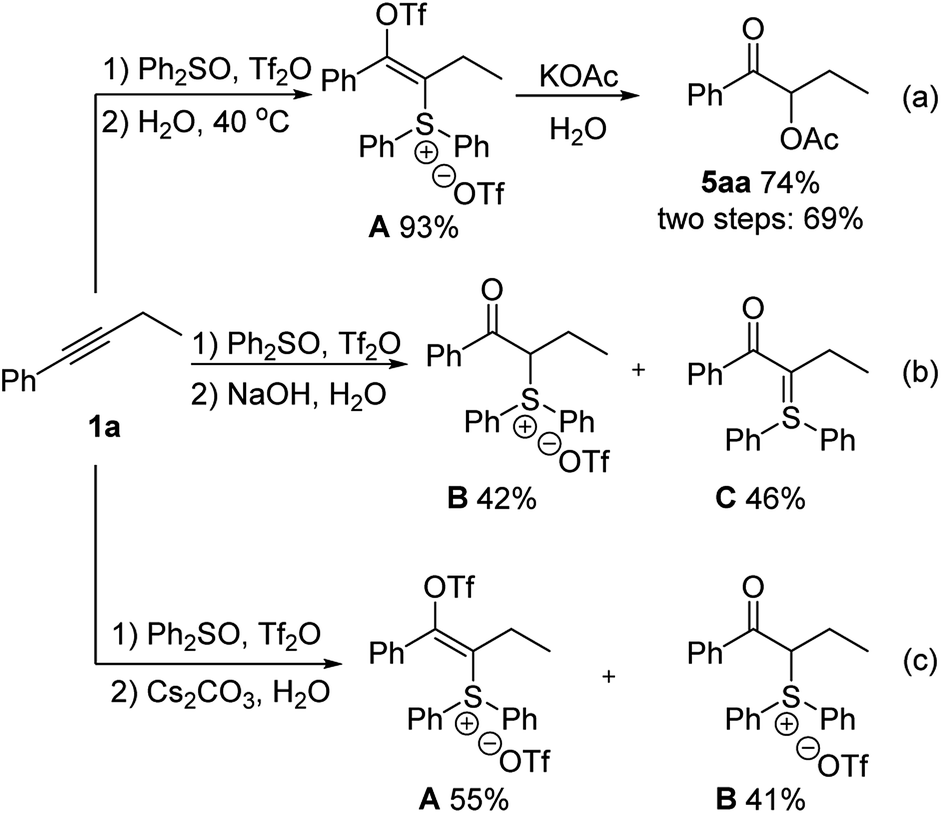 | ||
| Scheme 2 Isolation of reaction intermediates. | ||
To further explore the application of this reaction in the context of natural product derivatization, alkyne 10 was prepared in two steps from estrone (Scheme 3). Our difunctionalization reactions proceeded smoothly, and desired products 11, 12 and 13 were afforded in 55%, 69% and 65% yields (ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two epimers with the newly generated stereocenters), respectively.
:1 mixture of two epimers with the newly generated stereocenters), respectively.
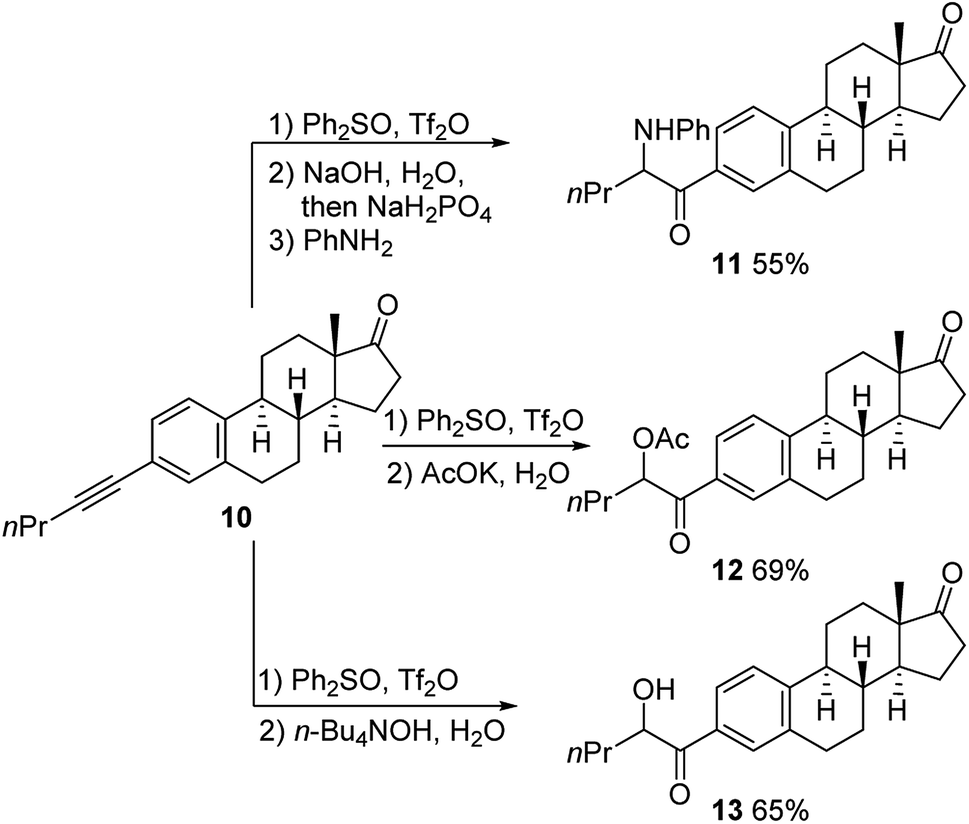 | ||
| Scheme 3 Functionalization of estrone derivative 10. | ||
Our preliminary results of a catalytic asymmetric version of this reaction are shown in Scheme 4. When a chiral PCCP catalyst22 was added in step 3 and the reaction temperature lowered to −20 °C, α-amino ketone 3ba was obtained in 67% yield and 50% ee, which indicates the feasibility to develop a chiral Brønsted acid catalyzed reaction to prepare enantioenriched α-amino ketones directly from alkynes.
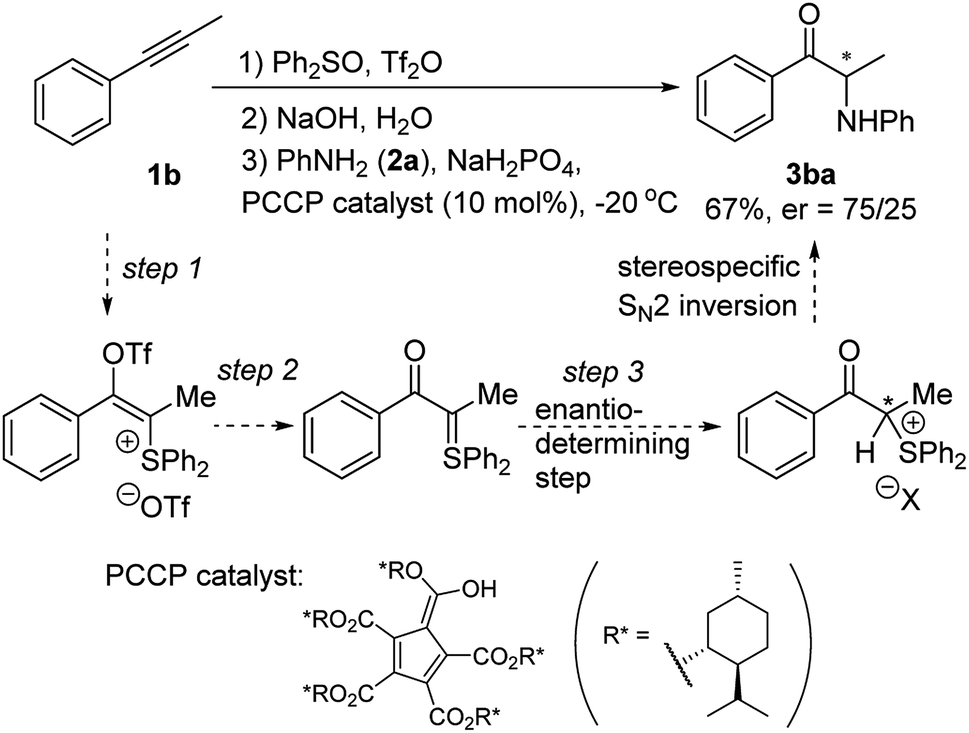 | ||
| Scheme 4 Preliminary results of a catalytic asymmetric version. | ||
Conclusions
In conclusion, we have developed a general procedure for the synthesis of α-heterosubstituted ketones through sulfur mediated difunctionalization of internal alkynes. A variety of α-substituted ketones could be prepared by using nitrogen, oxygen, sulfur and halogen nucleophiles. Applications for the synthesis of heterocycles and derivatization of natural products are also realized. We are currently exploring the catalytic asymmetric version of this reaction, and those results will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported in part by the National Natural Science Foundation of China (21402005, 21572017) and the Fundamental Research Funds for the Central Universities (XK-1802-6, 12060093063).Notes and references
- Product Class 6: α-Heterosubstituted Ketones, in Science of Synthesis, ed. J. Cossy, Georg Thieme Verlag, Stuttgart, 2005, vol. 26 Search PubMed.
- D. M. Perrine, J. T. Ross, S. J. Nervi and R. H. Zimmerman, J. Chem. Educ., 2000, 77, 1479–1480 CrossRef CAS.
- K. F. Foley and N. V. Cozzi, Drug Dev. Res., 2003, 60, 252–260 CrossRef CAS.
- (a) B. Maji and H. Yamamoto, Bull. Chem. Soc. Jpn., 2015, 88, 753–762 CrossRef CAS; (b) E. Erdik, Tetrahedron, 2004, 60, 8747–8782 CrossRef CAS; (c) J. M. Janey, Angew. Chem., Int. Ed., 2005, 44, 4292–4300 CrossRef CAS PubMed; (d) A. M. R. Smith and K. K. Hii, Chem. Rev., 2011, 111, 1637–1656 CrossRef CAS PubMed; (e) D. Sandoval, C. P. Frazier, A. Bugarin and J. Read de Alaniz, J. Am. Chem. Soc., 2012, 134, 18948–18951 CrossRef CAS PubMed; (f) P. Selig, Angew. Chem., Int. Ed., 2013, 52, 7080–7082 CrossRef CAS PubMed; (g) F. Zhou, F.-M. Liao, J.-S. Yu and J. Zhou, Synthesis, 2014, 46, 2983–3003 CrossRef CAS.
- (a) L. E. Fisher and J. M. Muchowski, Org. Prep. Proced. Int., 1990, 22, 399–484 CrossRef CAS; (b) P. A. Levene and A. Walti, Org. Synth. Coll. Vol. II, 1943, 2, 5 Search PubMed.
- (a) R. N. Reddi, P. V. Malekar and A. Sudalai, Org. Biomol. Chem., 2013, 11, 6477–6482 RSC; (b) R. N. Reddi, P. K. Prasad and A. Sudalai, Org. Lett., 2014, 16, 5674–5677 CrossRef CAS PubMed; (c) K. Muñiz, C. H. Hövelmann, A. Villar, R. Vicente, J. Streuff and M. Nieger, J. Mol. Catal. A: Chem., 2006, 251, 277–285 CrossRef; (d) P. K. Prasad, R. N. Reddi and A. Sudalai, Org. Lett., 2016, 18, 500–503 CrossRef CAS PubMed; (e) M. H. Shinde and U. A. Kshirsagar, Org. Biomol. Chem., 2016, 14, 858–861 RSC; (f) M. Venkat Ram Reddy, R. Kumareswaran and Y. D. Vankar, Tetrahedron Lett., 1995, 36, 6751–6754 CrossRef; (g) A. Villar, C. H. Hövelmann, M. Nieger and K. Muñiz, Chem. Commun., 2005, 0, 3304–3306 RSC.
- For bormohydration of alkynes see: (a) K. K. Rajbongshi, D. Hazarika and P. Phukan, Tetrahedron, 2016, 72, 4151–4158 CrossRef CAS; (b) C. Wu, X. Xin, Z.-M. Fu, L.-Y. Xie, K.-J. Liu, Z. Wang, W. Li, Z.-H. Yuan and W.-M. He, Green Chem., 2017, 19, 1983–1989 RSC; (c) W. He, L. Xie, Y. Xu, J. Xiang and L. Zhang, Org. Biomol. Chem., 2012, 10, 3168–3171 RSC.
- T. Miura, T. Biyajima, T. Fujii and M. Murakami, J. Am. Chem. Soc., 2012, 134, 194–196 CrossRef CAS PubMed.
- M. Li, L. Zhang, B. Zhao and F. Liang, RSC Adv., 2016, 6, 93325–93329 RSC.
- (a) D.-L. Mo, L.-X. Dai and X.-L. Hou, Tetrahedron Lett., 2009, 50, 5578–5581 CrossRef CAS; (b) G. Deng and J. Luo, Tetrahedron, 2013, 69, 5937–5944 CrossRef CAS.
- K. Ji, Y. Zhao and L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 6508–6512 CrossRef CAS PubMed.
- For carbohydration of alkynes see: (a) D. Kaiser, L. F. Veiros and N. Maulide, Chem.–Eur. J., 2016, 22, 4727–4732 CrossRef CAS PubMed; (b) T. Stopka, M. Niggemann and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 13270–13274 CrossRef CAS PubMed.
- (a) N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2003, 125, 6038–6039 CrossRef CAS PubMed; (b) N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 5360–5361 CrossRef CAS PubMed.
- R. W. Evans, J. R. Zbieg, S. Zhu, W. Li and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 16074–16077 CrossRef CAS PubMed.
- Z. T. He and J. F. Hartwig, Nat. Chem., 2019, 11, 177–183 CrossRef CAS PubMed.
- (a) G. Hu, J. Xu and P. Li, Org. Lett., 2014, 16, 6036–6039 CrossRef CAS PubMed; (b) Z. Zhang, H. Du, J. Xu and P. Li, Chem. Commun., 2016, 52, 11547–11550 RSC.
- (a) G. Hu, J. Xu and P. Li, Org. Chem. Front., 2018, 5, 2167–2170 RSC; (b) Z. Zhang, P. He, H. Du, J. Xu and P. Li, J. Org. Chem., 2019, 84, 4517–4524 CrossRef CAS PubMed.
- (a) L. H. S. Smith, S. C. Coote, H. F. Sneddon and D. J. Procter, Angew. Chem., Int. Ed., 2010, 49, 5832–5844 CrossRef CAS PubMed; (b) A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 9842–9860 CrossRef CAS PubMed; (c) A. Shafir, Tetrahedron Lett., 2016, 57, 2673–2682 CrossRef CAS; (d) H. Yorimitsu, Chem. Rec., 2017, 17, 1156–1167 CrossRef CAS PubMed; (e) T. Yanagi, K. Nogi and H. Yorimitsu, Tetrahedron Lett., 2018, 59, 2951–2959 CrossRef CAS; (f) L. Meng, J. Zeng and Q. Wan, Synlett, 2018, 29, 148–156 CrossRef CAS; (g) Z.-Y. Tian, Y.-T. Hu, H.-B. Teng and C.-P. Zhang, Tetrahedron Lett., 2018, 59, 299–309 CrossRef CAS; (h) V. G. Nenajdenko, P. V. Vertelezkij and E. S. Balenkova, Synthesis, 1997, 1997, 351–355 CrossRef; (i) A. J. Eberhart and D. J. Procter, Angew. Chem., Int. Ed., 2013, 52, 4008–4011 CrossRef CAS PubMed; (j) J. A. Fernández-Salas, A. J. Eberhart and D. J. Procter, J. Am. Chem. Soc., 2016, 138, 790–793 CrossRef PubMed.
- Note: diphenyl sulfide was obtained as the byproduct.
- H. Kobayashi, J. A. Eickhoff and A. Zakarian, J. Org. Chem., 2015, 80, 9989–9999 CrossRef CAS PubMed.
- R. M. P. Dias and A. C. B. Burtoloso, Org. Lett., 2016, 18, 3034–3037 CrossRef CAS PubMed.
- (a) C. D. Gheewala, B. E. Collins and T. H. Lambert, Science, 2016, 351, 961–965 CrossRef CAS PubMed; (b) C. D. Gheewala, J. S. Hirschi, W.-H. Lee, D. W. Paley, M. J. Vetticatt and T. H. Lambert, J. Am. Chem. Soc., 2018, 140, 3523–3527 CrossRef CAS PubMed; (c) C. Yuan, J. Li and P. Li, ACS Omega, 2018, 3, 6820–6826 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00568d |
| This journal is © The Royal Society of Chemistry 2019 |