
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing N-glycoprotein microheterogeneity by lectin affinity purification-mass spectrometry analysis†
Di
Wu
,
Jingwen
Li
,
Weston B.
Struwe
 and
Carol V.
Robinson
*
and
Carol V.
Robinson
*
Department of Chemistry, University of Oxford, South Parks Road, OX1 3QZ, Oxford, UK. E-mail: carol.robinson@chem.ox.ac.uk
First published on 16th April 2019
Abstract
Lectins are carbohydrate binding proteins that recognize specific epitopes present on target glycoproteins. Changes in lectin-reactive carbohydrate repertoires are related to many biological signaling pathways and recognized as hallmarks of several pathological processes. Consequently, lectins are valuable probes, commonly used for examining glycoprotein structural and functional microheterogeneity. However, the molecular interactions between a given lectin and its preferred glycoproteoforms are largely unknown due to the inherent complexity and limitations of methods used to investigate intact glycoproteins. Here, we apply a lectin-affinity purification procedure coupled with native mass spectrometry to characterize lectin-reactive glycoproteoforms at the intact protein level. We investigate the interactions between the highly fucosylated and highly branched glycoproteoforms of haptoglobin and α1-acid glycoprotein using two different lectins Aleuria aurantia lectin (AAL) and Phaseolus vulgaris leucoagglutinin (PHA-L), respectively. Firstly we show a co-occurrence of fucosylation and N-glycan branching on haptoglobin, particularly among highly fucosylated glycoproteoforms. Secondly, we analyze the global heterogeneity of highly branched glycoproteoforms of haptoglobin and α1-acid glycoprotein and reveal that while multi-fucosylation attenuates the lectin PHA-L binding to haptoglobin, it has no impact on AGP. Taken together, our lectin affinity purification native MS approach elucidates lectin specificities between intact glycoproteins, not achievable by other methods. Moreover, since aberrant glycosylation of Hp and AGP are potential markers for many diseases, including pancreatic, hepatic and ovarian cancers, understanding their interactions with lectins will help the development of carbohydrate-centric monitoring methods to understand their pathophysiological implications.
Introduction
Glycosylation is one of the most important protein post-translational modifications (PTMs) that controls protein conformation, localization and biological function.1,2 Aberrant protein N-glycosylation, namely abnormal levels of sialylation, fucosylation and glycan branching, are related to various cancers and diseases.1 Elucidating protein glycosylation is therefore critical for understanding regulatory features relevant to signaling pathways, disease onset and potential treatments. Glycoproteins are challenging biomolecules to characterize due to glycan microheterogeneity (monosaccharide composition and linkage information) as well as glycan macroheterogeneity (the presence or absence of glycans along the protein backbone).Lectins which are derived from microbes, plants and animals, are a large group of carbohydrate-binding proteins important in glycoprotein regulation, transport and signalling. Furthermore, lectins are extensively used to detect, characterize and quantify glycoprotein microheterogeneity in biochemical and clinical studies.3 The specificities of lectins are classically analysed by monosaccharides and haptens, such as polysaccharides and/or complex glycans, and then deduced at the glycoprotein level. Aleuria aurantia lectin (AAL) which specifically binds fucosylated carbohydrates4,5 and Phaseolus vulgaris leucoagglutinin (PHA-L) which recognizes β1-6 linked GlcNAc residues on branched N-glycan6,7 are used primarily to study glycoprotein fucosylation and N-glycan branching. Moreover the AAL and PHA-L reactive glycoproteins are vulnerable to metabolic stress and regulated by glycosyltransferases which are differentially expressed in various diseases.8 However, it is difficult to unpick binding mechanisms of lectins due to the presence of several glycan binding epitopes present at different sites on a given glycoprotein. Moreover steric restrains, arising from subtle changes in glycosylation, influence these interactions and cannot be resolved by classical biophysical methods such as isothermal titration calorimetry or surface plasmon resonance.
Mass spectrometry (MS) based glycoproteomics methods are used primarily to dissect glycoprotein micro- and macro-heterogeneity by providing compositional and/or structural information of enzymatically released glycans and glycopeptides.9 Recently, high-resolution native MS has advanced our ability to study intact glycoproteins, providing detailed information on the heterogeneity of intact complexes.10–13 Native MS while valuable in structural biology generally relies on complementary glycoproteomics methods to fully characterize glycan structures and to locate them within a given glycoprotein. Some combinations of monosaccharide residues cannot be easily distinguished by mass measurements of intact glycoproteins.14 For example, two fucose residues (Fuc2, 292.2829 Da) and one N-acetylneuraminic acid residue (Neu5Ac1, 291.2550 Da) differ by 1 Da, while the mass difference of Fuc5 (730.7072 Da) and two N-acetyllactosamine residues (LacNAc2, 730.6674 Da) is less than 0.1 Da. Glycosidase digestion to trim terminal sialic acid residues, by neuraminidase treatment, can simplify mass spectra and reduce ambiguous assignments.11,12 Nevertheless, relatively low digestion efficiencies of additional glycosidases limit their application in elucidating glycoprotein microheterogeneity.12,15
Here, we describe a coupled lectin affinity purification and high-resolution MS approach that is able to quantitatively characterize protein fucosylation, N-glycan branching and lectin specificities on intact glycoproteins. We use two human plasma glycoproteins: haptoglobin phenotype 1-1 (Hp) and alpha 1-acid glycoprotein (AGP). The tetrameric Hp is composed of two covalently linked α/β dimers that are heavily glycosylated at each β subunit (Asn180, Asn203, Asn207 and Asn237) with primarily bi- and tri-antennary N-glycans.16,17 AGP is monomeric and contains five highly branched complex type N-glycans at Asn15, Asn38, Asn54, Asn75, Asn85.18,19 Their inherent glycan modifications are extensively described at the glycomics and glycoproteomics levels.17,18,20
In this report, we first combine exoglycosidase digestion with affinity purification using the two lectins (AAL/PHA-L) to reduce the glycoprotein compositional heterogeneity and enrich highly fucosylated and highly branched glycoproteoforms of the two glycoproteins Hp and AGP. Secondly, by combining glycoproteomics and native MS, we define the microheterogeneity of highly fucosylated Hp and AGP and observe a co-occurrence of N-glycan branching and fucosylation on Hp, particularly among highly fucosylated glycoproteoforms. Lastly, we characterize the highly branched glycoproteoforms of Hp and AGP using two lectins, PHA-L and Concanavalin A (Con A)21,22 for affinity purification and MS analysis. We uncover multi-fucosylation on Hp and show how this attenuates binding to PHA-L. Moreover, we demonstrate how this lectin affinity purification-MS approach has the potential to become a generic method, capable of characterizing the inherent microheterogeneity of other complex glycoproteins.
Results and discussion
Analysis of glycoprotein microheterogeneity by native MS
The impact of terminal sialylation on glycoprotein binding to AAL and PHA-L is largely unknown.6,23,24 To facilitate unambiguous glycoproteoform assignments, and to limit the potential influence of terminal sialylation on lectin binding, we used asialo-Hp and asialo-AGP for lectin affinity purification and native MS analysis. We first documented the microheterogeneity of asialo-Hp and asialo-AGP by native MS analysis (Fig. 1A and D) and confirmed a complete removal of all sialic acid residues using glycoproteomics and glycomics (Fig. S1–S3†). As the complex type N-glycans are the only known PTM on Hp and AGP,17,18 we assigned the mass spectra of asialo-Hp and asialo-AGP based on the peptide backbone and monosaccharide residue masses (Fig. 1B and E, Table S1 and S2†) using UniDec software.25 We observed different proteoforms for AGP (F1 and S variants) and partially glycosylated glycoproteoforms for Hp (Fig. 1B and E), in agreement with our previous study.13 In order to reduce the complexity of lectin affinity purification-MS analysis, we focused on the main proteoform F1 variant for AGP and the fully glycosylated glycoproteoform for Hp.‡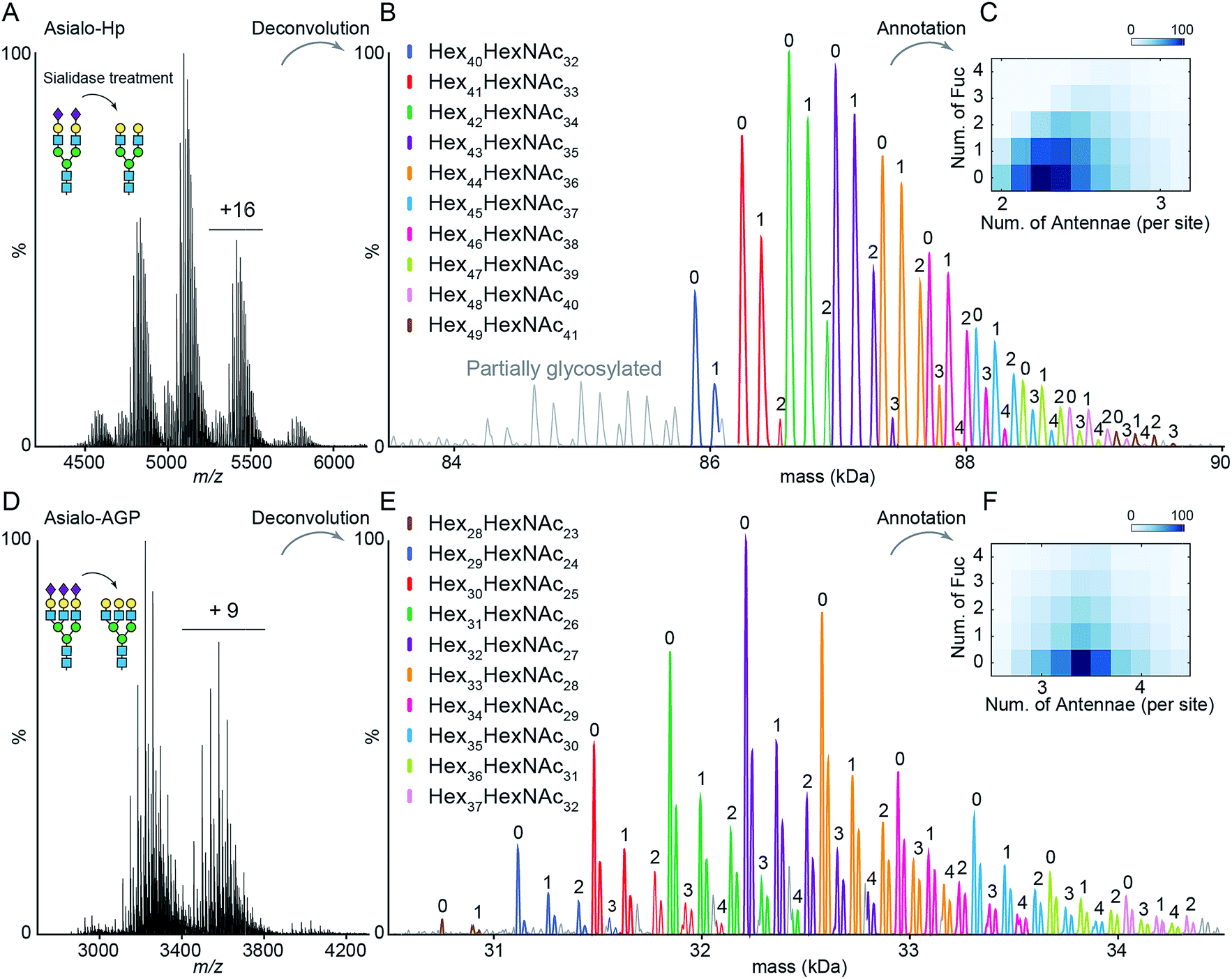 | ||
| Fig. 1 Native MS analysis of asialo-Hp and asialo-AGP. (A) Native mass spectrum of asialo-Hp. (B) Deconvoluted spectrum of asialo-Hp. The peaks of fully glycosylated Hp are assigned with the corresponding glycan compositions. The peaks with same hexose composition are annotated with same colors. The numbers of fucose residues are labelled on each peak. (C) Heatmap plots of the total number of fucose residues versus the average number of N-glycan antennae of each asialo-Hp glycoproteoform. (D) Native mass spectrum of asialo-AGP. (E) Deconvoluted spectrum of asialo-AGP. The peaks with same hexose composition are annotated with the same colors. The numbers of fucose residues are labelled on each peak. (F) Heatmap plots of the total number of fucose residues versus the average number of N-glycan antennae of each asialo-AGP glycoproteoform. | ||
To gain insight into N-glycan branching and fucosylation on Hp and AGP, we generated heatmap plots of the total number of fucose residues versus the average number of N-glycan antennae of each glycoproteoform from the native mass spectrum glycoform annotations (Fig. 1C and F). Hp carries mainly bi- and tri-antennary N-glycans (2.25 antennae per site), while AGP is more branched typically bearing tri- and tetra-antennary N-glycans (3.4 antennae per site) in agreement with the previous glycomics and glycoproteomics studies.17,18,20 Notably, we observed a positive correlation between the Hp N-glycan branching and fucosylation levels (Fig. 1C). Based on the native Hp spectrum, the Hex42HexNAc34 glycoproteoform of Hp (the base peak), which carries primarily six bi- and two tri-antennary N-glycans, can only be mono- and bi-fucosylated (Hex42HexNAc34Fuc1 and Hex42HexNAc34Fuc2) (Fig. 1B and C). For AGP, which is highly branched, we did not observe a correlation between N-glycan branching and fucosylation levels.
The Hp and AGP total fucosylation levels are similar (57% and 53%, respectively), but AGP has a higher multi-fucosylation level (Fig. 1C, F and S4†). Notably, the masses of Fuc5 and Hex2HexNAc2 are 730.715 Da and 730.6748 Da, respectively, and cannot be resolved at the intact protein level. Therefore, we can only resolve the glycoproteoform that carries less than five fucose residues. According to abundances of tetra-fucosylated glycoproteoforms of Hp and AGP, the highly fucosylated glycoproteoforms (Fucn, n > 4) are of relatively low-abundance.
Analysis of Hp fucosylation by AAL affinity purification-MS
To gain further insight into fucosylation on Hp, particularly highly fucosylated glycoproteoforms, we used affinity purification employing as AAL pull-down of asialo-Hp and analyzed AAL-bound and AAL-unbound fractions by native MS. The AAL-bound asialo-Hp exhibited a significantly different peak pattern to the AAL unbound fraction (Fig. 2A and S5†). The base peak of the AAL-bound Hp spectrum is 1312 Da larger than the base peak of the AAL-unbound Hp spectrum. This mass shift could be assigned to hyper-fucosylation (Fuc9 = 1315.287 Da; Hex40HexNAc32Fuc9) or to an increase in both N-glycan branching and fucosylation (Hex2HexNAc2Fuc4 = 1315.2468 Da; Hex42HexNAc34Fuc4) based on monosaccharide residue masses.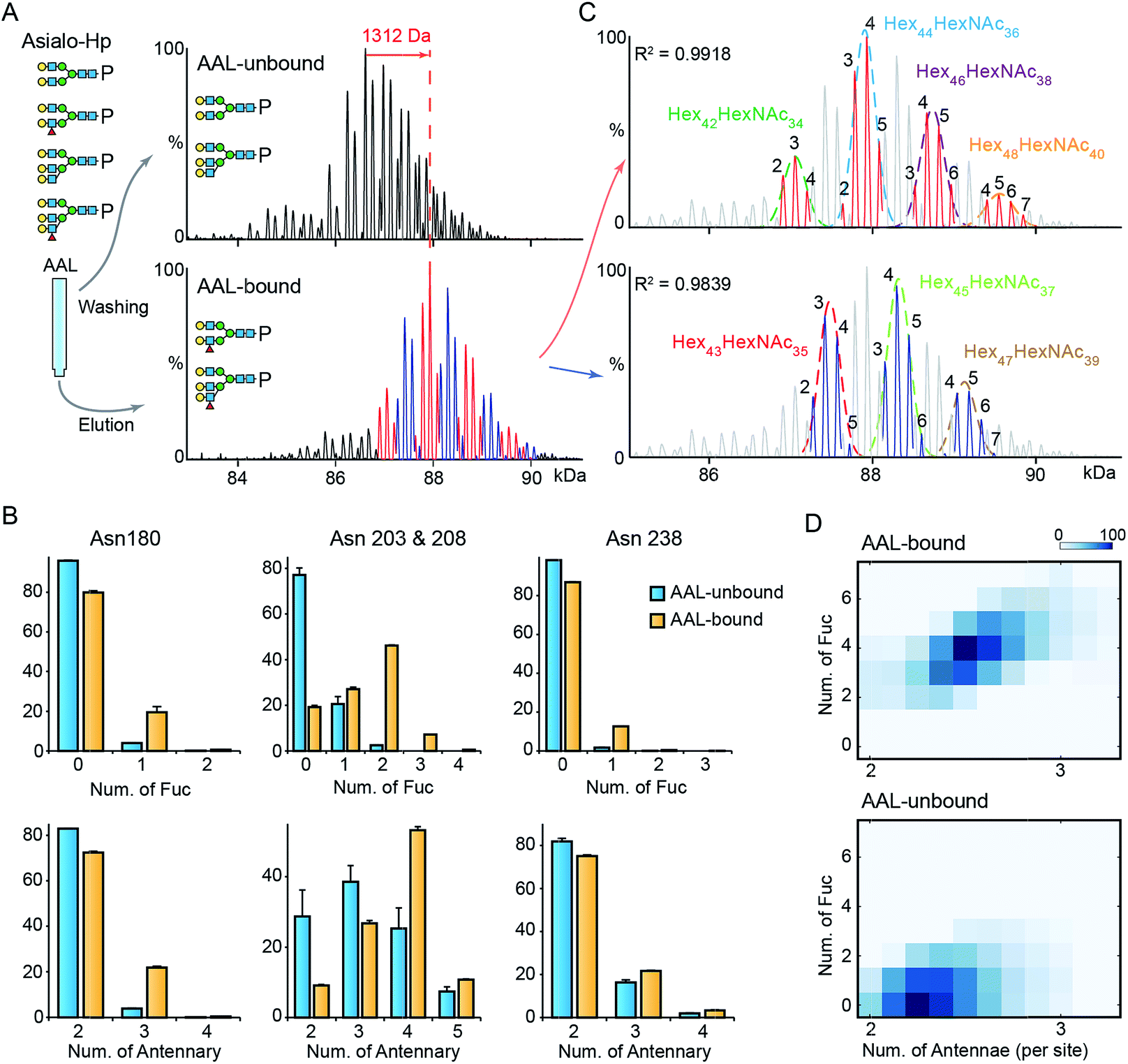 | ||
| Fig. 2 MS analysis of AAL fractionated Hp. (A) The deconvoluted spectrum of AAL-bound and unbound asialo-Hp. (B) The relative abundances of fucosylated and branched N-glycans on the three glycosylated tryptic peptides. The N-glycan on Asn203 & 208 are at one tryptic peptide. Error bars represent the standard error of three replicate experiments. (C) The AAL-bound Hp spectrum is divided to two series in which the peaks differ from 146 Da. The two peak envelopes were fitted with multiple Gaussian functions. The peaks under one Gaussian curve are assigned with the same hexose composition. The numbers of fucose residues are labelled on each peak. (D) The N-glycan branching and fucosylation levels of AAL-bound and AAL-unbound Hp are summarized and displayed on heatmaps. | ||
To distinguish the glycoproteoform composition associated with this mass shift we performed an LC-MS based glycoproteomic analysis. We assigned the site-specific microheterogeneity of the AAL-reactive asialo-Hp and obtained relative abundances of the glycoforms on each glycosylation site (Fig. 2B). We found that fucosylation levels on all sites were increased, although the non-fucosylated glycoforms were predominately located on Asn180 and Asn 237. Importantly, we also observed elevated N-glycan branching levels on all glycosylation sites in AAL-bound asialo-Hp, most notably on Asn180, Asn203 and Asn 208 (Fig. 2B). Therefore, the AAL-reactive Hp carries N-glycans with higher branching and fucosylation levels than AAL-unbound glycoproteoforms.
From these data, we attributed the mass shift of 1312 Da observed in the native mass spectrum to an elevation of both fucosylation and N-glycan branching (Hex2HexNAc2Fuc4). Therefore, the base peak of AAL-bound asialo-Hp corresponds to Hex44HexNAc36Fuc4 glycoforms. As described above, the Fuc5 and Hex2HexNAc2 peaks overlap, and therefore we cannot assign the adjacent +146 peak simply as an increase in fucosylation or N-glycan branching (e.g. Hex44HexNAc36Fuc6 or Hex46HexNAc38Fuc1). Therefore, we divided the main peaks in AAL-reactive Hp spectrum into two series, the adjacent peaks in each series differ by 146 Da (blue and red peak series, Fig. 2A). Then, we annotated these two peak series separately (red and blue peaks, Fig. 2C). We fitted two peak envelopes with the sum of multiple Gaussian functions and assigned the peaks under one Gaussian curve with the same hexose compositions (black numbering, Fig. 2C). We found the Hex42HexNAc34 glycoproteoforms, the major glycoproteoforms in unfractionated Hp, can have up to four fucose residues, and in the absence of the AAL affinity purification step, we only observe mono- and bi-fucosylated forms. We conclude that fucosylation levels are positively correlated to the extent of N-glycan branching of highly fucosylated asialo-Hp (AAL-bound fraction) with an additional two to seven fucose residues present with increased N-glycan branching (Fig. 2D).
Analysis of AGP fucosylated levels by AAL affinity purification-MS
Next, we explored the highly fucosylated AGP glycoproteoforms by AAL affinity purification-MS analysis. We observed the base peak of AAL-bound AGP increases by 877 Da which could be assigned to Fuc6 or Hex2HexNAc2Fuc1 (Fig. 3A and S6†). Notably, the N-glycan branching levels are generally unchanged on all detected four glycosylation sites, in contrast to the significantly elevated fucosylation levels across all AAL-bound asialo-AGP glycosylation sites (Fig. 3B). Therefore, AAL-reactive asialo-AGP carries fucosylated N-glycans without altered N-glycan branching level. We attributed the base peak of AAL-reactive AGP to Hex32HexNAc27Fuc6 which is 877 Da larger than that of AAL-unbound AGP (Hex32HexNAc27). | ||
| Fig. 3 MS analysis of AAL fractionated AGP. (A) The deconvoluted spectrum of AAL-bound and unbound asialo-AGP. (B) The relative abundances of fucosylated and branched N-glycans on the four glycosylated tryptic peptides. Error bars represent the standard error of three replicate experiments. (C) The AAL-bound AGP spectrum is divided to two series in which the peaks differ from 146 Da. The two peak envelopes were fitted with multiple Gaussian functions. The peaks under one Gaussian curve are assigned with the same hexose composition. The numbers of fucose residues are labelled on each peak. (D) The N-glycan branching and fucosylation levels of AAL-bound and AAL-unbound AGP are summarized and plotted on heatmaps. | ||
Due to the hyper-fucosylation on AAL-reactive AGP, it is still difficult to unambiguously assign the other peaks by monosaccharide residue masses, e.g. Hex32HexNAc27Fuc8 and Hex34HexNAc29Fuc3 glycoproteoforms are both highly fucosylated and overlap in native mass spectra. As above, we divided the peaks in AAL-bound AGP spectrum into two distinct peak series (red and blue peaks, Fig. 3A and C). Similarly, we fitted the peak envelopes with the sum of multiple Gaussian functions and assigned the compositions of the peaks under one Gaussian curve with the same hexose compositions (Fig. 3C). The highly fucosylated AGP carries six fucose residues on average and the glycans on Asn85 contribute most to the hyper-fucosylation. The most abundant glycoproteoforms (Hex32HexNAc27) can carry up to nine fucose residues, suggesting over the half of N-glycan antennae can be modified with terminal Lewis X (or sialyl-Lewis X) epitopes (Fig. 3C and D).
Notably, mono-/bi-fucosylated glycoproteoform in AAL-reactive AGP are not observed, albeit these two glycoproteoforms are the most abundant fucosylated isoforms (Fig. 1F). This indicates the multivalent interactions between fucosylated glycoproteins and AAL are essential for high avidity binding. Interestingly, the degeneracy pre-factor (Ω), a measure of multivalent interactions, of AAL to Hp with two fucose residues (Ω = 56) is similar to that of AAL binding to AGP with three fucose residues (Ω = 60), assuming the fucose residues on one glycosylation site can interact with one AAL molecule. These imply the multivalent interactions between AAL and fucosylated glycoprotein is sensitive to the fucose density on glycoproteins.
Hp and AGP N-glycan branching analysed by PHA-L and Con A affinity purification-MS
The inherent N-glycan branching levels of Hp and AGP are evident by native MS analysis (Fig. 1). To quantitatively analyze their highly branched glycoproteoforms, and to gain insight to their multivalent interactions with lectins, we used PHA-L and Con A to selectively purify highly branched (via PHA-L) and less branched (via Con A) glycoproteoforms. PHA-L is specific for N-glycans with β1-6 antenna and Con A only binds bi-antennary structures.The PHA-L-reactive Hp showed a significantly increased N-glycan branching level, resulting in three additional LacNAc units (1095 Da) to Hp on average (Fig. 4A and S7†). Thus, the highly branched Hp carries 2.6 N-glycan antennae on each glyco-site on average. Interestingly, we observed a decrease in the bi- and tri-fucosylated glycoproteoforms, suggesting the hyper-antennary fucosylation inhibits PHA-L binding to the branched antennae (Fig. 4A). On the contrary, the PHA-L bound AGP showed a subtle change in fucosylation level to the PHA-L unbound fraction (Fig. 4B and S7†). For Hp, which is a less branched glycoprotein, the presence of fucose reduces the availability of the β1-6 antennae to interact to PHA-L.26 The highly branched N-glycans on AGP have more PHA-L binding determinants, therefore the fucose residues only have limited influence of the multivalent interactions between AGP and PHA-L. Remarkably, the PHA-L reactive AGP does not show a significant alteration in N-glycan branching level (Fig. 4B), suggesting the numbers of β1-6 GlcNAc antennae on each glycoproteoform is sufficient for strong interactions with PHA-L and the extensions of polylactosamine unit on β1-6 GlcNAc branch is low on AGP in agreement with the glycoproteomics results (Fig. 3B). Unlike PHA-L specificity for highly branched N-glycan, Con A selectively recognizes bi-antennary N-glycans. We found the Con A-reactive Hp are similar to the unbound fraction (Fig. 4C and S8†), while the Con A-reactive AGP are significantly less branched than the unbound sample (the base peak of Con A-reactive AGP is 730 Da smaller) (Fig. 4D and S8†).
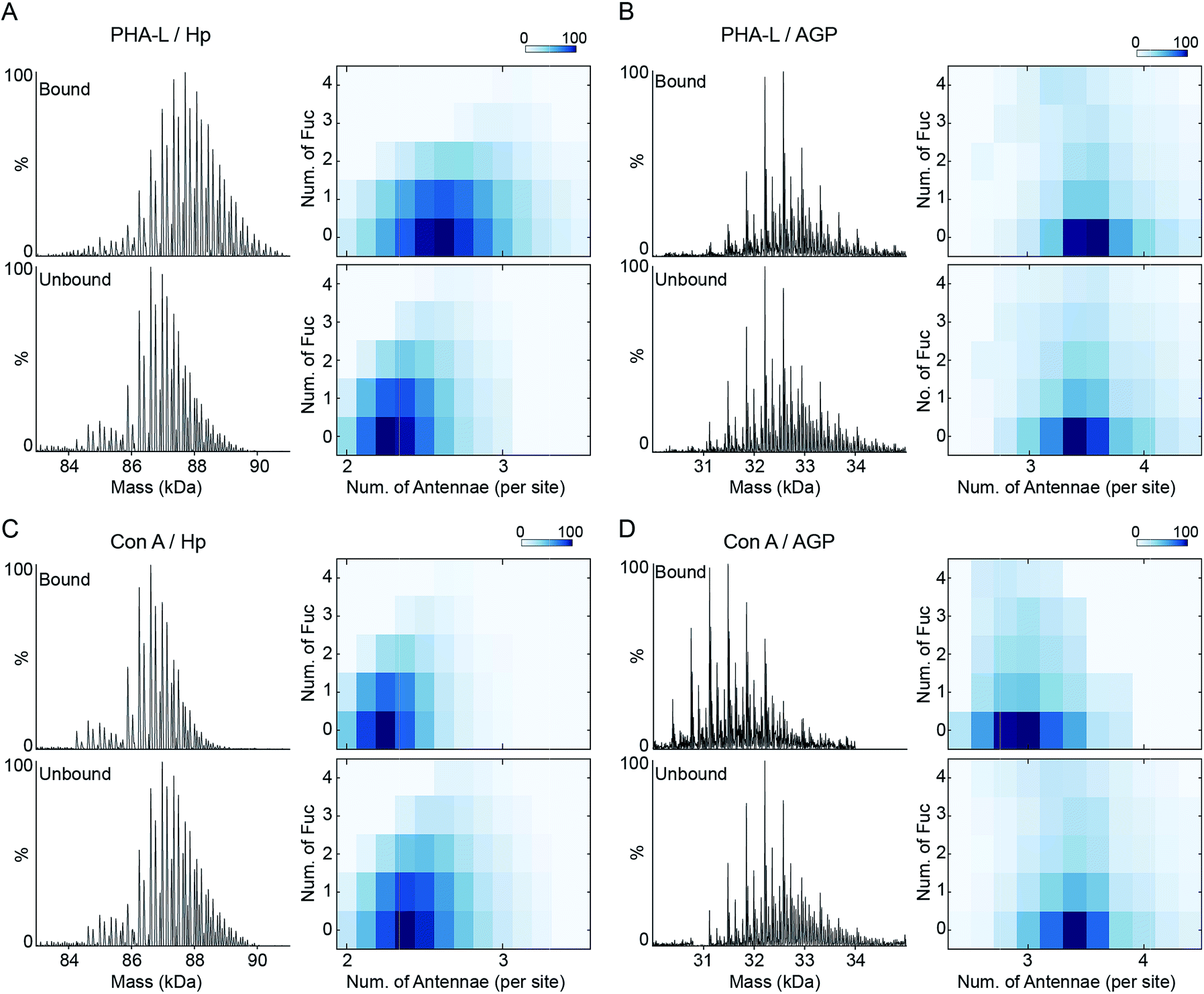 | ||
| Fig. 4 PHA-L and Con-A affinity purification-native MS analysis of Hp and AGP. Native spectra of PHA-L fractionated asialo-Hp and asialo-AGP (Fig. S7†) are deconvoluted to zero-charge spectra as (A) and (B). The N-glycan branching and fucosylation levels are summarized and plotted as heatmaps. The Con A-fractionated asialo-Hp (C) and asialo-AGP (D) are analyzed accordingly (Fig.S8†). | ||
The co-occurrence of fucosylation and N-glycan branching on Hp highlights that alteration of fucosylation levels can influence PHA-L based assays against less branched glycoproteins. A previous report described a relationship between up-regulated Mgat5 expression (which catalyses β1-6 branching) and decreased PHA-L reactivity of Hp during disease with increased fucosyltransferase levels.27 Hyper-fucosylation may occur on highly branched Hp and reduce its binding to PHA-L. Further screening of glycoprotein interactions with other lectins using the affinity purification MS approach described here will provide better interpretation and design for microarray detection by lectins.
Since Con A and PHA-L are the two most widely used lectins for probing glycoprotein N-glycan branching levels, we compared the abilities of PHA-L and Con A to differentiate the branched glycoproteoforms from the collective pools at the intact glycoprotein level (Fig. 5A and B). PHA-L effectively separates highly branched and larger mass glycoproteoforms from the total Hp. However, for AGP, which is already highly branched, it captures the glycoproteoforms containing β1-6 GlcNAc antenna which are structural isomers to the non-reactive glycoproteoforms which mainly carry β1-3 GlcNAc antennae. Conversely, Con A is more practical to discriminate N-glycan branching levels for highly branched AGP, rather than for Hp. Together, these also suggest an elevated level of β1-6 GlcNAc branching on Hp would give rise to a substantial change in its peak envelope in the native spectrum. Nevertheless, this is not the case for AGP, due to its intrinsic high branching level. For probing fucosylation level, AAL completely divides hyper-fucosylated Hp glycoproteoforms which cannot be observed directly in native MS from the non- and low fucosylated glycoproteoforms (Fig. 5C). More generally, PHA-L and Con A fractionations are better for less branched and highly branched glycoproteins, respectively, by unveiling more glycoproteomics for in-depth native MS analysis of glycoproteins.
 | ||
| Fig. 5 Lectin specificities to discriminate glycoprotein microheterogeneity. The N-glycan branching and fucosylation levels of PHA-L (A), Con A (B) and AAL (C) fractionated asialo-Hp and asialo-AGP are plotted as heatmaps, respectively. PHA-L and Con A show better separations for less branched Hp and highly branched AGP, respectively. AAL only captures highly fucosylated glycoproteomics. | ||
Fucosylation and N-glycan branching levels on Hp are both reported to be altered in various cancers, however, they are rarely verified experimentally by PHA-L based tests whereas AAL based confirmations are routine.16 We propose that the PHA-L based validation is limited by the attenuation effect of hyper-fucosylation on less branched N-glycoproteins binding to PHA-L. Interestingly, several previous studies report probing N-glycan branching alterations on Hp using Con A based tests in the disease states in which fucosylation levels are also changed.28,29 Our comparative analysis of PHA-L and Con A reactive glycoproteoforms suggests that Con A does not have a bias between non-fucosylated and fucosylated glycoproteoforms.
Collectively, the lectin affinity purification-MS analysis provides a deeper understanding of the Hp and AGP glycosylation and their multivalent interactions to lectins. Our data demonstrate that multi-fucosylation enhances multivalent interactions between AAL and glycoproteins, on the other hand, it attenuates PHA-L interactions with less branched glycoproteins (Fig. 6).
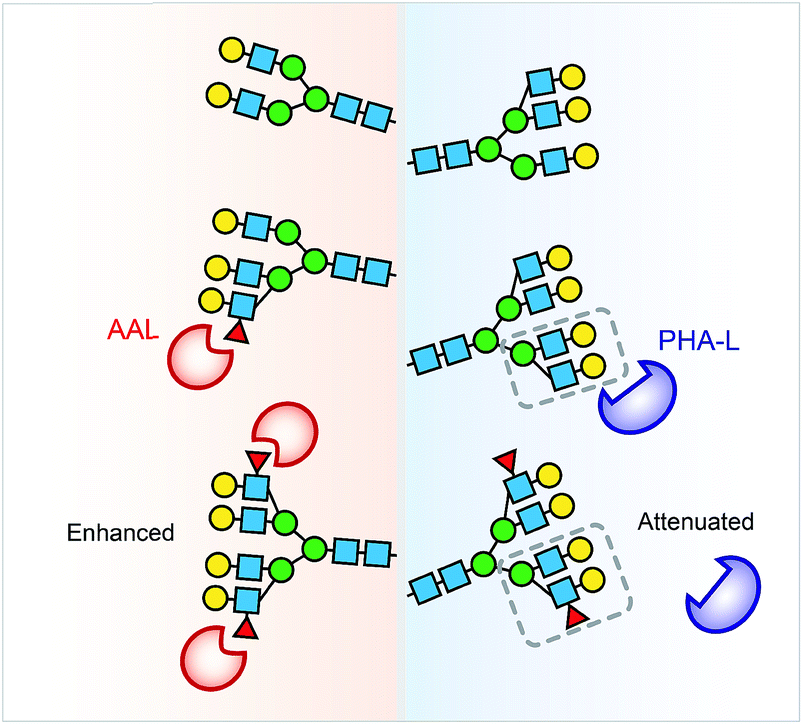 | ||
| Fig. 6 Multi-fucosylation enhances the interactions between AAL and glycoproteins, but attenuates PHA-L interactions with less branched glycoprotein. We propose that the PHA-L based lectin detection approach is limited by the attenuation effect of hyper-fucosylation on less branched N-glycoproteins binding to PHA-L. | ||
Conclusions
We have demonstrated a comprehensive approach to analyse subtle changes in complex N-glycosylation, namely terminal fucosylation and branching, providing global quantitative protein glycosylation, monosaccharide composition and insight into glycan-specific lectin interactions. We applied lectin affinity purification and MS approaches to define highly fucosylated and highly branched glycoproteins at the intact glycoprotein level. We identified highly fucosylated structures on Hp and AGP with four and six fucose residues on average as well as additional modifications up to six and nine fucose residues, respectively. We found that a co-occurrence of fucosylation and N-glycan branching was significant on Hp and more substantial on highly fucosylated glycoproteoforms. We revealed that multi-fucosylation on Hp attenuates binding to PHA-L, and conversely has no impact on AGP–PHA-L interactions. We further profiled highly branched Hp and AGP glycoproteoforms and compared their PHA-L- and Con A-reactive glycoproteoforms.Lectin specificity is normally investigated by assessing interaction to free carbohydrate determinants independent of global or site-specific glycoprotein microheterogeneity information. Our lectin affinity purification-MS analysis identified the carbohydrate determinant stoichiometry of an intact folded glycoprotein taking into account steric constraints arising from neighboring glycosylation and also microheterogeneity of the target N-glycan. Previous reports evaluated and criticized lectin affinities and specificities to glycoproteins and glycopeptides using lectin affinity fractionation coupled to MS-based glycomics30 and glycoproteomics.31 We also observed glycopeptides with fucose residues in AAL-unbound fraction using glycoproteomics approach (Fig. 2B and 3B). Interestingly, our native MS analysis illustrates that AAL captures only hyper-fucosylated forms without any compromise of hypo-fucosylated or non-fucosylated forms (Fig. 5C). The “leaking” fucosylated glycopeptides in AAL-unbound fraction are present on hypo-fucosylated glycoproteoforms that are less reactive to AAL. Our data inform that lectins are less efficient to probe/isolate all glycoprotein/glycopeptides with certain determinant from non-glycosylated form, because the binding avidity is determined by lectin–glycoprotein multivalent interactions.
The comprehensive MS analysis of lectin-reactive glycoproteoforms bridges the gap between the classic lectin-based detection of protein glycosylation, namely lectin blotting and enzyme-linked lectin assay (ELLA), and MS based glycoproteomics, both of which are popular pipelines of serological glycoprotein marker discovery for cancers and tumors.32 Nevertheless, combining and interpreting the results from these two orthogonal techniques is difficult, due to their differing levels of complexity. Herein we have resolved the AAL-reactive glycoproteoforms and related their microheterogeneities to hyper-fucosylation and/or hyper-N-glycan branching levels. Specifically we found the mono- or bi-fucosylated glycoproteoforms are absent in the AAL-reactive fraction following stringent elution protocols. These two abundant fucosylated forms may contribute to high baseline signals in lectin blotting/ELLA and obscure the signals from the diseases-relative hyper-fucosylated forms. For example, single core-fucosylation on antibody often induces a high background in ELLA.33
Native MS has already shown an unparalleled performance in analysing and comparing microheterogeneities of biopharmaceuticals.11,12 Due to the enormous number of N-glycan combinations on each glycoproteoform, assigning the composition of overlapping isobaric glycoproteoforms is however extremely challenging. As sialylation, N-glycan branching and fucosylation account for the major part of N-glycan complexity, we applied a combination of exoglycosidase digestion and lectin-affinity purification to reduce the inherent heterogeneity of human N-glycoproteins, unravel the minor, but functionally important glycoproteoforms, and related the peak composition to fucosylation and N-glycan branching from the corroboration of the comparative glycoproteomics. Several recent reports apply MS approach to study intact glycoprotein microheterogeneity variations and alterations in individuals and patients with diseases.34–36 Our data demonstrate the importance of interpreting the isobaric glycoproteoforms of intact glycoproteins using lectin affinity purification-native MS and glycoproteomics. Our comprehensive approach extends native MS to profile the complex glycoproteins with highly branched and fucosylated N-glycans and their interactions with lectins. The approach also has great potential not only to characterize disease related glycoproteins but also in the evaluation of recombinant glycosylated biopharmaceuticals.
Experimental
Materials
Human alpha 1-acid glycoprotein (AGP), human haptoglobin phenotype 1-1 (Hp) were from Sigma-Aldrich (Steinheim, Germany). Agarose bound AAL, PHA-L and Con A were from Vector Laboratories.Native MS of glycoproteins
The protein samples were loaded into an in-house made gold coated needle and analyzed on a modified Q-Exactive mass spectrometry (Thermo Fisher Scientific)37 with m/z range of 2500–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Th. The typical MS settings were spray voltage of 1.1–1.3 kV, source fragmentation of 50–100 V, source temperature at 30 °C, HCD collision energy of 0 V and resolution of 17500 at m/z 200. Backing pressure was maintained at ∼6 × 10−10 mbar and native MS data were analyzed using Xcalibur 2.2.
000 Th. The typical MS settings were spray voltage of 1.1–1.3 kV, source fragmentation of 50–100 V, source temperature at 30 °C, HCD collision energy of 0 V and resolution of 17500 at m/z 200. Backing pressure was maintained at ∼6 × 10−10 mbar and native MS data were analyzed using Xcalibur 2.2.
Lectin affinity purification
For AAL affinity purification, asialo-AGP was buffer exchanged to Tris-buffered saline (TBS, 50 mM Tris HCl, 150 mM NaCl and pH 7.4). Agarose bound AAL (0.5 ml settled gel) was washed with 5 ml TBS three times to remove free sugar bound to AAL, then incubated with 1 mg asialo-AGP at a final volume of 1 ml for 2 hours at room temperature. The agarose bound AAL and asialo-AGP mixture was transferred into a new empty Bio-Spin chromatography column and washed with 5 ml TBS. The flow through and wash fractions were collected as AAL-unbound asialo-AGP. AAL-bound asialo-AGP was eluted from agarose bound AAL with 2 ml TBS containing 400 mM fucose. The AAL-unbound and bound asialo-AGP was buffer exchanged into 200 mM ammonium acetate immediately for the following experiment. For PHA-L affinity purification, the general method is the same with minor modifications. TBS with 1 mM CaCl2 and 1 mM MnCl2 was used to condition agarose bound PHA-L, wash off PHA-L unbound asialo-AGP. To elute PHA-L-bound asialo-AGP, 100 mM acetic acid was used.Glycoproteomics analysis
Tryptic digested glycoprotein was analyzed on EASY-nLC 1000 coupled to LTQ-Orbitrap XL spectrometer (Thermo Fisher Scientific) via a dynamic nanospray source. The glycopeptides was firstly loaded onto a 75 μm × 2 cm pre-column and separated on a 75 μm × 15 cm Pepmap C18 analytical column (Thermo Fisher Scientific) with a binary buffer system. Buffer A was 0.1% formic acid in 100% H2O and buffer B was 0.1% formic acid in 80% acetonitrile with 20% H2O. A 100 min gradient (0% buffer B for 5 min, 0 to 60% buffer B for 60 min, 60% to 100% buffer B for 10 min, 100% buffer B for 10 min and 100 to 0% buffer B for 5 min, 0% buffer B for 10 min) was used. LTQ-Orbitrap XL was operated in data-dependent acquisition mode with one full MS scan followed by 5 MS/MS scans with collision-induced dissociation. For full MS scan, the mass range was 335 to 2000 m/z at a resolution of 60000. For MS/MS scan, the precursor isolation width was 2 Da and the CID normalized energy was 35%.
Extended experimental and method details can be found in the ESI.†
Author contributions
D. W. and C. V. R. designed the research. D. W. and J. L. performed PHA-L and Con A lectin-pull down experiments. D. W. and W. B. S. carried out native MS data analysis. D. W. performed all other experiments and data analysis. D. W. and C. V. R. wrote the manuscript with contributions from all other authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge funding from a Wellcome Trust Investigator Award (104633/Z/14/Z). We thank Mike Ferguson (University of Dundee) for discussions on glycobiology and Manman Guo (University of Oxford) for suggestions in proteomics data analysis.Notes and references
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS.
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS PubMed.
- A. Varki, Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, 2nd edn, 2009 Search PubMed.
- N. Kochibe and K. Furukawa, Biochemistry, 1980, 19, 2841–2846 CrossRef CAS PubMed.
- K. Matsumura, K. Higashida, H. Ishida, Y. Hata, K. Yamamoto, M. Shigeta, Y. Mizuno-Horikawa, X. Wang, E. Miyoshi, J. Gu and N. Taniguchi, J. Biol. Chem., 2007, 282, 15700–15708 CrossRef CAS PubMed.
- E. Green and J. Baenziger, J. Biol. Chem., 1987, 262, 12018–12029 CAS.
- Y. Kaneda, R. F. Whittier, H. Yamanaka, E. Carredano, M. Gotoh, H. Sota, Y. Hasegawa and Y. Shinohara, J. Biol. Chem., 2002, 277, 16928–16935 CrossRef CAS PubMed.
- G. Lauc, A. Essafi, J. E. Huffman, C. Hayward, A. Knežević, J. J. Kattla, O. Polašek, O. Gornik, V. Vitart, J. L. Abrahams, M. Pučić, M. Novokmet, I. Redžić, S. Campbell, S. H. Wild, F. Borovečki, W. Wang, I. Kolčić, L. Zgaga, U. Gyllensten, J. F. Wilson, A. F. Wright, N. D. Hastie, H. Campbell, P. M. Rudd and I. Rudan, PLoS Genet., 2010, 6, e1001256 CrossRef CAS PubMed.
- S. Pan, R. Chen, R. Aebersold and T. A. Brentnall, Mol. Cell. Proteomics, 2011, 10, R110.003251 CrossRef PubMed.
- W. B. Struwe, A. Stuckmann, A.-J. Behrens, K. Pagel and M. Crispin, ACS Chem. Biol., 2017, 12, 357–361 CrossRef CAS PubMed.
- Y. Yang, F. Liu, V. Franc, L. A. Halim, H. Schellekens and A. J. R. Heck, Nat. Commun., 2016, 7, 13397 CrossRef CAS.
- T. Wohlschlager, K. Scheffler, I. C. Forstenlehner, W. Skala, S. Senn, E. Damoc, J. Holzmann and C. G. Huber, Nat. Commun., 2018, 9, 1713 CrossRef.
- D. Wu, W. B. Struwe, D. J. Harvey, M. A. J. Ferguson and C. V. Robinson, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8763–8768 CrossRef CAS PubMed.
- P. Lössl, J. Snijder and A. J. R. Heck, J. Am. Soc. Mass Spectrom., 2014, 25, 906–917 CrossRef.
- K.-H. Khoo, in Comprehensive Natural Products II, Elsevier, 2010, pp. 123–156 Search PubMed.
- S. Zhang, S. Shang, W. Li, X. Qin and Y. Liu, Glycobiology, 2016, 26, 684–692 CrossRef CAS.
- J. Zhu, Z. Lin, J. Wu, H. Yin, J. Dai, Z. Feng, J. Marrero and D. M. Lubman, J. Proteome Res., 2014, 13, 2986–2997 CrossRef CAS PubMed.
- M. J. Treuheit, C. E. Costello and H. B. Halsall, Biochem. J., 1992, 283, 105–112 CrossRef CAS PubMed.
- J. L. Dage, B. L. Ackermann and H. B. Halsall, Glycobiology, 1998, 8, 755–760 CrossRef CAS PubMed.
- T. Imre, G. Schlosser, G. Pocsfalvi, R. Siciliano, É. Molnár-Szöllősi, T. Kremmer, A. Malorni and K. Vékey, J. Mass Spectrom., 2005, 40, 1472–1483 CrossRef CAS PubMed.
- D. N. Moothoo, B. Canan, R. A. Field and J. H. Naismith, Glycobiology, 1999, 9, 539–545 CrossRef CAS.
- J. H. Naismith and R. A. Field, J. Biol. Chem., 1996, 271, 972–976 CrossRef CAS PubMed.
- R. D. Cummings and S. Kornfeld, J. Biol. Chem., 1982, 257, 11230–11234 CAS.
- N. D. Pham, P. C. Pang, S. Krishnamurthy, A. M. Wands, P. Grassi, A. Dell, S. M. Haslam and J. J. Kohler, J. Biol. Chem., 2017, 292, 9637–9651 CrossRef CAS PubMed.
- M. T. Marty, A. J. Baldwin, E. G. Marklund, G. K. A. Hochberg, J. L. P. Benesch and C. V. Robinson, Anal. Chem., 2015, 87, 4370–4376 CrossRef CAS PubMed.
- M. F. A. Bierhuizen, H. Tedzes, W. E. C. M. Schiphorst, D. H. van den Eijnden and W. van Dijk, Glycoconjugate J., 1988, 5, 85–97 CrossRef CAS.
- K. L. Abbott, A. V. Nairn, E. M. Hall, M. B. Horton, J. F. Mcdonald, K. W. Moremen, D. M. Dinulescu and M. Pierce, Proteomics, 2008, 8, 3210–3220 CrossRef CAS PubMed.
- G. A. Turner, M. T. Goodarzi and S. Thompson, Glycoconjugate J., 1995, 12, 211–218 CrossRef CAS PubMed.
- B. Maresca, L. Cigliano, M. S. Spagnuolo, F. Dal Piaz, M. M. Corsaro, N. Balato, M. Nino, A. Balato, F. Ayala and P. Abrescia, PLoS One, 2012, 7, e52040 CrossRef CAS PubMed.
- A. Lee, M. Nakano, M. Hincapie, D. Kolarich, M. S. Baker, W. S. Hancock and N. H. Packer, OMICS: J. Integr. Biol., 2010, 14, 487–499 CrossRef CAS PubMed.
- F. Zhu, D. E. Clemmer and J. C. Trinidad, Analyst, 2017, 142, 65–74 RSC.
- S. A. Svarovsky and L. Joshi, Anal. Methods, 2014, 6, 3918–3936 RSC.
- O. H. Hashim, J. J. Jayapalan and C.-S. Lee, PeerJ, 2017, 5, e3784 CrossRef PubMed.
- M. Baerenfaenger and B. Meyer, J. Proteome Res., 2018, 17, 3693–3703 CrossRef CAS PubMed.
- J. J. H. Kim, S. J. H. Lee, S. Choi, U. Kim, I. S. Yeo, S. H. S. T. Kim, M. J. Oh, H. Moon, J. H. H. J. J. Lee, S. Jeong, M. G. Choi, J. H. H. J. J. Lee, T. S. Sohn, J. M. Bae, S. H. S. T. Kim, Y. W. Min, H. Lee, J. H. H. J. J. Lee, P.-L. Rhee, J. J. H. Kim, S. J. H. Lee, S. H. S. T. Kim, J. H. H. J. J. Lee, S. H. Park, J. O. Park, Y. S. Park, H. Y. Lim, W. K. Kang, H. J. An, J. J. H. Kim, J. J. H. Kim, S. Hyeon Lee, S. Choi, U. Kim, I. Seok Yeo, S. Hee Kim, M. Jin Oh, H. Moon, J. H. H. J. J. Lee, S. Jeong, M. Gew Choi, J. Ho Lee, T. Sung Sohn, J. Moon Bae, S. H. S. T. Kim, Y. Won Min, H. Lee, J. Haeng Lee, P.-L. Rhee, J. J. H. Kim, S. Jin Lee, S. Tae Kim, J. H. H. J. J. Lee, S. Hoon Park, J. Oh Park, Y. Suk Park, H. Yeong Lim, W. Ki Kang, H. Joo An, J. Hoe Kim, J. J. H. Kim, S. J. H. Lee, S. Choi, U. Kim, I. S. Yeo, S. H. S. T. Kim, M. J. Oh, H. Moon, J. H. H. J. J. Lee, S. Jeong, M. G. Choi, J. H. H. J. J. Lee, T. S. Sohn, J. M. Bae, S. H. S. T. Kim, Y. W. Min, H. Lee, J. H. H. J. J. Lee, P.-L. Rhee, J. J. H. Kim, S. J. H. Lee, S. H. S. T. Kim, J. H. H. J. J. Lee, S. H. Park, J. O. Park, Y. S. Park, H. Y. Lim, W. K. Kang, H. J. An and J. J. H. Kim, Oncotarget, 2017, 8, 11094–11104 Search PubMed.
- N. Abu Bakar, N. C. Voermans, T. Marquardt, C. Thiel, M. C. H. Janssen, H. Hansikova, E. Crushell, J. Sykut-Cegielska, F. Bowling, L. MØrkrid, J. Vissing, E. Morava, M. van Scherpenzeel and D. J. Lefeber, Transl. Res., 2018, 199, 62–76 CrossRef CAS PubMed.
- J. Gault, J. A. C. Donlan, I. Liko, J. T. S. Hopper, K. Gupta, N. G. Housden, W. B. Struwe, M. T. Marty, T. Mize, C. Bechara, Y. Zhu, B. Wu, C. Kleanthous, M. Belov, E. Damoc, A. Makarov and C. V. Robinson, Nat. Methods, 2016, 13, 333–336 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00360f |
| ‡ The raw mass spectra have been deposited on figshare (https://doi.org/10.6084/m9.figshare.7309022.v1). |
| This journal is © The Royal Society of Chemistry 2019 |