
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cationic polymethacrylate-copolymer acts as an agonist for β-amyloid and an antagonist for amylin fibrillation†
Bikash R.
Sahoo
 a,
Takuya
Genjo
a,
Takahiro W.
Nakayama
b,
Andrea K.
Stoddard
a,
Toshio
Ando
b,
Kazuma
Yasuhara
c,
Carol A.
Fierke
ad and
Ayyalusamy
Ramamoorthy
*a
a,
Takuya
Genjo
a,
Takahiro W.
Nakayama
b,
Andrea K.
Stoddard
a,
Toshio
Ando
b,
Kazuma
Yasuhara
c,
Carol A.
Fierke
ad and
Ayyalusamy
Ramamoorthy
*a
aBiophysics and Department of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA. E-mail: ramamoor@umich.edu
bBio-AFM Frontier Research Center, Kanazawa University, Kanazawa 920-1192, Japan
cGraduate School of Materials Science, Nara Institute of Science and Technology, Ikoma, Nara 6300192, Japan
dDepartment of Chemistry, Texas A&M University, College Station, TX 77843, USA
First published on 27th February 2019
Abstract
In humans, β-amyloid and islet amyloid polypeptide (IAPP, also known as amylin) aggregations are linked to Alzheimer's disease and type-2 diabetes, respectively. There is significant interest in better understanding the aggregation process by using chemical tools. Here, we show the ability of a cationic polymethacrylate-copolymer (PMAQA) to quickly induce a β-hairpin structure and accelerate the formation of amorphous aggregates of β-amyloid-1-40, whereas it constrains the conformational plasticity of amylin for several days and slows down its aggregation at substoichiometric polymer concentrations. NMR experiments and microsecond scale atomistic molecular dynamics simulations reveal that PMAQA interacts with β-amyloid-1-40 residues spanning regions K16-V24 and A30-V40 followed by β-sheet induction. For amylin, it binds strongly close to the amyloid core domain (NFGAIL) and restrains its structural rearrangement. High-speed atomic force microscopy and transmission electron microscopy experiments show that PMAQA blocks the nucleation and fibrillation of amylin, whereas it induces the formation of amorphous aggregates of β-amyloid-1-40. Thus, the reported study provides a valuable approach to develop polymer-based amyloid inhibitors to suppress the formation of toxic intermediates of β-amyloid-1-40 and amylin.
Introduction
Self-assembly of amyloidogenic proteins is involved in numerous neurodegenerative diseases and other diseases including Alzheimer's disease (AD) and type-2 diabetes (T2D).1,2 Nevertheless, our current understanding of the role of protein aggregation in the pathogenesis of such diseases remains elusive.3 Despite the recent advancements in high-throughput screening of several anti-amyloidogenic compounds,4 there is no treatment for protein aggregation based disorders including AD and T2D.5 β-amyloid and islet amyloid polypeptide (amylin) aggregations are linked to AD and T2D, respectively.6 Both β-amyloid and amylin follow a conserved aggregation pathway and form toxic oligomer intermediates. Thus, they have been the subject of intense research to establish a pathological correlation and develop small molecules that interfere with the aggregation pathways of β-amyloid and amylin.6 The sequential protein aggregation mechanism from a water-soluble monomer to insoluble amyloid fibers triggering AD and T2D still remains elusive. However, several studies have observed a conserved pathway where both β-amyloid and amylin monomers aggregate to form pre-fibrillar toxic oligomers followed by matured β-sheet rich fiber structures.7 Thus, the sequential aggregation products of β-amyloid or amylin have been targeted to design potential inhibitors to interrupt amyloid formation.4,8 Moreover, substantial research effort has been devoted to developing strategies to reduce the formation of toxic intermediates of β-amyloid or amylin. In response to this, several amyloid inhibitors or modulators have been clinically tested for AD or T2D treatment. However, small molecule inhibitors or modulators targeting amyloidosis have recently faced several clinical trial challenges.8,9 In addition, the complexities associated with amyloid aggregation such as biological pathways, involvement of other cellular key players, structural heterogeneity, etc., pose challenges to current therapeutic developments. Among several compounds, chemically conserved scaffold molecules characterized by multi-aromatic groups have been tested in vitro or in vivo.10–14 These compounds have been reported to modulate the amyloid aggregation pathways. But the poor solubility and bioavailability affect their therapeutic nature and recently have been shown to be overcome using nanocarriers.15–17 Although, their mechanism of action remains unknown, covalent bonding, hydrogen bonding, hydrophobic interactions and π–π stacking are thought to be the major driving forces for their inhibitory mechanism of action.18–21 Oxidation of amyloid peptides by chemical compounds can also modulate amyloid aggregation.22 Despite substantial efforts, no significant new drugs have been discovered against these most tenacious and unnerving medical disorders. The successive failures of small molecule compounds direct researchers to develop new anti-amyloidogenic molecules such as polymers, peptoids, macrocyclic peptides, nanoparticles, molecular chaperones, etc.8,23–27 Among them, several polymers characterized by their ionic properties have been tested to investigate their activities towards amyloidogenic aggregation. Notably, amine containing polymers, polyamino acids, cationic surfactants and cellular polyamines have been observed to modulate β-amyloid aggregation.28–30 Similarly, controlled aggregation kinetics and toxicity of amylin using star-polymers and β-amyloid using polymer-nanodiscs have been studied recently.19,20,25 Here we demonstrate the modulation of amyloid aggregation pathways for amylin and β-amyloid-1-40 using a polymethacrylate derived copolymer (PMAQA) that has been implicated in several biological studies including lipid-nanodisc formation, enhancement of drug delivery, modulation of bioavailability and microencapsulation.31–33Experimental section
Materials
The polymethacrylate quaternary ammonium copolymer (PMAQA, ∼4.7 kDa) was synthesized and purified as reported elsewhere.31 Unlabeled and uniform 15N isotope labeled full-length β-amyloid-1-40 (DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV) was recombinantly expressed in E. coli BL21 (DE3). The β-amyloid-1-40 expression followed previously described protocols34,35 and purified by loading the samples onto an ECOPLUS HPLC column packed with the reversed-phase separation material. Synthetic human amylin (KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY-NH2) was purchased from AnaSpec at >95% purity.Sample preparation
The sample preparation followed a previously described method.27 Briefly, 1 mg mL−1 β-amyloid-1-40 peptide was dissolved in 5% (v/v) NH4OH and aliquoted to 0.1 mg mL−1 followed by lyophilization. The β-amyloid-1-40 peptide powder was re-suspended in 10 mM sodium phosphate, pH 7.4, and sonicated for 30 s followed by centrifugation at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g for 15 min at 4 °C to remove small aggregates. 1 mg mL−1 amylin was treated with 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and kept on ice for 30 minutes. The peptide solutions were aliquoted to 0.1 mg mL−1 and lyophilized. The amylin powder was re-suspended in 30 mM sodium acetate buffer, pH 5.5, and sonicated for 30 s. The PMAQA powder (10 mg mL−1) was dissolved in deionized water.
000×g for 15 min at 4 °C to remove small aggregates. 1 mg mL−1 amylin was treated with 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and kept on ice for 30 minutes. The peptide solutions were aliquoted to 0.1 mg mL−1 and lyophilized. The amylin powder was re-suspended in 30 mM sodium acetate buffer, pH 5.5, and sonicated for 30 s. The PMAQA powder (10 mg mL−1) was dissolved in deionized water.
Thioflavin-T fluorescence assay
Thioflavin T (ThT) fluorescence assays were performed to monitor the aggregation kinetics of 5 μM β-amyloid-1-40 or amylin at 37 °C in the presence of PMAQA (0.1, 0.2, 1, 2.5, 5, 6, 7.5, 10, and 50 μM) and 10 μM ThT. Fisher 96-well polystyrene plates with a sample volume of 100 μL per well in triplicate were used for the ThT assay. The kinetics of amyloid formation was monitored at 3 min intervals under no-shaking conditions for 4 days using a microplate reader (Biotek Synergy 2) with excitation and emission wavelengths of 440 and 485 nm, respectively.Circular dichroism and Fourier transform-infrared spectroscopy
β-amyloid-1-40 or amylin secondary structural transition in the presence or absence of PMAQA was studied by Far-UV circular dichroism (CD) using a JASCO (J820) spectropolarimeter. A 1 mm light-path length cuvette containing 25 μM β-amyloid-1-40 or amylin solution titrated with an increasing concentration of PMAQA (ranging from 0.25 to 50 μM) was used to monitor the evolution of structural transition at 25 °C. The samples (peptide:polymer = 1:1.5) were stored at room temperature and the CD spectra were recorded for 5 days at different time intervals. The CD spectra were averaged and conveyed as the mean residue ellipticity [Θ] after subtracting the signal from a solution without peptide. Fourier transform-infrared (FT-IR) spectra were measured for 25 μM β-amyloid-1-40 or amylin mixed with 1.5 molar equivalent of PMAQA (incubated for 6 hours followed by lyophilization) in transmission mode within a range of 4000–400 cm−1 using a Thermos scientific ATR-FTIR instrument. 10 μM of β-amyloid-1-40 or amylin was incubated with 15 μM of PMAQA for ∼5 minutes and overnight, respectively, for size distribution analysis. The resultant solutions were purified using size exclusion chromatography (SEC) by passing through a Superdex 200 Increase 300/10 GL column operated on an AKTA purifier (GE Healthcare, Freiburg, Germany).
NMR
1D and 2D NMR spectra were recorded on a 600 MHz Bruker Avance III NMR spectrometer equipped with a z-axis gradient cryogenic probe. Unlabeled amylin (50 μM) dissolved in 30 mM sodium acetate, pH 5.5, and unlabeled β-amyloid-1-40 or uniformly 15N-labeled β-amyloid-1-40 peptides (60 μM) dissolved in 10 mM sodium phosphate, pH 7.4, and buffer containing 90% H2O/10% 2H2O were used for NMR measurements. The 2D 15N/1H SOFAST-HMQC36 NMR titration experiments of β-amyloid-1-40 (60 μM) with 0.6, 1.2 and 3 μM PMAQA were performed at 10 °C with 64 scans and 200 t1 increments. The NMR spectra were processed using TopSpin 3.5 (Bruker) and analyzed using Sparky.37High-speed AFM and transmission electron microscopy imaging
A high-speed atomic force microscopy (HS-AFM) instrument38,39 was operated in the tapping mode with a small cantilever (BL-AC10DS-A2, Olympus with spring constant k = 0.1 N m−1, and resonance frequency f = ∼400 kHz in water). Each cantilever has an amorphous carbon tip at its top. The tip was prepared by electron beam deposition with a scanning electron microscope (ELS-7500, Elionix). The free oscillation amplitude of the cantilever and the typical set-point were ∼1 nm and 80% of the free oscillation amplitude. For the HS-AFM sample stage, a freshly cleaved mica disc (of 1 mm in diameter) fixed on a glass rod (2 mm in diameter) was used. Before the deposition of amylin fibril seeds, each mica disc was freshly cleaved. For 3-aminopropyltriethoxysilane (APTES)-modification, mica was immersed in 2 μL of ten-thousand-fold dilution of APTES with water followed by the removal of unbound APTES with 60 μL of water. Amylin fibril seeds for the imaging were prepared by incubation of 60–100 μL of 5 μM amylin in 30 mM sodium acetate, pH 5.5, for 2–3 days followed by sonication with a handheld sonicator (UR-21P, TOMY). The validation of the sonication conditions was performed using AFM images (typical sonication conditions: ten times of ∼0.5 s at the output level 3). 2 μL of fibril seeds was deposited on bare mica or APTES-modified mica followed by incubation for about 5 minutes. After the removal of unbound seeds with 20 μL of 30 mM sodium acetate, pH 5.5, the scanner with the sample stage was set on the scanner holder so that the stage was immersed in 60 μL of 30 mM sodium acetate, pH 5.5, in the sample chamber with the cantilever. After the observation of the fibril seeds, the buffer solution in the chamber was replaced with 5 μM amylin with/without equimolar PMAQA. HS-AFM observation was started just after the replacement of the sample chamber solution. The HS-AFM movie analysis and kymographs were prepared using the ImageJ (NIH) plugin as described elsewhere.405 μM of β-amyloid-1-40 monomers dissolved in 10 mM sodium phosphate, pH 7.4, or 5 μM of amylin monomers dissolved in 30 mM sodium acetate, pH 5.5, were incubated for 24 hours under continuous agitation with/without an equimolar concentration of PMAQA. β-amyloid-1-40 or amylin fibers were prepared by incubating 5 μM of peptide monomers in respective buffers as described above at room temperature for 3 days under continuous agitation. Then, the fibers were sonicated using an ultrasonic bath sonicator for 2 hours at room temperature to generate short fibers, which were used as amyloid seeds. The sonicated fibers were incubated with 5 μM of PMAQA in respective peptide buffers at room temperature for 24 hours under continuous agitation to check the effect of the polymer on the fiber morphology. In addition, to monitor the effect of PMAQA on the self-seeding reaction, sample solutions containing sonicated fibers were incubated with 5 μM monomers of respective peptides for 24 hours in the presence or absence of 5 μM PMAQA. 10 μL of all sample solutions described above were added to a collodion-coated copper grid and incubated for 3 minutes at room temperature followed by three times rinsing with double deionized water. Then, the copper grid was stained with 3 μL of 2% (w/v) uranyl acetate and incubated for 2 minutes followed by three times rinsing with double deionized water. The sample grids were dried overnight under vacuum and used to obtain transmission electron microscopy (TEM) images using a HITACHI H-7650 transmission microscope (Hitachi, Tokyo, Japan) at 25 °C.
MD simulations
The 2D structure of PMAQA (Fig. S1†) was created using ChemDraw 16.0 and exported to Chem3D for energy minimization. The 3D structure of PMAQA and its topology files were created using ATB builder41 from the energy minimized 2D structure for all-atom molecular dynamics (MD) simulation. The solution NMR structures of β-amyloid-1-40 (PDB ID: 2LFM)42 and amylin (PDB ID: 5MGQ)43 were considered as the initial structures for PMAQA interaction analysis; the amino acid sequences of the peptides are shown in Fig. S1.† The MD system was built in the GROMACS44 software package, version 5.0.7 (GROMOS96 54A7 force field45), by placing β-amyloid-1-40 or amylin at the center of a cubic box and the polymer ∼1 nm away from the protein. The MD systems were solvated using SPC/E water (≈1000 kg m−3) and neutralized by adding counter ions followed by energy-minimization using the steepest-descent method. Short NVT (100 ps) followed by 1 ns NPT (310 K and 1 bar) was performed to equilibrate the MD systems. MD simulations were carried out using 3D periodic boundary conditions over a production run of 0.7 and 1 μs for amylin-PMAQA and β-amyloid-PMAQA systems, respectively. MD trajectories were interpreted using visual molecular dynamics46 and images were built using Discovery studio visualizer 3.5. The binding free energy was calculated for amylin-PMAQA and β-amyloid-PMAQA complexes using MM/PBSA by retrieving 500 structures from the last 100 ns MD simulation.47 The GMXAPBS tool was used for the free energy calculation as described elsewhere.48–50Results and discussion
The effect of PMAQA on β-amyloid-1-40 and amylin aggregation kinetics was monitored using ThT based fluorescence assay. ThT aggregation assays performed for 4 days showed opposite activities for β-amyloid-1-40 and amylin aggregation in the presence of PMAQA (Fig. 1 and S2†). As shown in Fig. 1a and b, PMAQA accelerated β-amyloid-1-40 aggregation and slowed down amylin aggregation. At a low polymer concentration, i.e. a peptide:PMAQA molar ratio of 1:0.02 or 1:0.04, a significant difference in the lag-times of β-amyloid-1-40 and amylin aggregation was ascertained (Fig. 1a and b). In the presence of 0.02 molar equivalent of PMAQA, the β-amyloid-1-40 fibrillation growth was observed after two days (blue) as compared to the aggregation after three days for the control sample (red). A slight increase in the polymer concentration, i.e. 0.04 molar equivalent of PMAQA (green), significantly accelerated β-amyloid-1-40 fibrillation with an increased ThT fluorescence intensity (Fig. 1a and S2a†), while a further increase in the polymer concentration to a 1:0.2 or 1:0.5 β-amyloid:PMAQA molar ratio also accelerated the peptide aggregation. But, a relatively lower ThT intensity was observed for 1:0.5 as compared to that obtained for 1:0.2 (or 1:0.04). As shown in Fig. 1a, the observed ThT intensities are higher for all the substoichiometric concentrations of PMAQA than the control (i.e., in the absence of PMAQA). At an equimolar or superstoichiometric concentration of PMAQA (Fig. 1a and S2b†), β-amyloid-1-40 fibrillation was ≈6 times faster than that observed for the control sample.
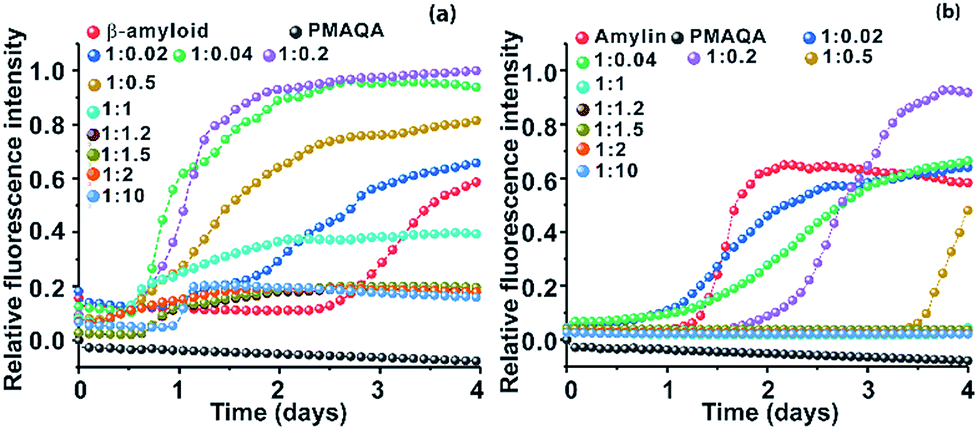 | ||
| Fig. 1 Effect of PMAQA on the aggregation kinetics of β-amyloid-1-40 or amylin. Relative ThT fluorescence of 5 μM β-amyloid-1-40 in 10 mM sodium phosphate, pH 7.4, (a) or amylin in 30 mM sodium acetate, pH 5.5, (b) in the presence of variable PMAQA concentrations at the indicated peptide to polymer molar ratios. The data in both graphs represent the average of ThT values performed in triplicate as shown in Fig. S2.† | ||
Unlike the effect of PMAQA on β-amyloid-1-40 fibrillation, amylin aggregation was slowed down at substoichiometric PMAQA concentrations, i.e. an amylin:PMAQA molar ratio of 1:0.02 or 1:0.04 (Fig. S2c†). Increasing the polymer concentration to a 1:0.2 amylin:PMAQA molar ratio further slowed down the aggregation with a lag-time of over 2 days as compared to its aggregation in ∼1 day in the absence of PMAQA (Fig. 1b). A further increase in PMAQA concentration, i.e. at equimolar polymer:peptide or superstoichiometric PMAQA concentrations, amylin aggregation was significantly slowed down as revealed by the negligibly small ThT fluorescence intensity (Fig. 1b and S2d†). The observed increase in the ThT intensity in both peptides at substoichiometric PMAQA concentrations (peptide:polymer 1:0.5 or lower) indicates the presence of a large number of β-amyloid or amylin fibers. On the other hand, at superstoichiometric PMAQA concentrations (peptide:polymer 1:1 or higher), a substantial decrease in the ThT intensity indicates the presence of a smaller number of fibers. Specifically, the low ThT intensity observed for β-amyloid-1-40 could also indicate the formation of morphologically distinct fibers, i.e. amorphous like aggregates that depict low ThT fluorescence intensity.51
Next, the conformational changes in both peptides in the presence and absence of PMAQA were investigated using far-UV CD experiments. The CD spectra revealed a gradual structural transition (from unfolded to folded) in β-amyloid-1-40 when titrated with a 1:0.01 or 1:0.04 β-amyloid:PMAQA molar ratio as indicated by a reduction in CD molar ellipticity [Θ] and a small change in CD minima at ≈200 nm (Fig. 2a). The reduction in [Θ] indicates β-amyloid-1-40 aggregation and is in line with the observed ThT aggregation (Fig. 1a). A partial helical CD spectrum containing α/β secondary contents of 9.4/26.1% (as estimated by BESTSEL52) was observed at a 1:0.2 β-amyloid:PMAQA molar ratio (Fig. 2a). A further increase in PMAQA concentration to a 1:1 β-amyloid:PMAQA molar ratio marginally increased the β-sheet contents in β-amyloid-1-40 (α/β content of 7.7/28.2%). The shift of CD minima at ≈200 nm and decrease in [Θ] with an increasing concentration of PMAQA indicated its activity in accelerating β-amyloid-1-40 aggregation as observed in the ThT assay (Fig. 1a). Remarkably, unlike β-amyloid-1-40, no significant conformational changes were observed in amylin when titrated with 0.2 to 2 equivalents of PMAQA with a CD minimum centered at ≈200 nm with a very little change in [Θ] that represents a partially folded random-coil rich conformation (Fig. 2b).
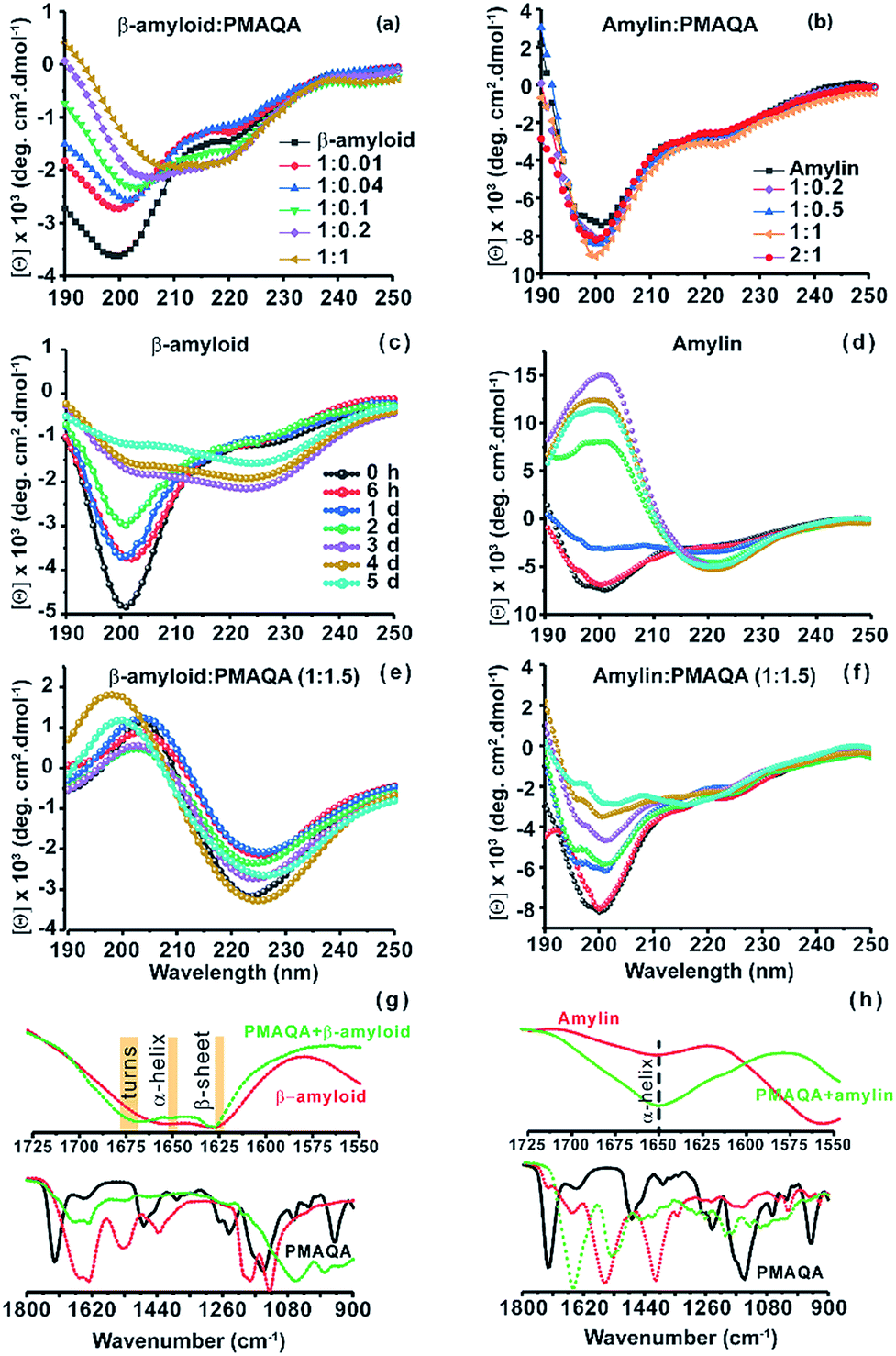 | ||
| Fig. 2 Conformational analysis of β-amyloid-1-40 and amylin with and without PMAQA. Far-UV CD measurements showing the structural changes in 25 μM of β-amyloid-1-40 in 10 mM sodium phosphate, pH 7.4, (a) and in 25 μM of amylin in 30 mM sodium acetate, pH 5.5, (b) in the presence of PMAQA incubated for ∼10 minutes at room temperature at the indicated peptide:PMAQA molar ratios (a and b). Time-lapsed structural changes in 25 μM β-amyloid-1-40 (c and e) and amylin (d and f) in the absence (c and d) and presence (e and f) of 1.5 molar equivalent PMAQA; time intervals used for results presented in c–f are indicated in (c). FT-IR spectra of β-amyloid-1-40 (g) and amylin (h) in the presence (green) and absence (red) of PMAQA obtained after 6 hours; peptide concentration and buffer conditions were as described for the samples used in CD measurements. Changes in the secondary structure of β-amyloid-1-40 are highlighted in yellow in the fingerprint regions (1550–1725 cm−1) (g, top). | ||
We next monitored the stability of the PMAQA induced β-amyloid-1-40 or amylin secondary structures for 5 days by incubating the peptides at room temperature in the absence or presence of 1.5 molar equivalents of PMAQA (Fig. 2c–f). The time-lapse CD spectra of β-amyloid-1-40 or amylin in the absence of PMAQA showed a sequential structural transition from a random-coil rich (negative peak ≈200 nm) to β-sheet structure (Fig. 2c and d). While β-amyloid-1-40 in solution is reported to exhibit a sequential structural change to form cross β-sheet structures over several days (Fig. 2c),53 in the presence of 1.5 molar equivalents of PMAQA a rapid β-sheet induction within several minutes was observed (Fig. 2e). The observed rapid structural change correlates with the ThT aggregation kinetics (Fig. 1a) and indicates a reduction in the level of potential β-amyloid-1-40 oligomers that are reported to be neurotoxic.53,54
Similar to the amyloidogenic properties of β-amyloid-1-40, amylin in solution after 2 days depicted a CD spectrum with a maximum at ≈200 and a minimum at ≈218 nm indicating its transitory state characterized by an increasing percentage of β-sheets (54%) (Fig. 2d). But, unlike β-amyloid-1-40, and as observed in ThT assays for amylin (Fig. 1b), CD spectra showed a relatively slow change in the secondary structure of amylin when incubated with a 1.5 molar excess of PMAQA. The CD minimum was observed at ≈200 nm up to day 3 and the spectra were quantified using BESTSEL (Fig. 2f and Table 1). Secondary structure assessment of PMAQA bound amylin from CD spectra showed a relative increase in α-helix (14.5%) and decrease in β-sheet (21.7%) content as compared to that observed in the absence of PMAQA (Table 1). In addition, an increased percentage of parallel β-sheets (13.4% on day 5) was observed for amylin in the presence of PMAQA over time as compared to 0% (on day 5) in the absence of PMAQA. This observation indicates that PMAQA bound amylin could have both anti-parallel and parallel β-structures. Such observations have been found previously using X-ray crystallography in different segments of amylin (segments 13–18 with parallel and anti-parallel β-structures for segments 16–21, 22–28, and 23–29).55 On the other hand, β-amyloid-1-40 showing a positive CD band at ≈200 nm and a negative CD band at ≈225 nm in the presence of PMAQA indicates the formation of a predominant β-sheet structure (Fig. 2e). The observed CD minimum with a red-shift (from ≈222 on day 1 to ≈225 nm on day 5) in β-amyloid-1-40 correlates with previously observed β-sheet rich supramolecular structures in modified β-amyloid-1-40 and other small peptide aggregates.56,57
| Time | 0 d | 1 d | 2 d | 3 d | 4 d | 5 d |
|---|---|---|---|---|---|---|
| a Antiparallel β-sheet. b Parallel β-sheet. | ||||||
| Amylin in 30 mM sodium acetate, pH 5.5 | ||||||
| α-helix | 0.7 | 1.4 | 0 | 0 | 1.3 | 0.4 |
| βa | 42.1 | 37.5 | 54.0 | 59.0 | 53.1 | 53.0 |
| βb | 0 | 3.9 | 0 | 0 | 0.2 | 0 |
|
||||||
| Amylin+PMAQA in 30 mM sodium acetate, pH 5.5 | ||||||
| α-helix | 14.5 | 9.1 | 10.6 | 5.3 | 5.3 | 6.6 |
| βa | 21.7 | 29.7 | 28.1 | 30.3 | 33.4 | 25.4 |
| βb | 0 | 0 | 0 | 5.7 | 3.7 | 13.4 |
Next, we carried out FT-IR experiments on samples after 6 hours of incubation under similar experimental conditions used for CD measurements to further confirm the observations from CD experiments. As shown by the FT-IR spectra in Fig. 2g, predominant β-sheet structures of β-amyloid-1-40 were observed as indicated by a sharp amide I peak at 1628 cm−1 along with an increasing percentage of turns (at 1675 cm−1) in the presence of PMAQA, which are in qualitative agreement with CD results shown in Fig. 2e. In the case of amylin, FT-IR spectra showed a minor change in the characteristics of the helical band at 1650 cm−1 in the presence of PMAQA (Fig. 2h). This indicates that PMAQA binding restrains the conformation of partially folded amylin.43 The β-sheet rich β-amyloid and partially folded amylin conformations in the presence of PMAQA (Fig. 2) coupled with ThT observations (Fig. 1) indicated the presence of distinct peptide species such as fibers or low-order aggregates. To probe this, SEC profiling was performed to quantify the size distribution of the PMAQA bound peptide species. SEC analysis of β-amyloid-1-40 incubated with a 1.5 molar excess of PMAQA for ∼5 minutes at room temperature showed two different elution profiles. The fractions collected at ∼5 to 12 mL correspond to amyloid fibers or protofibers and those collected at ∼20 to 25 mL are free polymers or low-order or monomeric β-amyloid-1-40 (Fig. S3a†).58 Remarkably, the SEC profile for PMAQA-amylin mixed solution, incubated for a relatively longer time (overnight) at room temperature, exhibited low-order or monomeric amylin and free polymers eluted at ≈20 to 25 mL (Fig. S3b†).59 Taken together, the above described experimental results present a counteractive role of PMAQA in β-amyloid-1-40 and amylin aggregation.
Next, we studied the binding mechanism of PMAQA with β-amyloid-1-40 or amylin using an integrated NMR and MD simulation approach. The amide-NH region of 1H NMR spectra obtained in the absence of PMAQA showed monomer or low-order aggregates for both β-amyloid-1-40 and amylin as indicated by the dispersed NMR peaks (Fig. 3). A substantial change in the 1H NMR spectra of β-amyloid-1-40 was observed at a 1:0.02 β-amyloid:PMAQA molar ratio (Fig. 3, pink trace). Remarkably, the loss of amide-1H peaks of β-amyloid-1-40 was observed when the PMAQA concentration (1:0.05 β-amyloid:PMAQA ratio) was slightly increased (Fig. 4d). This agrees with the PMAQA induced β-amyloid-1-40 aggregation observed in ThT assays. In contrast, the amide peaks of amylin were observed even when titrated with a 1.2 molar equivalent of PMAQA (Fig. 3, blue trace). These NMR findings are in good agreement with the observed conformational transition from CD and ThT based aggregation results (Fig. 1b and 2b). Interestingly, a significant line broadening was observed for the proton peak of the –NR3+ group of PMAQA (at 2.97 ppm) in the presence of β-amyloid-1-40 which indicates the interaction of –NR3+ with β-amyloid-1-40. In contrast, a sharp proton peak for –NR3+ of PMAQA was observed in amylin solution (Fig. 3). This observation most likely indicates the formation of an electrostatic interaction between the cationic PMAQA and anionic β-amyloid-1-40, as β-amyloid-1-40 (at pH = 7.4) and amylin (at pH = 5.5) carry negative (−3) and positive (+4) charges, respectively.60,61 Thus, while the cationic group of PMAQA binds strongly to anionic β-amyloid-1-40, a repulsive force could be expected in the presence of cationic amylin.
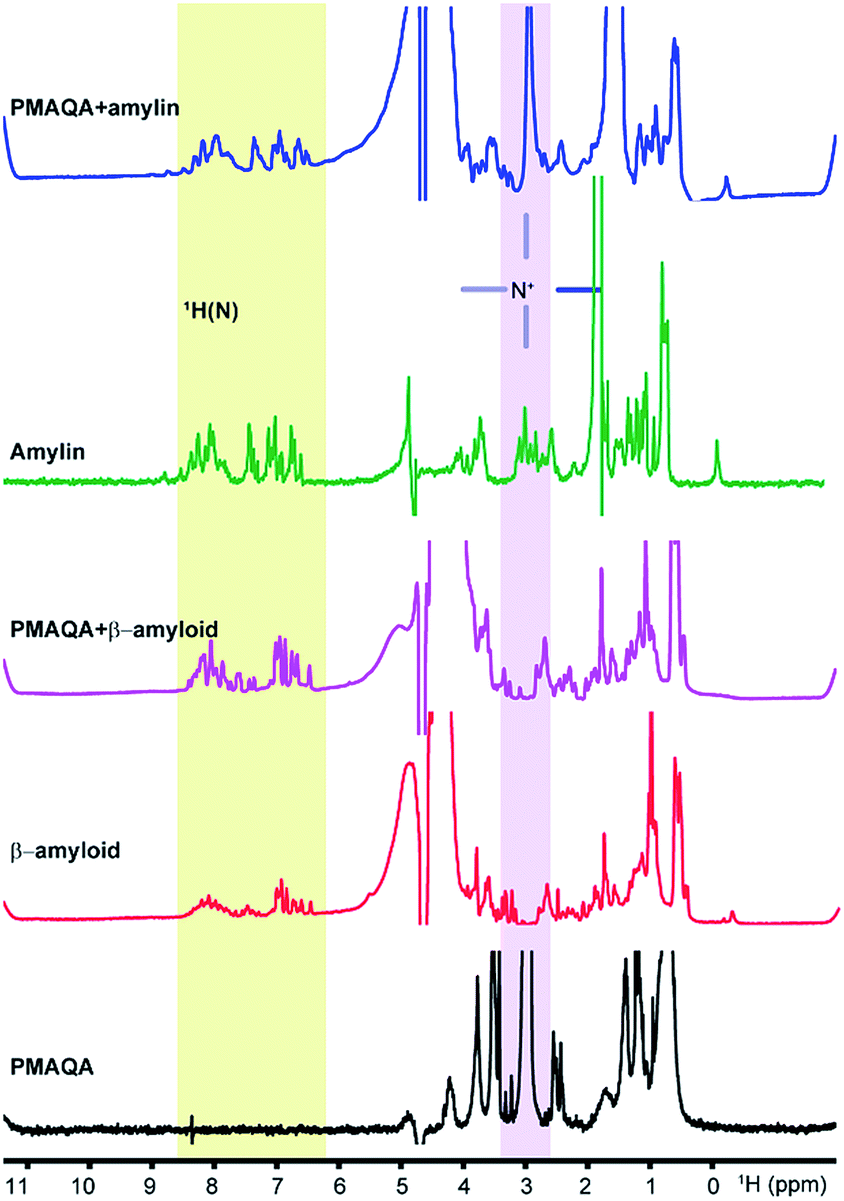 | ||
| Fig. 3 1H NMR spectra of β-amyloid-1-40 or amylin. NMR spectra showing the interaction of PMAQA with β-amyloid-1-40 (60 μM β-amyloid-1-40, 1.2 μM PMAQA dissolved in 10 mM sodium phosphate, pH 7.4) or amylin (50 μM amylin, 60 μM PMAQA dissolved in 30 mM sodium acetate, pH 5.5). The change in the NMR signal intensity of the –NR3+ proton in the PMAQA or protein amide region (H–N) is highlighted. | ||
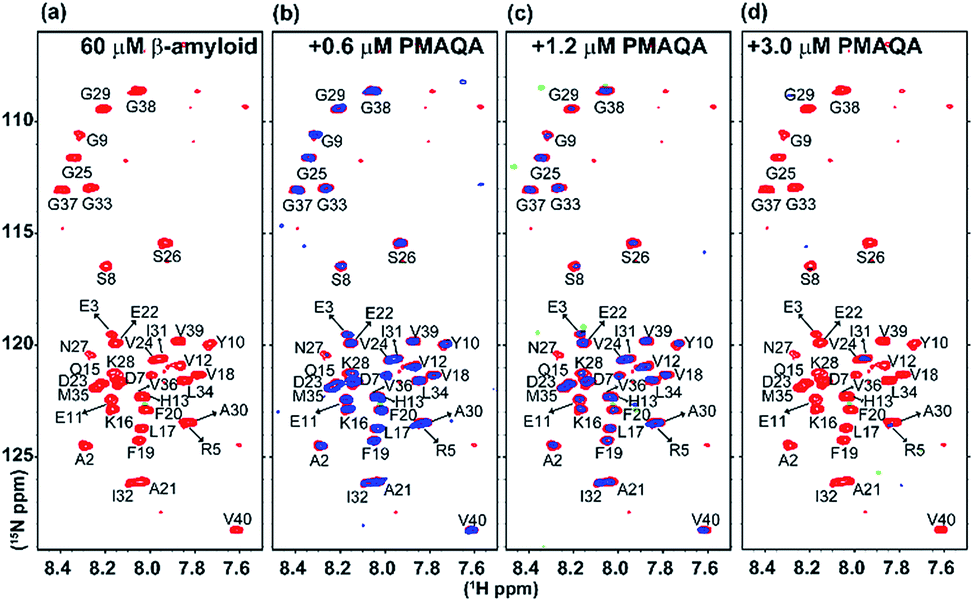 | ||
| Fig. 4 SOFAST-HMQC NMR titration of PMAQA with β-amyloid-1-40. 2D 1H/15N SOFAST-HMQC NMR spectra of β-amyloid-1-40 (60 μM) dissolved in 10 mM sodium phosphate, pH 7.4, (a) in the absence (red) or (b–d) presence of PMAQA (blue) at the indicated concentration (top). NMR spectra were recorded at 10 °C on a 600 MHz Bruker NMR spectrometer. Blue contours are presented with 1.5 times higher values as compared to red contours (in Sparky). | ||
To gain further atomistic insight into the mechanism, β-amyloid-1-40 aggregation was monitored using a 2D 15N/1H SOFAST-HMQC experiment at variable substoichiometric polymer concentrations; since 1H NMR of amylin showed very little change in the amide region at superstoichiometric PMAQA concentrations, further NMR experiments were not carried out on amylin. As shown in Fig. 4a–d, intensity reduction and chemical shift changes were observed for β-amyloid-1-40 residues indicating a PMAQA induced structural rearrangement for β-amyloid-1-40. A substantial loss of signal intensity was observed in β-amyloid-1-40 spanning regions K16-V24 and A30-V40 (as per previously published assignments62) indicates potential PMAQA binding sites on β-amyloid-1-40 at 1:0.01 and 1:0.02 β-amyloid:PMAQA molar ratios (Fig. 4b and c). A further increase in PMAQA concentration (1:0.05 β-amyloid:PMAQA molar ratio) increased the line-broadening and also resulted in the loss of 15N/1H resonances (Fig. 4d). This indicates that PMAQA interaction with β-amyloid-1-40 induces the aggregation of the peptide.
To further explore the binding mechanism of PMAQA with amylin or β-amyloid-1-40 at the structural level, we performed all-atom MD simulation on a time scale of 0.7 or 1 μs, respectively. Structural analysis showed the formation of a substantial number of hydrogen bonds between PMAQA and β-amyloid-1-40 over a time-scale of 1 μs (Fig. S4a†). All-atom MD simulation revealed potential electrostatic interactions or hydrogen bonds between the Glu22 and Asp23 residues of β-amyloid-1-40 and –NR3+ of the polymer (Fig. 5a), whereas no interaction with the charged residues of β-amyloid-1-40 in the N-terminal (Asp1, Glu3 and Asp7) was observed. Other residues such as Leu17, Phe20, Val24, Ser26, Asn27, Ile31, and Ile32 were observed to interact with PMAQA through hydrogen bonding or hydrophobic interactions (Fig. 5a and Table S1†). This correlates with the NMR results that showed a substantial decrease in the NMR signal intensity in β-amyloid-1-40 regions spanning K16-V24 and A30-V40 (Fig. 5c). MD calculations also revealed several intermolecular hydrogen bonds and hydrophobic interactions between amylin and PMAQA (Fig. S4a†). Amylin residues such as Asn21, Ile26, Ser28, Thr30, Asn31, Ser34 and Tyr37 were identified to be involved in intermolecular hydrogen bonding interactions with PMAQA (Fig. 5b and Table S2†). Overall, PMAQA exhibited a greater number of hydrogen bonds with amylin than with β-amyloid-1-40 indicating a relatively stronger binding affinity of PMAQA to amylin (Fig. S4a†).
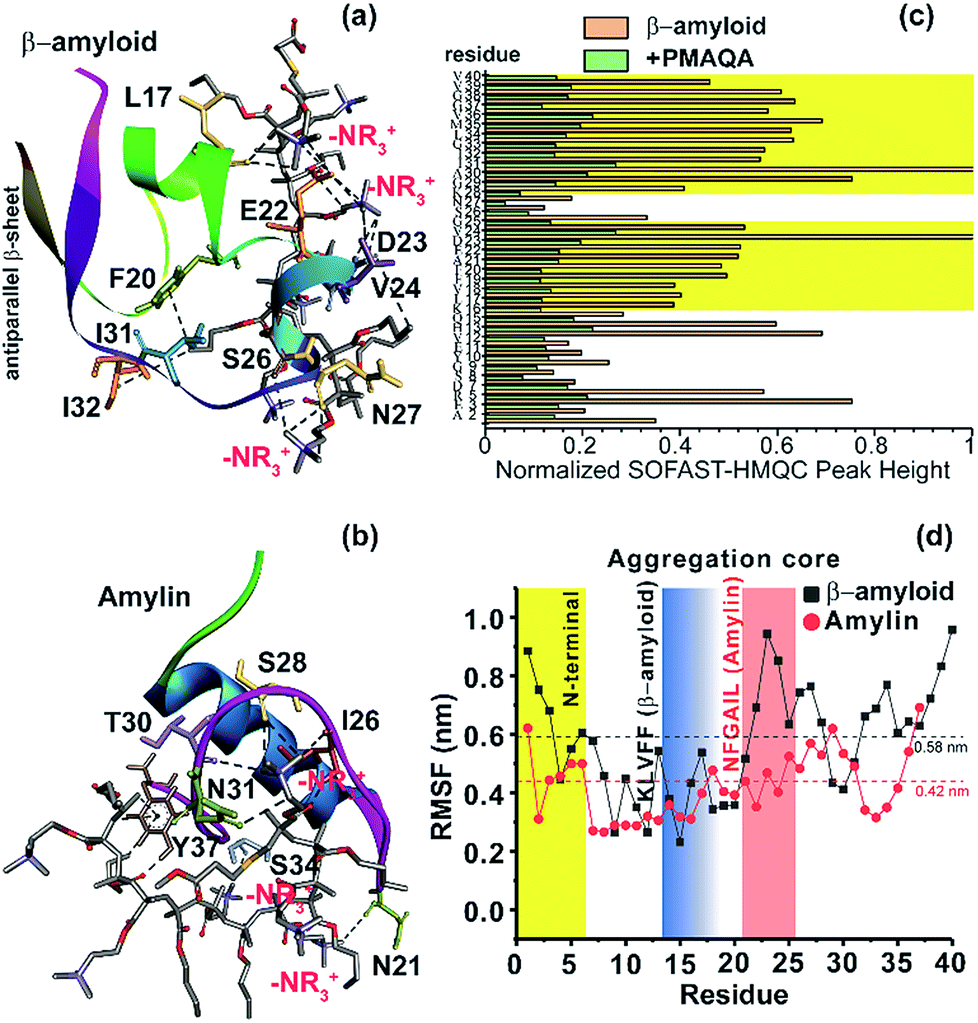 | ||
| Fig. 5 Structural insights into the PMAQA interaction with β-amyloid-1-40 or amylin. MD snapshots showing PMAQA (shown in balls and sticks) interaction with β-amyloid-1-40 (a) or amylin (b) as a cartoon. The PMAQA binding peptide amino acids (shown in sticks) are labelled and hydrogen bonds are shown in black dashed lines in Discovery Studio Visualizer. (c) Peak intensities measured from the 2D SOFAST-HMQC spectra (Fig. 4) of 60 μM β-amyloid-1-40 in the absence or presence of 0.6 μM PMAQA. The yellow area highlights the β-amyloid-1-40 residues with significantly reduced signal intensities. (d) Root mean square fluctuation (RMSF) of residues in β-amyloid-1-40 (grey) or amylin (red) interacting with PMAQA derived from 1 or 0.7 μs MD simulations, respectively. The yellow region indicates a comparatively flexible β-amyloid-1-40 N-terminal domain. The blue and orange regions indicate aggregation core domains of amylin and β-amyloid-1-40, respectively. The average RMSF value is shown using dashed horizontal lines. | ||
Computation of binding energy using MM/PBSA presented distinct energetic parameters that favor PMAQA binding to β-amyloid-1-40 or amylin. The binding energy estimated for PMAQA-β-amyloid and PMAQA-amylin complexes was −16.5 ± 10.1 and −27.4 ± 11.7 kcal mol−1, respectively. The energetic parameter analysis shown in Table 2 indicates that coulombic and van der Waals interactions play a crucial role in PMAQA binding to β-amyloid-1-40, while the polar solvation energy restricts the interaction. On the other hand, PMAQA binding to amylin is favored by the nonpolar component of the solvation free energy and van der Waals interaction, while coulombic interaction disfavors the complex formation (Table 2). This indicates that the opposite charges in β-amyloid-1-40 and amylin are the major factors responsible for their distinguished amyloid aggregation behavior in the presence of PMAQA. The electrostatic interaction or repulsion in β-amyloid-1-40 or amylin interaction with PMAQA, respectively, could contribute to the acceleration or deceleration of amyloid aggregation as observed in previous studies using differently charged nanoparticles.63,64
| Polar contribution | Non-polar contribution | ||||||
|---|---|---|---|---|---|---|---|
| ΔGbinda | ΔGcoulb | ΔGpsc | ΔGpod | ΔGvdwe | ΔGnpsf | ΔGnpg | |
| a Binding free energy. b Coulombic term. c Polar solvation. d Polar contribution. e van der Waals energy. f Nonpolar solvation. g Nonpolar contribution. | |||||||
| β-amyloid + PMAQA | −16.5 ± 10.1 | −521.4 ± 132.7 | 560.0 ± 137.0 | 38.5 | −48.0 ± 7.2 | −7.0 ± 0.8 | −55.1 |
| Amylin + PMAQA | −27.4 ± 11.7 | 84.9 ± 106.2 | −50.0 ± 103.6 | 34.8 | −54.7 ± 7.8 | −7.6 ± 0.9 | −62.3 |
The root mean square deviation (RMSD) of the backbone atoms calculated from 1 μs MD simulation of the β-amyloid-PMAQA system showed an RMSD plateau with an average value ≈11 Å (Fig. S4b†). A relatively small backbone RMSD (average value ≈8.8 Å) was observed for the amylin-PMAQA system indicating a comparatively stable complex formation (Fig. S4c†). The RMSD of PMAQA calculated from all-atom MD simulation depicted a nearly equal RMSD value of ≈6.5 Å for both MD systems (Fig. S4d†). Overall, the stable peptide backbone with an average RMSD of <2 Å over several hundreds of nanoseconds indicates a strong coupling between PMAQA and amylin or β-amyloid-1-40 over the ∼μs time scale MD simulation. Furthermore, the root mean square fluctuation (RMSF) analysis of individual amino acids in β-amyloid-1-40 or amylin highlighted the potential PMAQA interaction regions. The amyloid aggregation core domains in both β-amyloid-1-40 (KLVFF) and amylin (NFGAIL) depicted an RMSF value lower than their corresponding average values as indicated in Fig. 5d. In amylin, residues 7–16 showed the lowest RMSF values that folded to a stable α-helical conformation. This indicates that the PMAQA interaction restrains the structural and dynamic properties of amylin. The tight coupling of PMAQA close to the proximity of the NFGAIL region in amylin followed by restriction in protein structural rearrangement reveals mechanistic insights into the antagonist properties of PMAQA in amylin aggregation. Furthermore, the secondary structure analysis performed using MD trajectories in the amylin-PMAQA complex showed no significant secondary structural change during the MD simulation in amylin (Fig. S5,† bottom). On the other hand, a substantial secondary structural change was observed in β-amyloid-1-40 complexed with PMAQA including the induction of an antiparallel β-sheet along the terminal residues (Fig. 5a and S5,† top). The interaction of PMAQA with the centrally located residues of β-amyloid-1-40 such as Glu22, Asp23, Val24, Ser26 and Asn27 induced a random-coil and helix conformation in β-amyloid-1-40. Structural changes in β-amyloid-1-40 by affecting these central residues by mutation, disruption of the D23-K28 salt-bridge or binding of the hexapeptide of genetic β-amyloid-1-40 variants has been reported previously to be crucial in accelerating β-amyloid-1-40 aggregation and induction of the terminal β-structure.65–68 The charge–charge interaction between PMAQA (–NR3+) and Glu22/Asp23 in β-amyloid-1-40 could interfere with the early oligomer morphology and distribution of large β-amyloid-1-40 aggregates as observed in SEC (Fig. S3†).69 The binding of PMAQA to Glu22 and Val24 also affects the hydrophobic interaction between Val24 and Lys28 and electrostatic interaction between Lys28 and Glu22/Asp23.70
We next monitored the aggregation kinetics of amylin in real time using HS-AFM as the results presented above indicated an antagonistic behavior of PMAQA in amylin aggregation (Fig. 6). The fibrillation kinetics of freshly prepared amylin monomers was accelerated by adding preformed sonicated amylin amyloid seeds as described in the Experimental section. As shown in Fig. 6a (top trace), the addition of amylin monomers to pre-incubated amylin amyloid seeds (at t = 0 s) induced a steady growth of fibers (Movie S1†). The seeding reaction (or fibril growth) and de novo nucleation (binding of additional fibril seeds to the stage) were observed (Fig. 6a). At 1000 s, we observed substantial growth in the fiber morphology; de novo nucleated amylin fibers and several fibers stopped growing after encountering other fibers at their growing end (Fig. 6a and Movie S1†). The growth of the selected amylin fiber (Fig. 6a, rectangle) in the absence of PMAQA is represented as kymographs as a function of time in one direction. The kymograph showed a substantial growth on the free end of the selected amylin fiber (Fig. 6b). These observations agree with the reported observations for other amyloidogenic peptides including β-amyloid.71,72 Remarkably, in the presence of equimolar PMAQA, both amylin fibril growth and de novo nucleation were found to be blocked (Fig. 6c and Movie S2†). In addition, no significant difference in the fiber length and population was observed between 0 s and 1350 s (Fig. 6c). Kymographs of amylin fibrillation in the presence of PMAQA showed no fiber growth. This indicates that PMAQA blocks the recruitment of amylin monomers to the growing ends of amyloid seeds that is needed to proceed the seeding reaction (Fig. 6d). These distinct morphological features observed for both bare mica and APTES-mica suggest that the electrostatic interaction between the sample and the stage did not affect amylin aggregation and interactions with PMAQA. The HS-AFM experimental results, therefore, revealed the inhibitory activity of PMAQA towards amylin aggregation at a molecular-level, which is consistent with other experimental results reported in this study.
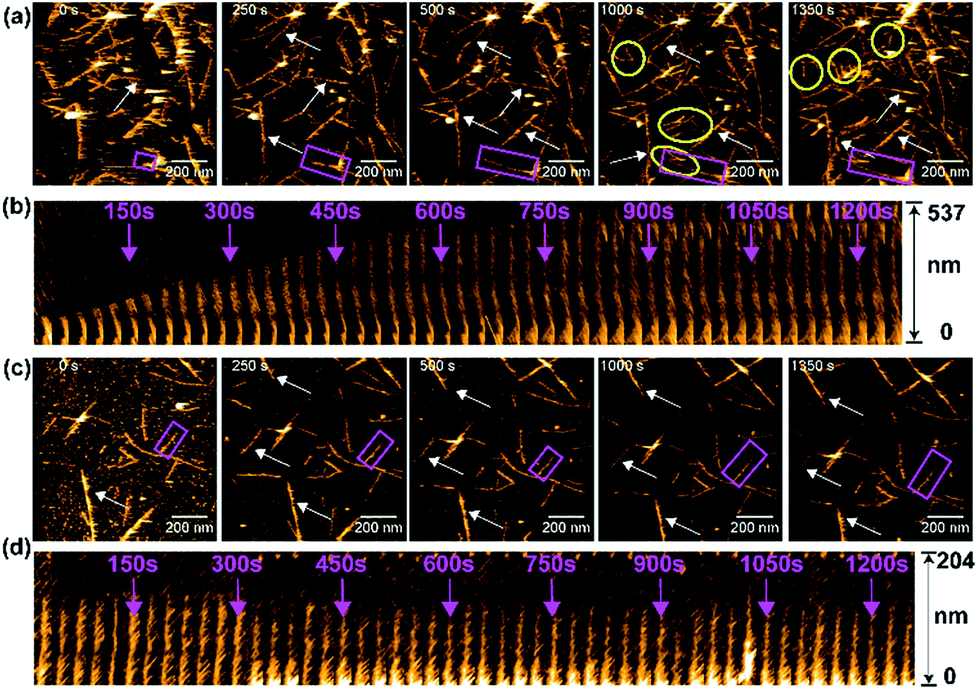 | ||
| Fig. 6 Real-time monitoring of amylin fibrillation using HS-AFM. HS-AFM images show the seeding reaction (fibril growth) of 5 μM amylin monomers in the absence (a) or presence (c) of equimolar PMAQA. These images were extracted at the indicated time intervals from movies S1 and S2 (included in the ESI†). The growth of fibers as a function of time is indicated by arrows. The de novo nucleated amylin fibrils in (a) are shown inside the yellow circles. The kymographs for an individual selected fiber (shown inside pink rectangles in a and c) are shown for amylin in the absence (b) and presence of PMAQA (d). The kymographs show the growth of amylin fibers as a function of time (indicated with pink arrows) in the absence of PMAQA, whereas PMAQA blocks amylin nucleation and seeding processes. The scale bar is 200 nm. | ||
We further characterized the morphology of β-amyloid-1-40 and amylin species present under various sample conditions (see the Experimental section). The TEM image of 5 μM PMAQA showed very small particles (Fig. 7a), whereas freshly dissolved β-amyloid-1-40 monomers (5 μM) after continuous agitation at room temperature for 24 hours showed a fibril morphology (Fig. 7b). On the other hand, β-amyloid-1-40 monomers (5 μM) incubated with an equimolar concentration of PMAQA exhibited amorphous-like aggregates (Fig. 7c). This correlates with the observed ThT fluorescence results that showed a substantial reduction in fluorescence intensity (Fig. 1a). We next examined the effect of PMAQA on β-amyloid-1-40 aggregation in the presence of amyloid seeds. TEM confirmed the formation of short fibrils when sonicating matured fibers for 2 hours (Fig. 7d). We did not observe a significant change in the sonicated fiber morphology incubated with equimolar PMAQA for 24 hours in the absence of peptide monomers (Fig. 7e). However, a remarkable difference in the β-amyloid-1-40 morphology was observed when a self-seeding reaction was performed in the absence or presence of PMAQA (Fig. 7f and g). The self-seeding reaction presented amyloid fiber growth in the absence of PMAQA (Fig. 7f), whereas in the presence of PMAQA short fibers and an amorphous-like fiber morphology were identified (Fig. 7g). Unlike β-amyloid-1-40, amylin monomers (5 μM) incubated with equimolar PMAQA exhibited no fibers (Fig. 7i) which correlates with the ThT and CD results (Fig. 1b and 2b). Moreover, sonicated amylin fibers incubated with 5 μM PMAQA for 24 hours in the absence of monomers showed a relatively thick fibril morphology as compared to the short-sized sonicated fibers (Fig. 7j–l). Interestingly, sonicated amylin fibers in the presence of equimolar peptide monomers and PMAQA showed no fibril growth which correlates with the HS-AFM observations (Fig. 7k). A quantitative analysis of the widths of sonicated fibers (Fig. 7j) using the ImageJ plugin presented an average fiber width of ∼10.79 nm. In the presence of PMAQA, the sonicated fibers depicted an average width of ∼15.69 nm indicating the possibility of PMAQA binding (Fig. 7l). Overall, the TEM analysis showed the formation of distinct morphological species of β-amyloid-1-40 and amylin in the presence or absence of PMAQA. These results further highlighted the opposite behaviours of PMAQA in the aggregation of β-amyloid-1-40 and amylin as observed in other biophysical experiments.
 | ||
| Fig. 7 TEM images of β-amyloid-1-40 and amylin interacting with PMAQA. TEM images of 5 μM PMAQA (a) and 5 μM β-amyloid-1-40 in the absence (b) or presence (c) of an equivalent concentration of PMAQA incubated for 24 hours at room temperature under continuous agitation. TEM images of sonicated β-amyloid-1-40 fibers (d) prepared from matured fibers (see the Experimental section) in the presence of 5 μM PMAQA (e); 5 μM β-amyloid-1-40 monomers (f); and 5 μM PMAQA and 5 μM β-amyloid-1-40 monomers (g) incubated for 24 hours at room temperature under continuous agitation. TEM images of 5 μM amylin in the absence (h) or presence (i) of 5 μM PMAQA incubated for 24 hours at room temperature under continuous agitation. TEM images of sonicated amylin fibers (j) prepared from matured fibers (see the Experimental section) in the presence of 5 μM PMAQA and 5 μM amylin monomers (k) and 5 μM PMAQA (l) incubated for 24 hours at room temperature under continuous agitation. The superscripts “M” and “F” denote the initial species of the peptide as monomers and fibers, respectively, used for the sample preparation. The inset in (l) shows the average width of amylin fibers that were calculated from 10 distinct short amylin fibers selected randomly using the ImageJ plugin from (j) and (l). | ||
Conclusions
In conclusion, we have demonstrated the counter-activities of a cationic PMAQA polymer towards two different amyloidogenic peptides that are linked to AD and T2D. At substoichiometric concentrations, PMAQA showed significant inhibitory activity in amylin aggregation, whereas it significantly accelerated β-amyloid-1-40 aggregation by quickly altering the equilibrium state of β-amyloid-1-40 from an unfolded structure to a β-sheet structure. Our mechanistic study provides insights into the binding of PMAQA to β-amyloid-1-40 or amylin at the atomic level that could be helpful in understanding the modulation of peptide self-assembly and could aid in potential inhibitor designing. We believe that the opposite aggregation kinetics of the two different amyloidogenic proteins in the presence of a cationic polymer delineated in this study are likely to open avenues to test their potential therapeutic activities against an array of amyloid proteins involved in other human amyloid diseases by controlling functionalization of the chemical properties of the polymer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by funds from NIH (AG048934 to A.R.). This work was (in part) performed under the International Collaborative Research Program of Institute for Protein Research, Osaka University, ICR-18-02.References
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 496 CrossRef CAS.
- S. A. Kotler, P. Walsh, J. R. Brender and A. Ramamoorthy, Chem. Soc. Rev., 2014, 43, 6692–6700 RSC.
- C. A. Ross and M. A. Poirier, Nat. Med., 2004, 10, S10–S17 CrossRef PubMed.
- L. M. Young, A. E. Ashcroft and S. E. Radford, Curr. Opin. Chem. Biol., 2017, 39, 90–99 CrossRef CAS PubMed.
- J. C. Sacchettini and J. W. Kelly, Nat. Rev. Drug Discovery, 2002, 1, 267–275 CrossRef CAS PubMed.
- J. Luo, S. K. T. S. Wärmländer, A. Gräslund and J. P. Abrahams, J. Biol. Chem., 2016, 291, 714576 Search PubMed jbc.R116.
- A. Abedini, A. Plesner, P. Cao, Z. Ridgway, J. Zhang, L. H. Tu, C. T. Middleton, B. Chao, D. J. Sartori, F. Meng, H. Wang, A. G. Wong, M. T. Zanni, C. B. Verchere, D. P. Raleigh and A. M. Schmidt, eLife, 2016, 5, e12977 CrossRef PubMed.
- A. Spanopoulou, L. Heidrich, H.-R. Chen, C. Frost, D. Hrle, E. Malideli, K. Hille, A. Grammatikopoulos, J. Bernhagen, M. Zacharias, G. Rammes and A. Kapurniotu, Angew. Chem., Int. Ed., 2018, 57, 14503–14508 CrossRef CAS PubMed.
- Z. Liu, A. Zhang, H. Sun, Y. Han, L. Kong and X. Wang, RSC Adv., 2017, 7, 6046–6058 RSC.
- S.-Y. Hung and W.-M. Fu, J. Biomed. Sci., 2017, 24, 47 CrossRef PubMed.
- A. J. Doig, M. P. Del Castillo-Frias, O. Berthoumieu, B. Tarus, J. Nasica-Labouze, F. Sterpone, P. H. Nguyen, N. M. Hooper, P. Faller and P. Derreumaux, ACS Chem. Neurosci., 2017, 8, 1435–1437 CrossRef CAS PubMed.
- H. Kroth, A. Ansaloni, Y. Varisco, A. Jan, N. Sreenivasachary, N. Rezaei-Ghaleh, V. Giriens, S. Lohmann, M. P. López-Deber, O. Adolfsson, M. Pihlgren, P. Paganetti, W. Froestl, L. Nagel-Steger, D. Willbold, T. Schrader, M. Zweckstetter, A. Pfeifer, H. A. Lashuel and A. Muhs, J. Biol. Chem., 2012, 287, 34786–34800 CrossRef CAS PubMed.
- A. Pithadia, J. R. Brender, C. A. Fierke and A. Ramamoorthy, J. Diabetes Res., 2016, 2016, 204632 Search PubMed.
- A. J. Doig and P. Derreumaux, Curr. Opin. Struct. Biol., 2015, 30, 50–56 CrossRef CAS PubMed.
- A. Granja, I. Frias, A. R. Neves, M. Pinheiro and S. Reis, BioMed Res. Int., 2017, 2017, 5813793 Search PubMed.
- Q. Song, H. Song, J. Xu, J. Huang, M. Hu, X. Gu, J. Chen, G. Zheng, H. Chen and X. Gao, Mol. Pharm., 2016, 13, 3976–3987 CrossRef CAS PubMed.
- M. Robinson, B. Yasie Lee and Z. Leonenko, AIMS Mol. Sci., 2015, 2, 332–358 CAS.
- M. Stefani and S. Rigacci, Int. J. Mol. Sci., 2013, 14, 12411–12457 CrossRef PubMed.
- J. Bieschke, M. Herbst, T. Wiglenda, R. P. Friedrich, A. Boeddrich, F. Schiele, D. Kleckers, J. M. Lopez Del Amo, B. A. Grüning, Q. Wang, M. R. Schmidt, R. Lurz, R. Anwyl, S. Schnoegl, M. Fändrich, R. F. Frank, B. Reif, S. Günther, D. M. Walsh and E. E. Wanker, Nat. Chem. Biol., 2012, 8, 93–101 CrossRef CAS PubMed.
- S. Derrick, R. A. Kerr, Y. Nam, S. B. Oh, H. J. Lee, K. G. Earnest, N. Suh, K. L. Peck, M. Ozbil, K. J. Korshavn, A. Ramamoorthy, R. Prabhakar, E. J. Merino, J. Shearer, J. Lee, B. T. Ruotolo and M. H. Lim, J. Am, Chem. Soc., 2015, 137, 14785–14797 CrossRef PubMed.
- M. Sato, K. Murakami, M. Uno, Y. Nakagawa, S. Katayama, K. I. Akagi, Y. Masuda, K. Takegoshi and K. Irie, J. Biol. Chem., 2013, 288, 23212–23224 CrossRef CAS PubMed.
- J. Kang, S. J. C. Lee, J. S. Nam, H. J. Lee, M. G. Kang, K. J. Korshavn, H. T. Kim, J. Cho, A. Ramamoorthy, H. W. Rhee, T. H. Kwon and M. H. Lim, Chem.–Eur. J., 2017, 23, 1645–1653 CrossRef CAS PubMed.
- B. R. Sahoo, T. Genjo, M. Bekier II, S. N. Cox, A. Stoddard, M. Ivanova, K. Yasuhara, C. Fierke, Y. Wang and A. Ramamoorthy, Chem. Commun., 2018, 54, 12883–12886 RSC.
- Y. Song, E. G. Moore, Y. Guo and J. S. Moore, J. Am. Chem. Soc., 2017, 139, 4298–4301 CrossRef CAS PubMed.
- E. H. Pilkington, M. Lai, X. Ge, W. J. Stanley, B. Wang, M. Wang, A. Kakinen, M.-A. Sani, M. R. Whittaker, E. N. Gurzov, F. Ding, J. F. Quinn, T. P. Davis and P. C. Ke, Biomacromolecules, 2017, 18, 4249–4260 CrossRef CAS PubMed.
- C. Cabaleiro-Lago, O. Szczepankiewicz and S. Linse, Langmuir, 2012, 28, 1852–1857 CrossRef CAS PubMed.
- B. R. Sahoo, T. Genjo, S. J. Cox, A. K. Stoddard, G. M. Anantharamaiah, C. Fierke and A. Ramamoorthy, J. Mol. Biol., 2018, 430, 4230–4244 CrossRef CAS PubMed.
- J. Luo, C. H. Yu, H. Yu, R. Borstnar, S. C. L. Kamerlin, A. Gräslund, J. P. Abrahams and S. K. T. S. Wärmländer, ACS Chem. Neurosci., 2013, 4, 454–462 CrossRef CAS PubMed.
- A. Assarsson, S. Linse and C. Cabaleiro-Lago, Langmuir, 2014, 30, 8812–8818 CrossRef CAS PubMed.
- Y. Li, M. Cao and Y. Wang, J. Phys. Chem. B, 2006, 110, 18040–18045 CrossRef CAS PubMed.
- K. Yasuhara, J. Arakida, T. Ravula, S. K. Ramadugu, B. Sahoo, J.-I. Kikuchi and A. Ramamoorthy, J. Am. Chem. Soc., 2017, 139, 18657–18663 CrossRef CAS PubMed.
- W. Wang, A. Shao, N. Zhang, J. Fang, J. J. Ruan and B. H. Ruan, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- D. Paolino, A. Vero, D. Cosco, T. M. G. Pecora, S. Cianciolo, M. Fresta and R. Pignatello, Front. Pharmacol., 2016, 7, 1–9 Search PubMed.
- K. Garai, S. L. Crick, S. M. Mustafi and C. Frieden, Protein Expr. Purif., 2009, 66, 107–112 CrossRef CAS PubMed.
- M. Dasari, A. Espargaro, R. Sabate, J. M. Lopez Del Amo, U. Fink, G. Grelle, J. Bieschke, S. Ventura and B. Reif, ChemBioChem, 2011, 12, 407–423 CrossRef CAS PubMed.
- P. Schanda and B. Brutscher, J. Am. Chem. Soc., 2005, 127, 8014–8015 CrossRef CAS PubMed.
- T. D. Goddard and D. G. Kneller, Univ. California, San Fr., 2004, vol. 14, p. 15, http://https://www.cgl.ucsf.edu/home/sparky/.
- T. Ando, N. Kodera, E. Takai, D. Maruyama, K. Saito and A. Toda, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12468–12472 CrossRef CAS PubMed.
- T. Uchihashi, N. Kodera and T. Ando, Nat. Protoc., 2012, 7, 1193–1206 CrossRef CAS PubMed.
- T. Watanabe-Nakayama and K. Ono, Methods Mol. Biol., 2018, 1814, 201–212 CrossRef PubMed.
- A. K. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger, P. C. Nair, C. Oostenbrink and A. E. Mark, J. Chem. Theory Comput., 2011, 7, 4026–4037 CrossRef CAS PubMed.
- S. Vivekanandan, J. R. Brender, S. Y. Lee and A. Ramamoorthy, Biochem. Biophys. Res. Commun., 2011, 411, 312–316 CrossRef CAS PubMed.
- D. C. Rodriguez Camargo, K. Tripsianes, K. Buday, A. Franko, C. Göbl, C. Hartlmüller, R. Sarkar, M. Aichler, G. Mettenleiter, M. Schulz, A. Böddrich, C. Erck, H. Martens, A. K. Walch, T. Madl, E. E. Wanker, M. Conrad, M. H. De Angelis and B. Reif, Sci. Rep., 2017, 7, 44041 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- N. Schmid, A. P. Eichenberger, A. Choutko, S. Riniker, M. Winger, A. E. Mark and W. F. Van Gunsteren, Eur. Biophys. J., 2011, 40, 843–856 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- D. Spiliotopoulos, A. Spitaleri and G. Musco, PLoS One, 2012, 7, e46902 CrossRef CAS PubMed.
- B. R. Sahoo, J. Maharana, M. C. Patra, G. K. Bhoi, S. K. Lenka, P. K. Dubey, S. Goyal, B. Dehury and S. K. Pradhan, Colloids Surf., B, 2014, 121, 307–318 CrossRef CAS PubMed.
- B. R. Sahoo, J. Maharana, G. K. Bhoi, S. K. Lenka, M. C. Patra, M. R. Dikhit, P. K. Dubey, S. K. Pradhan and B. K. Behera, Mol. BioSyst., 2014, 10, 1104–1116 RSC.
- Y. Yoshimura, Y. Lin, H. Yagi, Y.-H. Lee, H. Kitayama, K. Sakurai, M. So, H. Ogi, H. Naiki and Y. Goto, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14446–14451 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- Q. Wang, N. Shah, J. Zhao, C. Wang, C. Zhao, L. Liu, L. Li, F. Zhou and J. Zheng, Phys. Chem. Chem. Phys., 2011, 13, 15200 RSC.
- K. Ono, M. M. Condron and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14745–14750 CrossRef CAS PubMed.
- A. B. Soriaga, S. Sangwan, R. MacDonald, M. R. Sawaya and D. Eisenberg, J. Phys. Chem. B, 2016, 120, 5810–5816 CrossRef CAS PubMed.
- V. Castelletto, I. W. Hamley, P. J. Harris, U. Olsson and N. Spencer, J. Phys. Chem. B, 2009, 113, 9978–9987 CrossRef CAS PubMed.
- T. Guterman, M. Kornreich, A. Stern, L. Adler-Abramovich, D. Porath, R. Beck, L. J. W. Shimon and E. Gazit, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- T. J. Esparza, N. C. Wildburger, H. Jiang, M. Gangolli, N. J. Cairns, R. J. Bateman and D. L. Brody, Sci. Rep., 2016, 6, 1–16 CrossRef PubMed.
- Y. Bram, A. Frydman-Marom, I. Yanai, S. Gilead, R. Shaltiel-Karyo, N. Amdursky and E. Gazit, Sci. Rep., 2014, 4, 1–9 Search PubMed.
- A. Assarsson, E. Hellstrand, C. Cabaleiro-Lago and S. Linse, ACS Chem. Neurosci., 2014, 5, 266–274 CrossRef CAS PubMed.
- P. J. Marek, V. Patsalo, D. F. Green and D. P. Raleigh, Biochemistry, 2012, 51, 8478–8490 CrossRef CAS PubMed.
- J. Miguel, U. Fink, M. Dasari, G. Grelle, E. E. Wanker, J. Bieschke and B. Reif, J. Mol. Biol., 2012, 421, 517–524 CrossRef PubMed.
- A. Gladytz, B. Abel and H. J. Risselada, Angew. Chem., Int. Ed., 2016, 55, 11242–11246 CrossRef CAS PubMed.
- M. Wang, A. Kakinen, E. H. Pilkington, T. P. Davis and P. C. Ke, Biomater. Sci., 2017, 5, 485–493 RSC.
- B. K. Yoo, Y. Xiao, D. McElheny and Y. Ishii, J. Am. Chem. Soc., 2018, 140, 2781–2784 CrossRef CAS PubMed.
- Y. Miller, B. Ma and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9490–9495 CrossRef CAS PubMed.
- V. S. Mithu, B. Sarkar, D. Bhowmik, M. Chandrakesan, S. Maiti and P. K. Madhu, Biophys. J., 2011, 101, 2825–2832 CrossRef CAS PubMed.
- S. Chakraborty and P. Das, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed.
- M. Guo, P. M. Gorman, M. Rico, A. Chakrabartty and D. V. Laurents, FEBS Lett., 2005, 579, 3574–3578 CrossRef CAS.
- M. A. Grant, N. D. Lazo, A. Lomakin, M. M. Condron, H. Arai, G. Yamin, A. C. Rigby and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16522–16527 CrossRef CAS PubMed.
- T. Watanabe-Nakayama, K. Ono, M. Itami, R. Takahashi, D. B. Teplow and M. Yamada, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5835–5840 CrossRef CAS PubMed.
- P. E. Milhiet, D. Yamamoto, O. Berthoumieu, P. Dosset, C. le Grimellec, J. M. Verdier, S. Marchal and T. Ando, PLoS One, 2010, 5, e13240 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05771k |
| This journal is © The Royal Society of Chemistry 2019 |