
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic carbanion generation – benzylation of aliphatic aldehydes to secondary alcohols†
Karsten
Donabauer
,
Mitasree
Maity
 ,
Anna Lucia
Berger
,
Gregory S.
Huff
,
Stefano
Crespi
and
Burkhard
König
*
,
Anna Lucia
Berger
,
Gregory S.
Huff
,
Stefano
Crespi
and
Burkhard
König
*
Institute of Organic Chemistry, Faculty of Chemistry and Pharmacy, University of Regensburg, Universitätsstraße 31, 93053 Regensburg, Germany. E-mail: burkhard.koenig@ur.de
First published on 16th April 2019
Abstract
We present a redox-neutral method for the photocatalytic generation of carbanions. Benzylic carboxylates are photooxidized by single electron transfer; immediate CO2 extrusion and reduction of the in situ formed radical yields a carbanion capable of reacting with aliphatic aldehydes as electrophiles giving the Grignard analogous reaction product.
Photocatalysis is a fast growing field in chemistry, enabling novel transformations previously unattainable under thermal conditions.1 The formation of carbon–carbon bonds is at the core of organic synthesis2 and many photocatalytic methods have been reported over the last two decades, the majority of which involve the generation of one or more radical species as key intermediates.3 In contrast, photocatalytic C–C bond formations involving anionic species as key intermediates are rare,4 although the reaction between a carbanion and a carbon electrophile, e.g. the Grignard reaction,5 is the most typical C–C bond forming reaction. Thus, photocatalytic methods to generate und utilize carbanion intermediates are desired in order to expand the limits of photocatalytic transformations from open shell to closed shell reactivity.
Commonly used photocatalysts, however, are only known to transfer a single electron to a substrate at once (SET), forming the corresponding radical and are not able to transfer two electrons in one step to generate the corresponding carbanion.3 Thus, the most intuitive way to photocatalytic carbanion generation is by two subsequent SETs, i.e. a consecutive two-fold reduction.6 There are a few literature examples illustrating the synthetic applicability of this strategy.4a,b These include the carbanion formation from 1,2-dibromomalonates giving cyclopropanes after addition to electron poor alkenes4a and the carbanion formation from tetraalkyl ammonium salts followed by their addition to aromatic aldehydes (Scheme 1a).4b The latter transformation reported by Yu et al. is especially interesting, as it is similar to the commonly used Grignard reaction. However, this method seems to be limited to aromatic aldehydes.
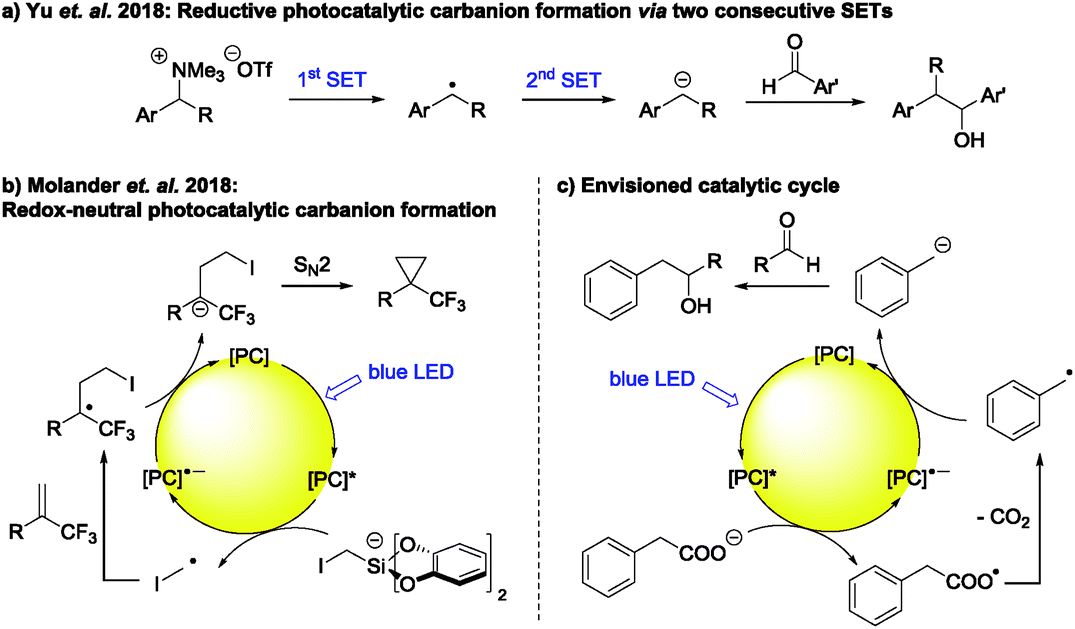 | ||
| Scheme 1 (a) Photocatalytic carbanion generation via two consecutive SETs. (b) Redox-neutral carbanion generation followed by intramolecular SN2 reaction. (c) Envisioned catalytic cycle. | ||
Beside the generation of a carbanion via a consecutive two-fold reduction, redox-neutral carbanion formations are proposed in several reports as well, typically by the reduction of a radical intermediate during the regeneration of the photocatalyst. However, this carbanion is in most cases simply protonated7 and examples where it is synthetically used are scarce.4c–i,8 These include the formation of a C–S bond with benzenesulfonothioates as electrophiles4e and intramolecular ring closures via an SN2 reaction (Scheme 1b).4f–i
To the best of our knowledge, there is so far no report for a redox-neutral photocatalytic carbanion generation followed by its intermolecular reaction with an aliphatic aldehyde or ketone as electrophile (Scheme 1c). As mentioned above, aldehydes and ketones are in this regard especially interesting electrophiles, as the corresponding transformation is analogous to the widespread Grignard reaction. Additionally, substituted carbonyl compounds are poor radical traps for an intermolecular radical addition and forming the desired product using established photocatalytic protocols for radical addition to double bonds is generally not successful.9 An exception to this is the photocatalytic radical addition enabled by in situ Brønsted acid activation yielding 3-alkoxy alcohols as reaction products reported by Glorius et al.10 Mainly aromatic carbonyl compounds could be used as radical traps.
Inspired by the above mentioned reports, we envisioned a photocatalytic cycle, in which a carbanion is formed from readily available carboxylic acids. Here, the in situ formed carboxylate is oxidized to the corresponding radical. This intermediate is prone to CO2 exclusion, forming the carbon centered radical, which may be converted to the corresponding carbanion by SET from the reduced photocatalyst. The desired product is then formed by addition of the carbanion to an aldehyde as electrophile (Scheme 1c). Herein we describe our efforts to realize this catalytic cycle.
With the envisioned photocatalytic cycle in mind, the coupling between phenylacetic acid (1a) and n-pentanal (2a) was chosen as model reaction. Compound 1a was selected as carbanion precursor, since the corresponding benzylic radical as well as the desired carbanion intermediate are stabilized by the aromatic moiety. As the photocatalyst is supposed to engage in a single electron oxidation as well as reduction, employing a catalyst with both a strong oxidation as well as reduction power is crucial. 4CzIPN is an in this regard very attractive organic photocatalyst, exhibiting an excited state redox potential of E1/2(P*/P˙−) = +1.35 V vs. SCE and a ground state reduction potential of Ered1/2(P/P˙−) = −1.21 V vs. SCE in MeCN.11 With 4CzIPN as catalyst and Cs2CO3 as base, the reaction proceeded smoothly yielding the desired addition product 3a in 73% GC-yield after 16 h (Table 1). Toluene (4a) was observed as a second product in 15% yield resulting from the reaction with protons as electrophile.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Additive (eq.) | Yield 3ab [%] | Yield 4ab [%] |
| a Reactions were performed with 1a (150 μmol, 1 eq.), 2a (3 eq.) and Cs2CO3 (1 eq.) in degassed solvent (2 mL) under a nitrogen atmosphere. b GC-yield determined with n-decane as internal standard. c Isolated yield in parentheses. d Reaction performed in absence of 4CzIPN. e Reaction performed in the dark. f Reaction preformed without base. g The preformed NBu4+ carboxylate salt (NBu4PA, 5) was used instead of 1a in absence of Cs2CO3. | ||||
| 1 | Dry DMF | — | 73 | 15 |
| 2 | Dry DMA | — | 75 | 11 |
| 3 | DMA | — | 75 (63) | 12 |
| 4 | DMA | H2O (3 eq.) | 34 | 55 |
| 5d | DMA | — | Not detected (n.d.) | n.d. |
| 6e | DMA | — | n.d. | n.d. |
| 7f | DMA | — | n.d. | n.d. |
| 8g | DMA | — | 64 (48)c | 12 |
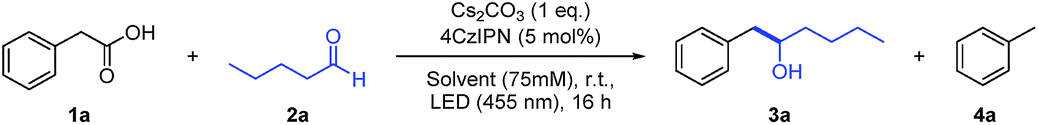
To increase the reaction yield, several parameters were optimized (Table 1, for the full optimization process see ESI†). A slight improvement could be realized by using DMA as solvent. Interestingly, prior drying of the solvent over 4 Å molecular sieve did not improve the yield significantly (entries 2 and 3), whereas the addition of H2O gave more toluene (4a) (entry 4). Control experiments confirmed that photocatalyst, light and base (entries 5–7) are necessary for the reaction conversion. However, the reaction can be performed in absence of base when using the preformed NBu4+ carboxylate salt (5) (entry 8), suggesting that the base merely serves to generate the carboxylate.
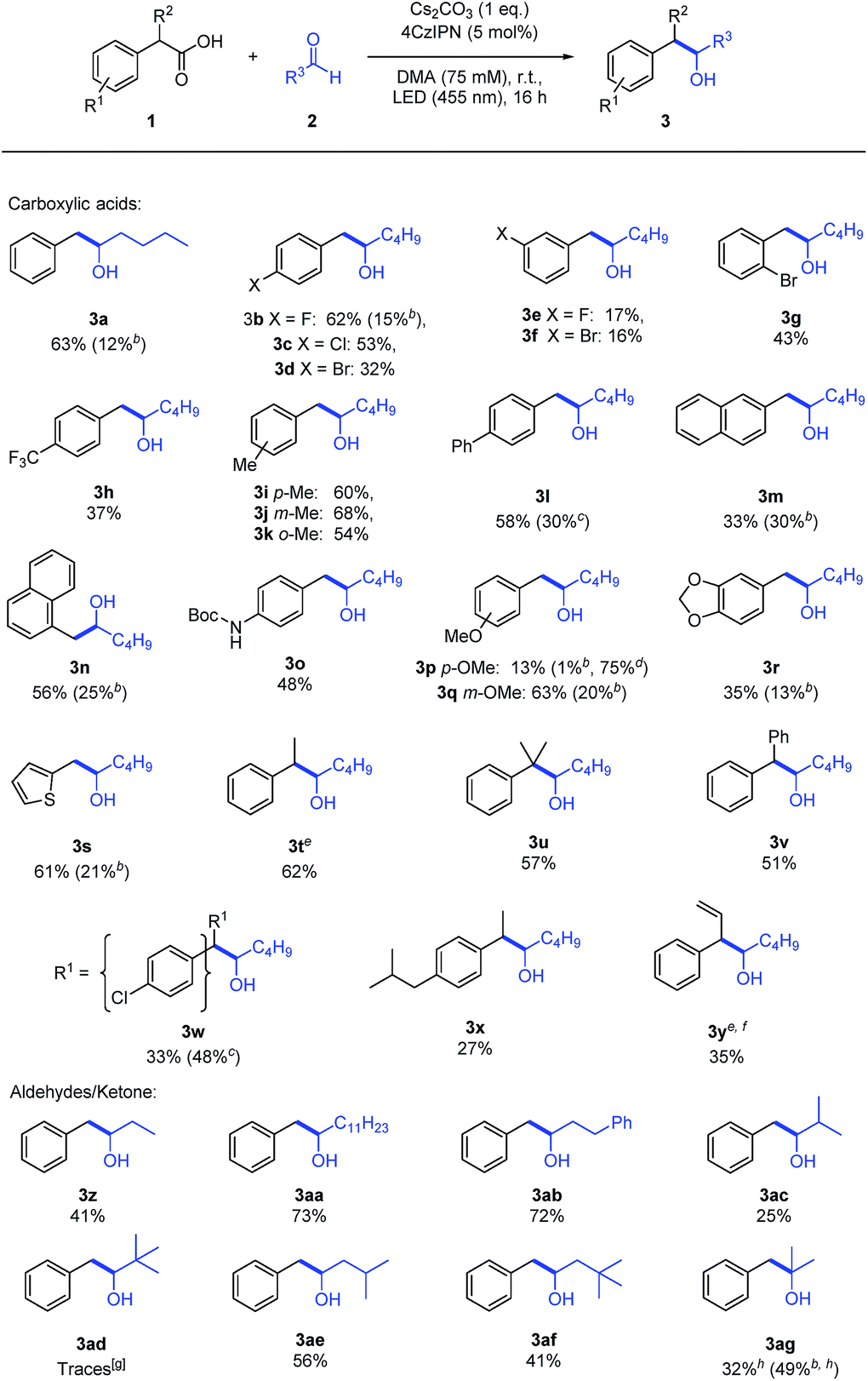
The reaction proceeds with various carboxylic acids (Table 2) as carbanion precursors in moderate to good yields. Halogen or –CF3 substituted phenyl acetic acid derivatives give the desired products (3b–h). The presence of an additional methyl or phenyl group on the aromatic ring (3i–l), extended aromatic systems (3m–n), a Boc-protected amine (3o) and methoxy (3p–r) functional groups or additional substituents on the benzylic carbon (3t–v) are tolerated.
a Reactions were performed with 1 (150 μmol, 1 eq.) and 2 (3 eq.) in degassed DMA (2 mL) under a nitrogen atmosphere. If not noted otherwise, the numbers indicate isolated yields.
b GC-yield of the corresponding decarboxylated side-product 4 determined by GC-FID analysis with n-decane as internal standard.
c Isolated yield of the corresponding decarboxylated side-product 4.
d Recovered starting material 1 after complete reaction time.
e Mixture of syn- and anti-product was obtained.
f
trans-Styrylacetic acid rather than α-vinylphenylacetic acid was used as starting material.
g Observed by GC-MS, not isolated.
h Acetone/DMA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was used as solvent. :1) was used as solvent.
|
|---|
|
The presence of a stabilizing aromatic moiety is necessary. Aliphatic and allylic carboxylic acids yield only traces of the desired product or none at all.
Aliphatic aldehydes bearing short or long chains (3z–ab) react well as electrophiles. The presence of an additional methyl group at the α-carbon decreased the yield to 25% (3ac) and only traces of the product could be observed when pivalaldehyde was employed (3ad). Substituents in the β-position (3ae) are tolerated and even adding a second methyl group showed only a minor effect (3af). Aromatic aldehydes, e.g. benzaldehyde (2i), gave the corresponding product as well, but a radical–radical cross coupling of the benzyl radical and the ketyl radical similar to our previous report12 instead of a carbanion generation cannot be excluded (Section 8, ESI†).
The formation of byproducts was investigated for selected examples (3a–b, 3l–n, 3p–s, 3w) and an almost complete mass balance could be obtained in most cases when combining the yield of the desired (3) and the decarboxylated (4) product (Table 2). If this was not the case, e.g. for 3p, an incomplete conversion of 1 was observed. Ketones, e.g. acetone (6), yield only small amounts of the tertiary alcohol (Table S9, ESI†). Using acetone (6) as co-solvent (1:1 mixture of DMA and acetone) increased the yield and 3ag could be isolated in 32% (Table 2).
Further, several electrophiles for an intermolecular SN2 reaction were tested (Table S11, ESI†), however an efficient system could not be found and only product traces were detected in some cases.
Starting the mechanistic investigation, a photo-conversion of 4CzIPN was observed during the course of the reaction (Fig. S1, ESI†). The degradation product could be isolated and identified as 4CzBnBN (7a) by X-ray crystal structure analysis (Scheme 2). Compound 7a is likely formed by radical addition of the benzyl radical to the radical anion of 4CzIPN, followed by cyanide elimination similar to reactions reported by different groups.13 4CzBnBN seems to be significantly more photo-stable and the product resulting from a second cyanide elimination could only be detected in traces. Performing the benzylation reaction with 4CzBnBN as catalyst gave the desired product as well (Scheme S4, ESI†), showing that 4CzBnBN is contributing and likely the main active catalyst for the carbanion formation. Ground state potentials of Eox1/2(P˙+/P) = +1.48 V vs. SCE and Ered1/2(P/P˙−) = −1.72 V vs. SCE in DMF were measured by CV.
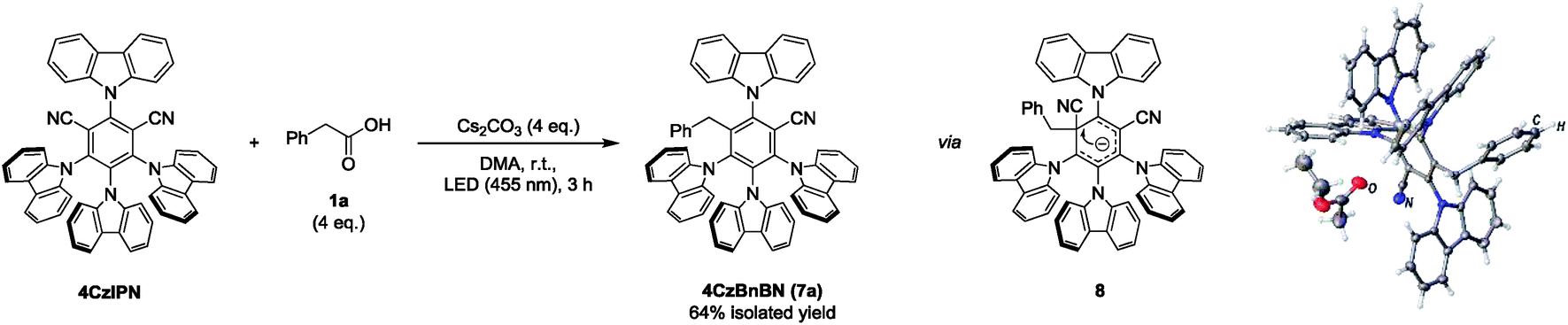 | ||
| Scheme 2 Isolation of 4CzIPN photo-conversion product 4CzBnBN (7a). The reaction was performed with 4CzIPN (30 μmol, 1 eq.) and 1a (4 eq.) in degassed DMA (2 mL) under a nitrogen atmosphere. | ||
The first step in our mechanistic hypothesis is the oxidation of the carboxylate followed by the extrusion of CO2 to generate a benzyl radical. There are several reports describing this process with various photoredox catalysts.14 Accordingly, the emission of 4CzBnBN could be quenched upon addition of NBu4PA (5) resulting in a linear Stern–Volmer plot (Fig. S7, ESI†), confirming the interaction between the excited photocatalyst and the substrate. The following CO2 elimination could be monitored by in situ IR spectroscopy (Fig. 1) together with the depletion of the aldehyde in course of the reaction.
 | ||
| Fig. 1 In situ FT-IR studies. Irradiation of a solution containing NBu4PA (5) (75 mM), 2a (75 mM) and 4CzBnBN (3.75 mM) in dry DMA lead to the formation of CO2 (2338 cm−1) and the depletion of 2a (1722 cm−1). | ||
Next, deuterium labeling experiments were conducted to support the formation of an anionic intermediate during the later course of the reaction (Scheme 3a). With D2O as electrophile, the corresponding deuterated decarboxylated starting material was isolated (4v-d). As a control experiment, the non-deuterated product (4v) was obtained when the reaction was performed in deuterated DMF in absence of D2O. Addition of D2O to a reaction mixture after completed irradiation did not yield any 4v-d from 4vvia base-induced (Cs2CO3) H/D-exchange (Scheme S5, ESI†).
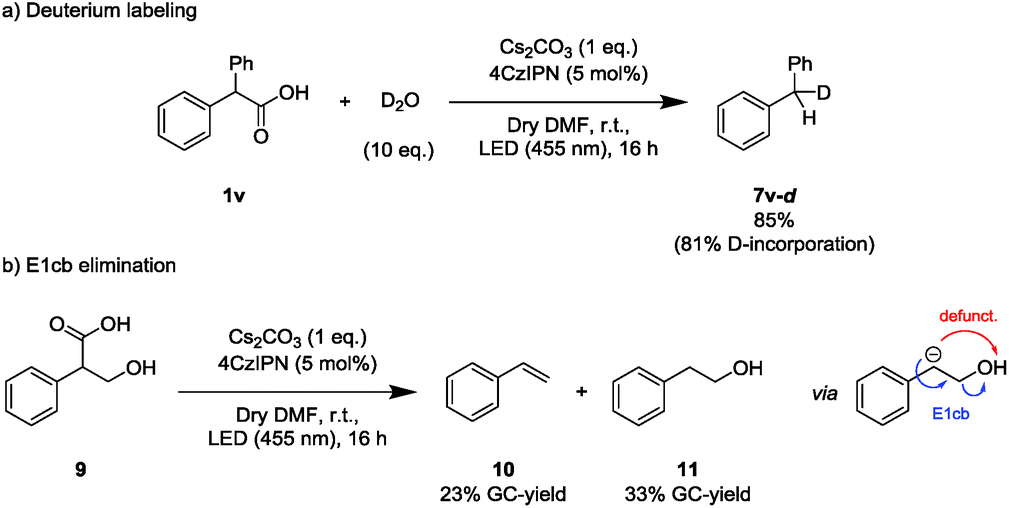 | ||
| Scheme 3 Experiments supporting the formation of a reactive anionic intermediate. | ||
In addition to the incorporation of deuterium, a carbanion intermediate is expected to engage in an E1cb elimination if an appropriate leaving group is present in the homobenzylic position. Hence, tropic acid (9) was subjected to the reaction conditions (Scheme 3b). Indeed, styrene (10) together with the decarboxylated byproduct (11) was detected. To exclude styrene formation from 11 by a simple E2 elimination induced by Cs2CO3, 11 was directly subjected to the applied reaction conditions, yielding no styrene (Scheme S6, ESI†).
In order to thoroughly study the feasibility and selectivity of the addition of the benzyl anion (15) to the electrophile, a computational analysis was performed. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O addition and the acid–base reaction of 15 and the Cα–H was proven at the SMD(DMF)-ωB97X-D/TZVP level of theory (see Section 7.9 in the ESI†). Indeed, the reaction of the benzyl anion with the aldehydes results to be exothermal (ΔG = −6.9 kcal mol−1) and slightly kinetically favored (ΔG* = +6.2 kcal mol−1) compared to the abstraction of the acidic proton in the α position (ΔG* = +6.8 kcal mol−1). Ketones were found to be less selective towards the CO addition of the benzyl anion compared to the aldehydes (see compound 3ag). As a parallelism, the reaction of the benzyl anion with the ketonic CO is almost thermoneutral (ΔG = +1.5 kcal mol−1). The competing acid–base reaction is exothermal (ΔG = −12.1 kcal mol−1), with comparable barriers (ΔG* ca. 10 kcal mol−1). The barriers for the reactions of 15 with DMA are higher compared to the ones of aldehydes and ketones.
O addition and the acid–base reaction of 15 and the Cα–H was proven at the SMD(DMF)-ωB97X-D/TZVP level of theory (see Section 7.9 in the ESI†). Indeed, the reaction of the benzyl anion with the aldehydes results to be exothermal (ΔG = −6.9 kcal mol−1) and slightly kinetically favored (ΔG* = +6.2 kcal mol−1) compared to the abstraction of the acidic proton in the α position (ΔG* = +6.8 kcal mol−1). Ketones were found to be less selective towards the CO addition of the benzyl anion compared to the aldehydes (see compound 3ag). As a parallelism, the reaction of the benzyl anion with the ketonic CO is almost thermoneutral (ΔG = +1.5 kcal mol−1). The competing acid–base reaction is exothermal (ΔG = −12.1 kcal mol−1), with comparable barriers (ΔG* ca. 10 kcal mol−1). The barriers for the reactions of 15 with DMA are higher compared to the ones of aldehydes and ketones.
Considering the experimental observations, computational results and cited literature reports, the following mechanism is proposed (Scheme 4): the carboxylic acid 1 is deprotonated by the base (Cs2CO3) to give carboxylate 12. The carboxylate (Eox1/2 (NBu4PA 5) = +1.27 vs. SCE14a) can be oxidized by the excited photocatalyst 4CzBnBN (Eox1/2(P*/P˙−) = +1.21 V vs. SCE) and the generated radical species 13 is transformed to the benzylic radical 14 by elimination of CO2. The radical anion of 4CzBnBN is with a reduction potential of Ered1/2(P/P˙−) = −1.72 V vs. SCE not able to reduce aliphatic aldehydes (Ered1/2(3-methylbutanal 2g) = −2.24 V vs. SCE15) and hence transfers an electron to the more easily reducible benzyl radical (Ered1/2 = −1.43 V vs. SCE16) to form carbanion 15. This species is capable of adding to aldehydes (2) forming the desired product 3 after protonation of the alcoholate.
 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In summary, a redox-neutral procedure to benzylate aliphatic aldehydes via the photocatalytic generation of a carbanion intermediate is presented, rendering the desired Grignard analogous products in moderate to good yields. The proposed mechanism is supported by emission quenching, in situ UV/VIS and in situ IR studies, while the presence of the reactive anionic intermediate is shown by deuterium labeling, E1cb elimination and DFT calculation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the German Science Foundation (DFG, KO 1537/18-1). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (grant agreement No. 741623). We thank Dr Rudolf Vasold for GC-MS measurements, Nele Berg for in situ NMR-measurements, the X-Ray department for crystal structure analysis and Regina Hoheisel for CV measurements.Notes and references
- M. H. Shaw, J. Twilton and D. W. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- G. Brahmachari, RSC Adv., 2016, 6, 64676–64725 RSC.
- (a) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (b) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (c) C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (e) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed; (f) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS PubMed.
- (a) Y. Zhang, R. Qian, X. Zheng, Y. Zeng, J. Sun, Y. Chen, A. Ding and H. Guo, Chem. Commun., 2015, 51, 54–57 RSC; (b) L. L. Liao, G. M. Cao, J. H. Ye, G. Q. Sun, W. J. Zhou, Y. Y. Gui, S. S. Yan, G. Shen and D. G. Yu, J. Am. Chem. Soc., 2018, 140, 17338–17342 CrossRef CAS PubMed; (c) Y. Kumagai, T. Naoe, K. Nishikawa, K. Osaka, T. Morita and Y. Yoshimi, Aust. J. Chem., 2015, 68, 1668 CrossRef CAS; (d) V. R. Yatham, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 10915–10919 CrossRef CAS PubMed; (e) W. Kong, H. An and Q. Song, Chem. Commun., 2017, 53, 8968–8971 RSC; (f) J. P. Phelan, S. B. Lang, J. S. Compton, C. B. Kelly, R. Dykstra, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2018, 140, 8037–8047 CrossRef CAS PubMed; (g) J. A. Milligan, J. P. Phelan, V. C. Polites, C. B. Kelly and G. A. Molander, Org. Lett., 2018, 20, 6840–6844 CrossRef CAS PubMed; (h) C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 15430–15434 CrossRef CAS PubMed; (i) C. Shu, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 3870–3874 CrossRef CAS PubMed.
- (a) V. Grignard, C. R. Acad. Sci., 1900, 130, 1322–1324 CAS; (b) G. S. Silverman and P. E. Rakita, Handbook of Grignard Reagents, Marcel Dekker, Inc., New York, 1996 CrossRef.
- (a) K. Hironaka, S. Fukuzumi and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1984, 1705–1709 RSC; (b) J.-M. Kern and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1987, 546–548 RSC; (c) D. Li, C.-M. Che, H.-L. Kwong and V. W.-W. Yam, J. Chem. Soc., Dalton Trans., 1992, 3325–3329 RSC.
- (a) H. Yokoi, T. Nakano, W. Fujita, K. Ishiguro and Y. Sawaki, J. Am. Chem. Soc., 1998, 120, 12453–12458 CrossRef CAS; (b) H. Huang, C. Yu, Y. Zhang, Y. Zhang, P. S. Mariano and W. Wang, J. Am. Chem. Soc., 2017, 139, 9799–9802 CrossRef CAS PubMed; (c) Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414–3420 CrossRef CAS; (d) A. J. Musacchio, L. Q. Nguyen, G. H. Beard and R. R. Knowles, J. Am. Chem. Soc., 2014, 136, 12217–12220 CrossRef CAS PubMed; (e) R. Zhou, H. Liu, H. Tao, X. Yu and J. Wu, Chem. Sci., 2017, 8, 4654–4659 RSC; (f) A. Gualandi, D. Mazzarella, A. Ortega-Martínez, L. Mengozzi, F. Calcinelli, E. Matteucci, F. Monti, N. Armaroli, L. Sambri and P. G. Cozzi, ACS Catal., 2017, 7, 5357–5362 CrossRef CAS; (g) X. Dong, P. Hu, W. Shen, Z. Li, R. Liu and X. Liu, Polymers, 2017, 9, 400 CrossRef PubMed; (h) A. Noble, R. S. Mega, D. Pflasterer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155–2159 CrossRef CAS PubMed.
- S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073–15077 CrossRef CAS PubMed.
- (a) A. Clerici, O. Porta and P. Zago, Tetrahedron, 1986, 42, 561–572 CrossRef CAS; (b) A. Clerici and O. Porta, J. Org. Chem., 1989, 54, 3872–3878 CrossRef CAS; (c) S. Wilsey, P. Dowd and K. N. Houk, J. Org. Chem., 1999, 64, 8801–8811 CrossRef CAS PubMed; (d) C. Che, Z. Qian, M. Wu, Y. Zhao and G. Zhu, J. Org. Chem., 2018, 83, 5665–5673 CrossRef CAS PubMed.
- L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, J. Am. Chem. Soc., 2017, 139, 13652–13655 CrossRef CAS PubMed.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef CAS.
- A. L. Berger, K. Donabauer and B. König, Chem. Sci., 2018, 9, 7230–7235 RSC.
- (a) T. Itou, Y. Yoshimi, T. Morita, Y. Tokunaga and M. Hatanaka, Tetrahedron, 2009, 65, 263–269 CrossRef CAS; (b) M. T. Pirnot, D. A. Rankic, D. B. Martin and D. W. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS PubMed.
- (a) L. Capaldo, L. Buzzetti, D. Merli, M. Fagnoni and D. Ravelli, J. Org. Chem., 2016, 81, 7102–7109 CrossRef CAS PubMed; (b) T. Patra and D. Maiti, Chem.–Eur. J., 2017, 23, 7382–7401 CrossRef CAS PubMed; (c) J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC; (d) H. Huang, X. Li, C. Yu, Y. Zhang, P. S. Mariano and W. Wang, Angew. Chem., Int. Ed., 2017, 56, 1500–1505 CrossRef CAS PubMed.
- D. Nicewicz, H. Roth and N. Romero, Synlett, 2015, 27, 714–723 CrossRef.
- (a) B. A. Sim, D. Griller and D. D. M. Wayner, J. Am. Chem. Soc., 1989, 111, 754–755 CrossRef CAS; (b) D. D. M. Wayner, D. J. McPhee and D. Griller, J. Am. Chem. Soc., 1988, 110, 132–137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1884950. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01356c |
| This journal is © The Royal Society of Chemistry 2019 |