DOI:
10.1039/C9SC00545E
(Edge Article)
Chem. Sci., 2019,
10, 3987-3993
Rhodium(III)-catalyzed diverse [4 + 1] annulation of arenes with 1,3-enynes via sp3/sp2 C–H activation and 1,4-rhodium migration†
Received
30th January 2019
, Accepted 21st February 2019
First published on 26th February 2019
Abstract
Nitrogen-rich heterocyclic compounds have a profound impact on human health. Despite the numerous synthetic methods, diversified, step-economic, and general synthesis of heterocycles remains limited. C–H bond functionalization catalyzed by rhodium(III) cyclopentadienyls has proven to be a powerful strategy in the synthesis of diversified heterocycles. Herein we describe rhodium(III)-catalyzed sp2 and sp3 C–H activation-oxidative annulations between aromatic substrates and 1,3-enynes, where alkenyl-to-allyl 1,4-rhodium(III) migration enabled the generation of electrophilic rhodium(III) π-allyls via remote C–H functionalization. Subsequent nucleophilic trapping of these species by various sp2-hybridized N-nucleophiles delivered three classes (external salts, inner salts, and neutral azacycles) of five-membered azacycles bearing a tetrasubstituted saturated carbon center, as a result of [4 + 1] annulation with the alkyne being a one-carbon synthon. All the reactions proceeded under relatively mild conditions with broad substrate scope, high efficiency, and excellent regioselectivity. The synthetic applications of this protocol have also been demonstrated, and experimental studies have been performed to support the proposed mechanism.
Introduction
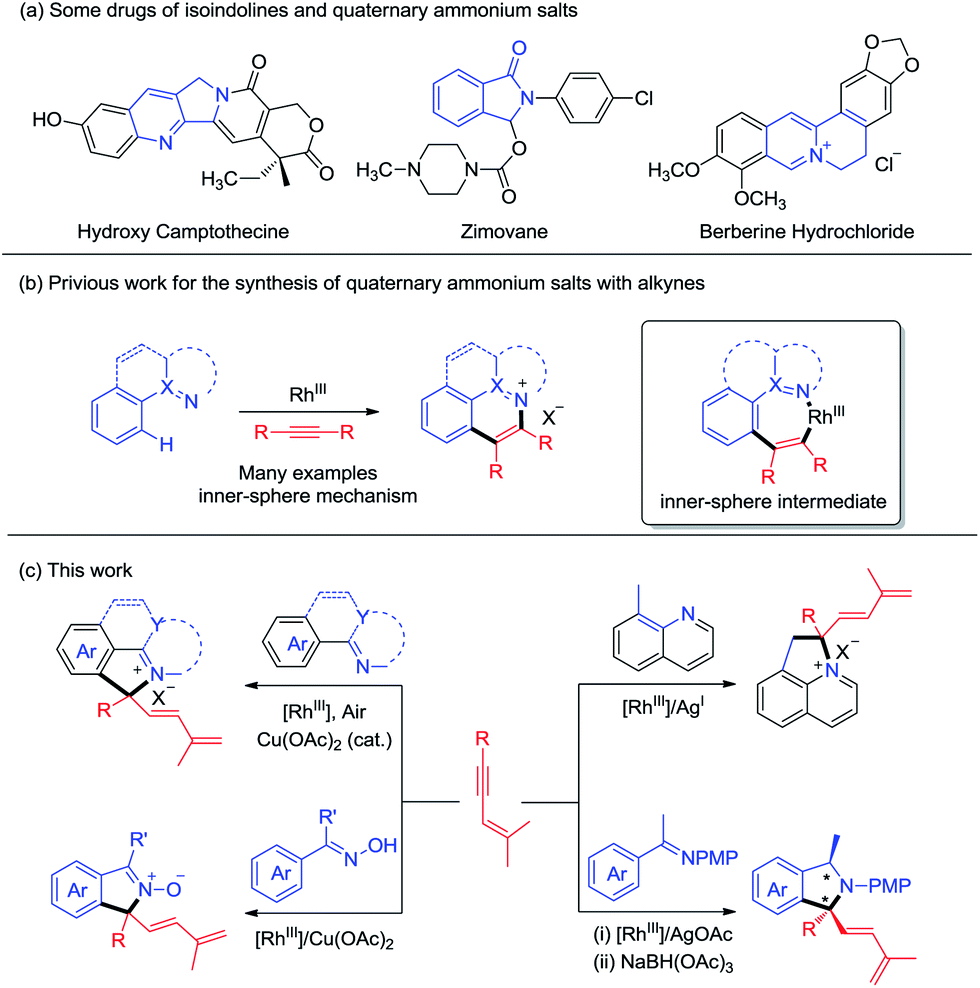
Nitrogen-containing heterocyclic compounds are prevalent in natural products and bioactive molecules. They are also found in a large number of drugs used to combat a broad range of diseases (Scheme 1a). Advances in transition-metal catalyzed C–H functionalization have significantly streamlined the synthesis of a plethora of heterocyclic molecules.1 In many cases, the heteroatom directing group participated in the annulation reaction besides offering chelation assistance, which calls for sufficient interactions between the metal center and the directing group. In this context, C–H bond activation catalyzed by high valent Rh(III) complexes has stood out as a powerful strategy for step-economical construction of C–C bonds and has become one of the increasingly important strategies in organic syntheses.2 In these systems, a large scope of directing groups can be accommodated to ensure high activity and selectivity of the C–H bond. In annulation reactions, development of mild and straightforward C–H activation processes assisted by a multifunctional directing group is especially attractive, allowing realization of molecular diversity and synthetic versatility.3 On the other hand, alkynes are widely used as a typical unsaturated coupling partner in numerous annulation reactions, and in most cases they react as a two-carbon synthon.4 To move beyond this limitation, the Lam group pioneered excellent work in oxidative coupling of arenes bearing a nucleophilic directing group and 1,3-enynes,5,6 which occurred via 1,4-rhodium migration and Rh(III) allyl intermediates, leading to [n + 3] and [n + 1] annulation in most cases. Still, the [n + 1] type annulation with the alkyne being a C1 synthon is still rare,5a,c where the nucleophilic directing group has been limited to 1,3-dione, acidic OH groups, and imide groups, and the corresponding C(sp3)–H activation has not been realized.
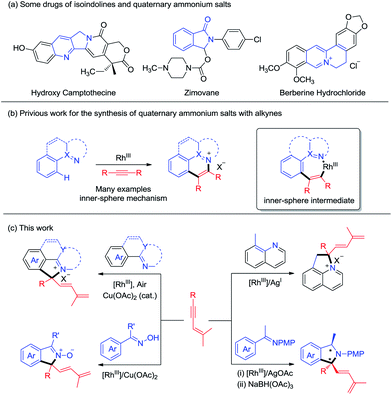 |
| | Scheme 1 Rh(III)-catalyzed oxidative annulation of 1,3-enynes or alkynes. (a) Some drugs with isoindoline or the quaternary ammonium motif. (b) Quaternary ammonium salts formed via coupling with alkynes. (c) Synthesis of diversified heterocycles bearing a quaternary carbon center via coupling of arenes and 1,3-enynes. | |
On the other hand, quaternary ammonium salts are versatile building blocks for a number of naturally occurring products and functional materials.7Although a number of methods for the synthesis of such ionic compounds are available, many are limited by the lack of generality, limited functional group tolerance, and lengthy synthetic procedures.8 Recently, quaternary ammonium salts have been synthesized through sp2 C–H activation of arenes and annulations with alkynes or diazo substrates in the presence of stoichiometric amounts of metal oxidants/additives, as has been pioneered by Cheng and others. While quaternary ammonium salts could be synthesized through the [n + 2] reaction with alkynes, the synthesis of quaternary ammonium salts9 bearing a quaternary carbon center is limited because of the challenge of C(tertiary)–N reductive elimination,4,10 an inner-sphere reaction pathway (Scheme 1b). However, it is challenging to construct a quaternary carbon center through an outer-sphere mechanism. Herein, we report diverse synthesis of four classes of nitrogen-containing heterocycles via C–H activation, 1,4-rhodium(III) migration, and allyl-to-allyl rearrangement. Importantly, quaternary ammonium salts can be readily accessed via Rh(III)-catalyzed sp2 and sp3 C–H functionalization with Cu(OAc)2/air as the oxidant. Besides ionic products, zwitterionic and neutral N-containing heterocycles bearing a quaternary carbon can also be accessed when oximes and ketimines are employed as the arene source. All the coupling systems have been realized with high regio- and chemoselectivity (Scheme 1c).
Results and discussion
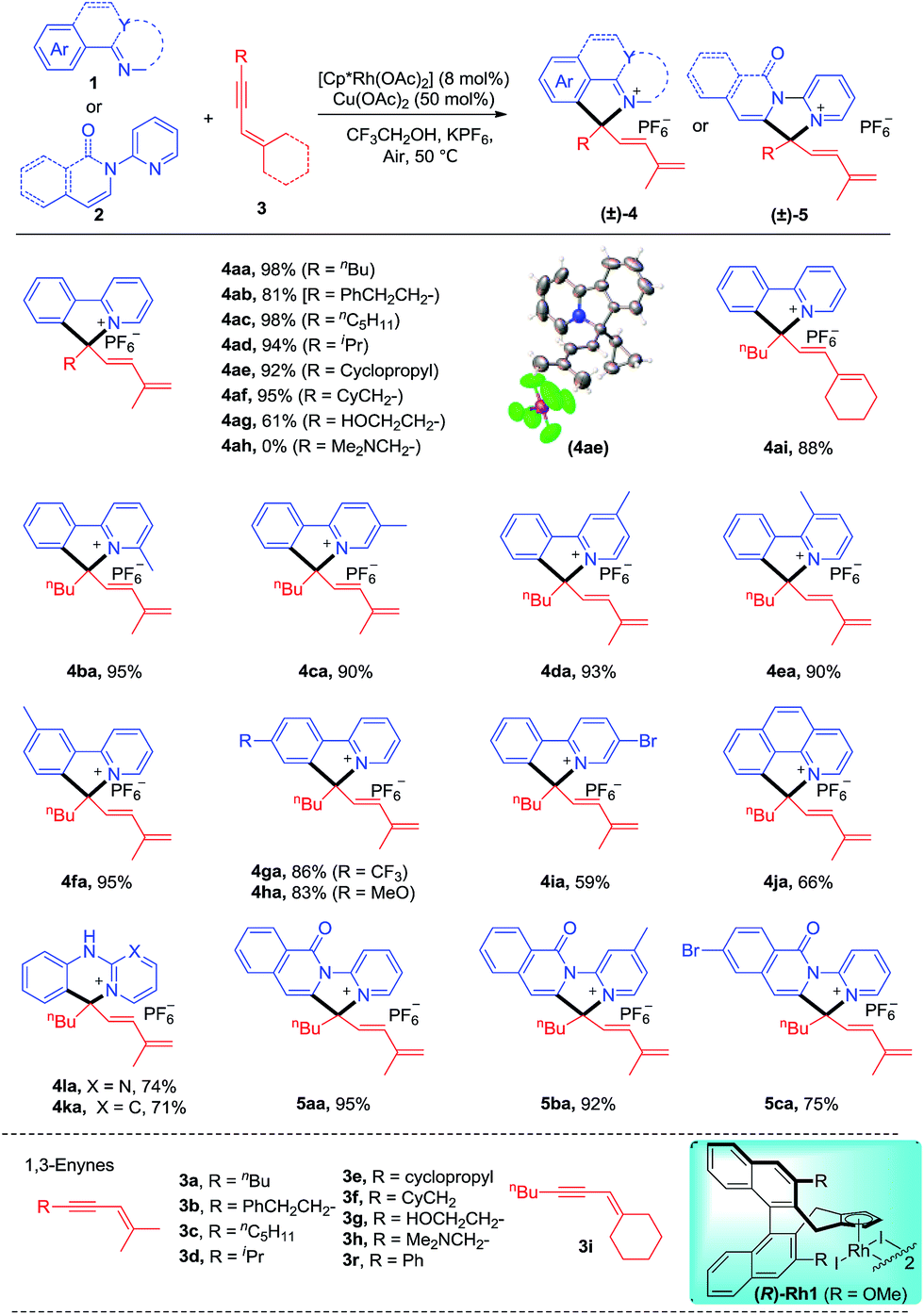
We commenced our studies by exploring the reaction parameters of the coupling of 2-phenylpyridine (1a) and 1,3-enyne (3a, see ESI Table 1†). An excellent yield of quaternary ammonium salt 4aa was obtained with Cp*Rh(OAc)2 as a catalyst, Cu(II)/air as the oxidant, and KPF6 as the anion source under mild conditions. With the optimized conditions in hand, we next investigated the scope and generality of this coupling system (Scheme 2). Various 1,3-enynes were tolerated (4aa-4ai, 61–98%), and the identity of product 4ae has been confirmed by X-ray crystallography (CCDC 1861504†). However, 1,3-enyne 3h containing an alkylamino moiety showed poor reactivity. The phenyl-substituted 1,3-enyne 3r reacted with poor regio- and chemoselectivity, and the [4 + 2] annulation side product was isolated in 45% yield.4m 2-Phenylpyridines bearing Me, electron-donating, electron-withdrawing, and bromo groups were fully tolerated (4ba-4ia, 59–95%). We also investigated preliminarily the enantioselectivity of the reactions of 1a and 3a using chiral (R)-Rh1, and so far, only <5% of 4aa was obtained under standard conditions. The reaction also worked well for benzo[h]quinoline, and the product 4ja was isolated in 66% yield. The arene was not limited to 2-arylpyridines. Thus, the coupling for N-phenylpyrimidin-2-amine and N-phenylpyridin-2-amine afforded the ionic products 4la and 4ka in good yields, respectively. Moreover, several N-pyridylisoquinolones were also viable substrates, providing the desired products (5aa-5ca) in good to excellent yields (75–95%). In all cases, the well-explored [n + 2] annulation side products were not detected.
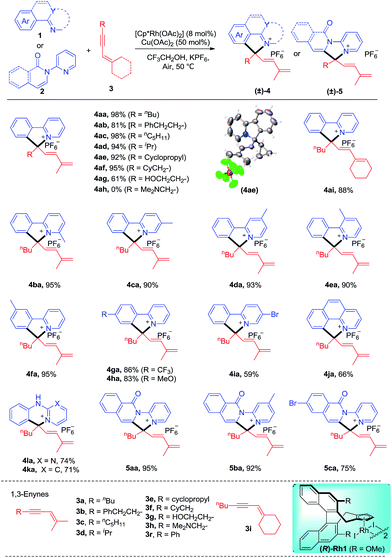 |
| | Scheme 2 Scope of the coupling of 2-phenylpyridines with 1,3-enynes. Reaction conditions: arene 1 or 2 (0.2 mmol), enyne 3 (0.22 mmol), [Cp*Rh(OAc)2] (8 mol%), Cu(OAc)2 (50 mol%), and KPF6 (0.5 mmol) in CF3CH2OH (2.0 mL) under air, 50 °C, monitored by TLC, isolated yields. See general procedure A. | |
To better define the scope of this reaction system, we next extended the reaction system to 8-methylquinolines (Scheme 3). To our delight, the quaternary ammonium salt 7aa was obtained in 82% yield via benzylic C–H activation, and AgOAc and AgSbF6 were identified as the optimal oxidant and anion source (see ESI Table 2†). The scope of this sp3 C–H coupling system was examined under the optimal conditions. The scope of the 1,3-enynes is reasonably broad (7aa-7af, 52–82%) although several heteroatom-functionalized enynes (3g and 3h) were inactive. Various substituted 8-methylquinolines bearing methyl, halogen, and methoxy groups at the 5- and 6-positions all reacted with moderate to good yields (7ba-7ka, 53–83%). 7-Substituted and 3,5-disubstituted quinolines also reacted smoothly but with slightly lower yields (7la-7ma, 46–52%)
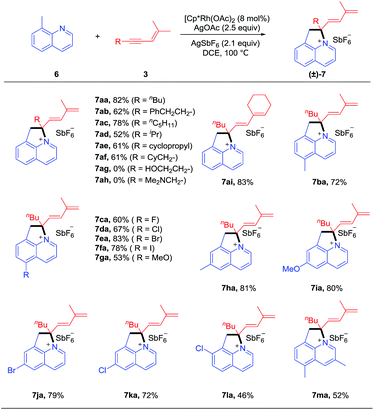 |
| | Scheme 3 Scope of the coupling of 8-methylquinolines with 1,3-enynes. Reaction conditions: 6 (0.2 mmol), 3 (0.3 mmol), [Cp*Rh(OAc)2] (8 mol%), AgSbF6 (0.2 mmol), AgOAc (0.3 mmol), DCE (2.0 mL), monitored by TLC, isolated yields. see general procedure B. | |
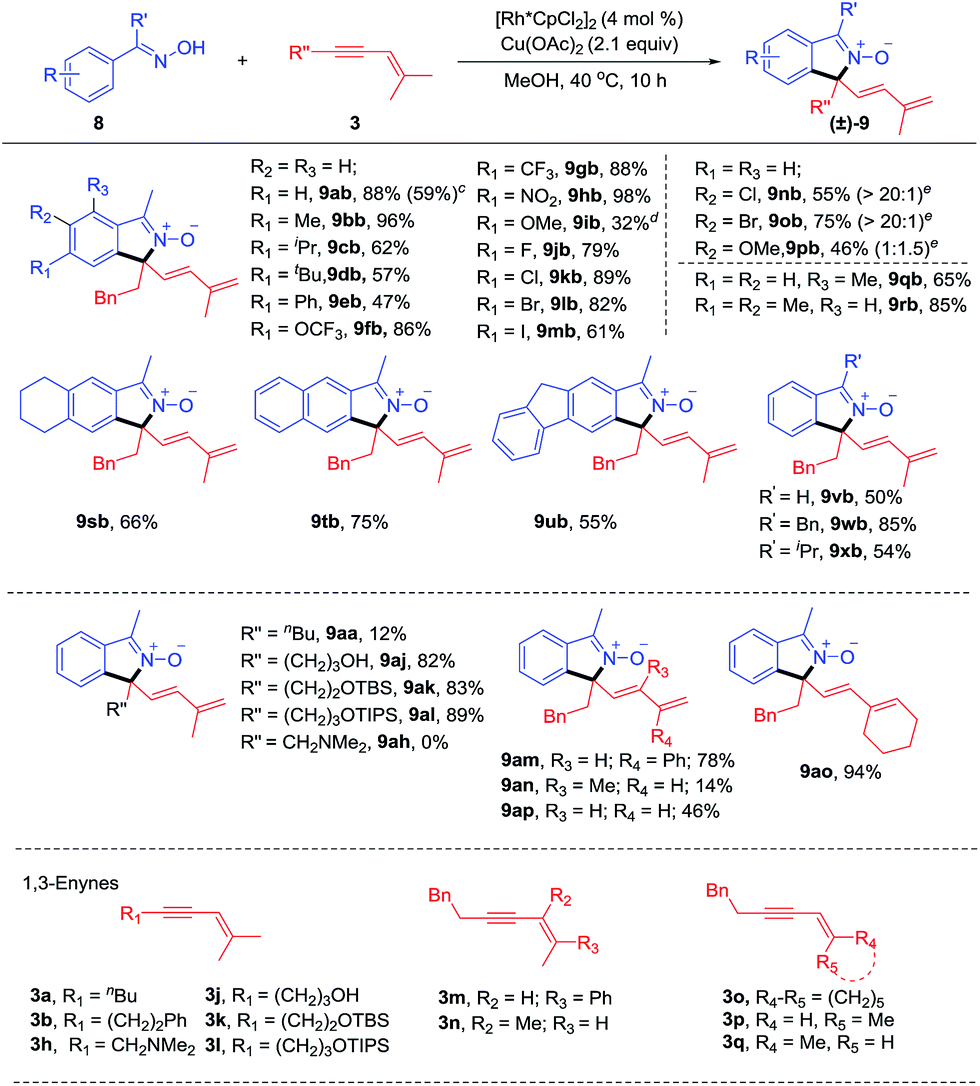
To our delight, the arene substrates of this [4 + 1] coupling reaction system could be smoothly extended to oximes (Scheme 4) and ketimines (Scheme 5, see ESI Table 3†).11 The coupling of readily available ketoximes afforded zwitterionic isoindole 2-oxides. The generality of this system was examined for oximes with different substituents at the para position using enyne 3b as a coupling reagent. Alkyl-substituted oximes gave good to excellent yields (57–96%, 9ab-9db) while relatively lower efficiency (9ib) was found when an electron-donating group (OMe) was present. Furthermore, an isoquinoline 9ib′, resulting from redox-neutral [4 + 2] annulation, was also detected as a by-product.4g Introduction of electron-withdrawing groups, such as –OCF3 (9fb), –CF3 (9gb), and –NO2 (9hb) all resulted in high efficiency, indicative of tolerance of electronic effect. We next evaluated the compatibility of halogen groups, and 9jb-9mb were delivered in good yields. Moreover, the reaction also proceeded smoothly when various meta substituents were present (9nb-9pb, 46–75%). High regioselectivity at the less hindered site was obtained for meta Cl- and Br-substituted oximes, while nearly equal amounts of two regioisomeric products were obtained for the meta MeO-substituent. Besides, introduction of ortho Me (9qb) was also tolerated. Doubly substituted arenes at the meta and para positions were also tolerated, providing products 9rb-9ub in 55–85% yields. Other oximes 8v, 8w and 8x also worked well to yield 9vb-9xb in good yields (50–85%). The ketoxime (8a) was next evaluated in reaction with a range of 1,3-enynes. 1,3-Enynes with the n-butyl substituent at the alkyne terminus gave an inferior result (9aa, 12%), probably due to the poor reactivity of such substrates, while unprotected or silyl-protected alcohol substituents were tolerated, affording the N-oxide in high yields (9aj-9al, 82–89%). However, alkylamino-substituted 1,3-enyne 3h showed poor reactivity. Variation of the group trans to the alkyne from phenyl (3m) to methyl (3a) was also successful. We also examined 1,3-enyne 3n that contains a trans butenyl group, giving high E selectivity albeit with lower yield (9an, 14%). Furthermore, 1,3-enyne 3o containing a cyclohexyl group was also an effective coupling partner with 94% yield (9ao). To confirm whether the allylic hydrogen atom cis to the alkyne is necessary, ketoxime 8a was allowed to react with 1,3-enynes 3p and 3q. It was found that reaction with 3p gave the [4 + 1] annulation product in 46% yield. However, no product was obtained when 3q was employed as a coupling partner. Those results demonstrated that allylic hydrogen cis to the alkyne in the 1,3-enyne is crucial for this reaction, which is consistent with a 1,4-Rh migration pathway.
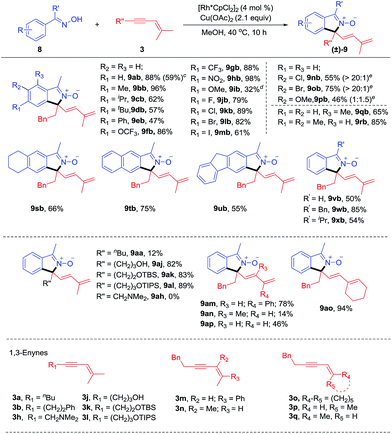 |
| | Scheme 4 Scope of the coupling of oximes with 1,3-enynes.a,b aReaction conditions A: 8 (0.20 mmol), 3 (0.22 mmol), [Rh*CpCl2]2 (4.0 mol%), Cu(OAc)2 (2.1 equiv.), MeOH (2 mL) at 40 °C under N2 for 10 h. bIsolated yields. cReaction was performed with 4 mmol of 8 at 2.5 mol% catalyst loading. dIsoquinoline 9ib′, a [4 + 2] annulation product, was also isolated in 10% yield (see ESI†). eregioselectivity. See general procedure C. | |
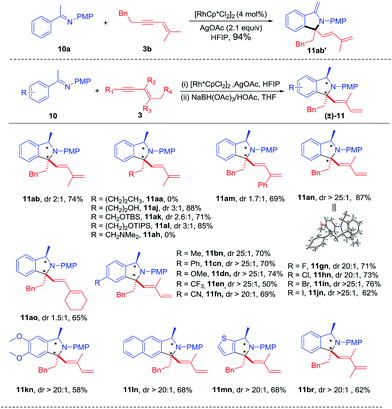 |
| | Scheme 5 Oxidative annulation of ketimines with 1,3-enynes and sequential hydrogenation. Reaction conditions: imine 10 (0.20 mmol), enyne 3 (0.22 mmol), [RhCp*Cl2]2 (4.0 mol%), AgOAc (2.1 equiv.), HFIP (2 mL) at 100 °C under N2 for 10 h; NaBH(OAc)3 (2.5 equiv.)/HOAc (20 equiv.) was sequentially added and was kept for 30 min at room temperature, followed by quenching with NaOH solution. Isolated yields. See general procedure D. | |
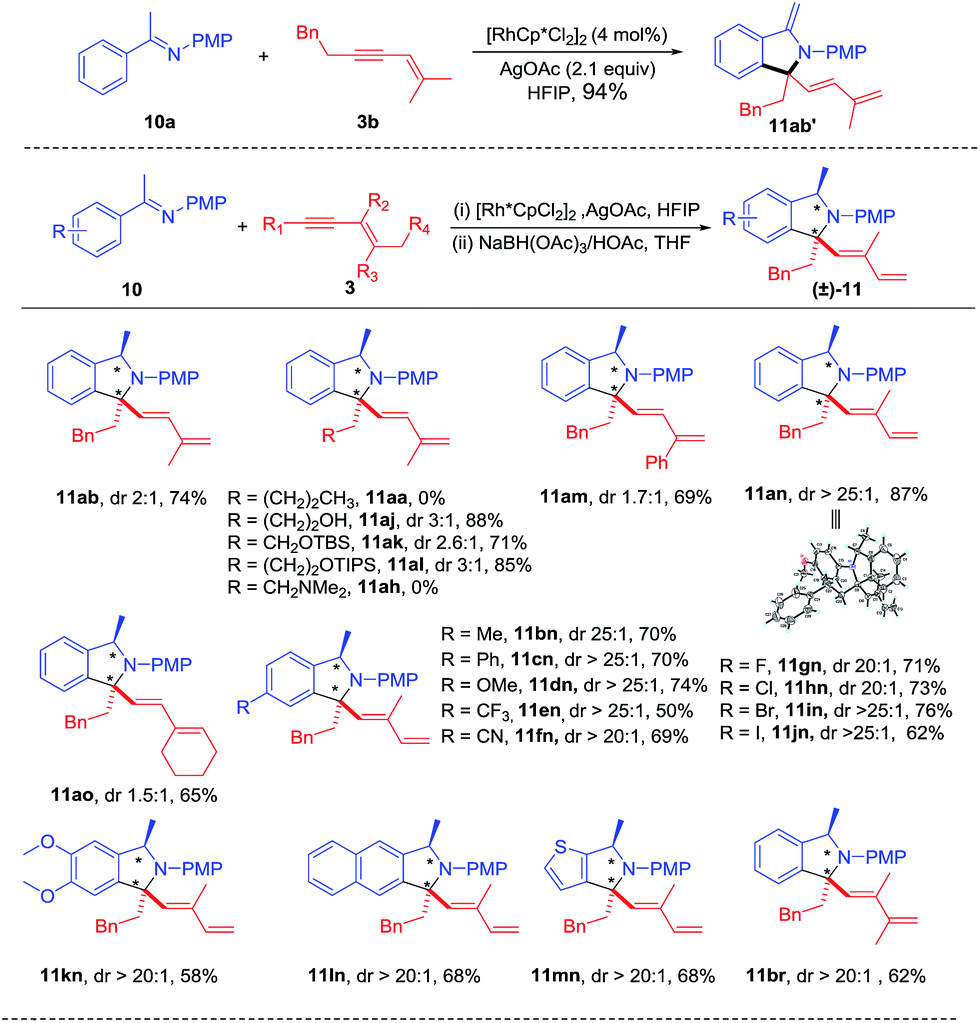
Extension of the oxime to ketimine substrate was next explored. We reasoned that the oxidative [4 + 1] annulation may afford an iminium salt. However, using a ketimine of acetophenone in the presence of a base may lead to further deprotonation of the putative iminium species, generating an enamine that is probably not air-stable. Indeed, reaction of ketoimine 10a and 1,3-enyne 3b under streamlined conditions delivered 11ab′ in excellent yield as an air-sensitive oil (see ESI Fig. 9†). To facilitate product isolation, intermediate 11ab′ was hydrogenated using NaBH(OAc)3,12 which provided stable amine 11ab in 74% total yield albeit with low diastereoselectivity (Scheme 5). Next, various 1,3-enynes were tested through this two-step process. It was found that 1,3-enynes terminated with an n-butyl or alkylamino group failed to react (11aa and 11ah), while several other 1,3-enynes coupled in good yields but with low diastereoselectivity (11aj-11am, 11ao). Delightfully, the coupling with 1,3-enyne containing a trans butenyl group (3n) provided excellent diastereoselectivity and high yield (11an). Therefore, the introduction of a methyl substituent at the alkenyl carbon had a positive effect on the diastereoselectivity in the hydrogenation step. Meanwhile, the identity of 11an was unambiguously confirmed by X-ray crystallography (CCDC 1457168†). Next, various electron-donating, electron-withdrawing, and halogen substituents at the para position of phenyl were examined by coupling with 3n, from which >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and moderate to good yields were obtained (11bn-11jn). In addition, the arene substrates have been extended to disubstituted and heteroaryl ketimines, leading to 11kn-11mn in high diastereoselectivity and good yields. Besides, a trimethyl-substituted 1,3-enyne was also evaluated in the reaction with ketamine 10a, affording 11br with good yield and diastereoselectivity.
:1 dr and moderate to good yields were obtained (11bn-11jn). In addition, the arene substrates have been extended to disubstituted and heteroaryl ketimines, leading to 11kn-11mn in high diastereoselectivity and good yields. Besides, a trimethyl-substituted 1,3-enyne was also evaluated in the reaction with ketamine 10a, affording 11br with good yield and diastereoselectivity.
Synthetic applications
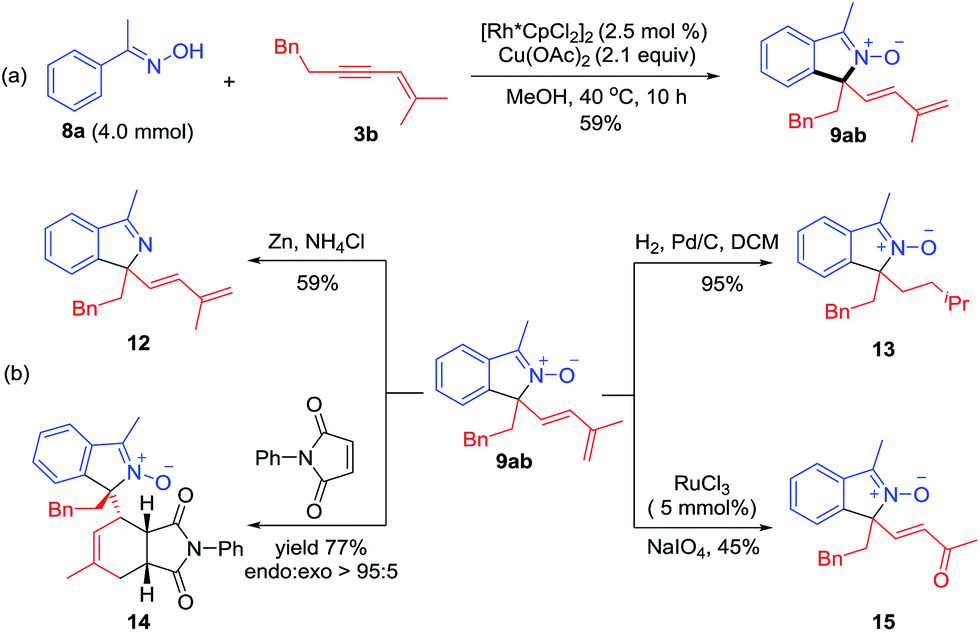
Several derivatization reactions of an N-oxide product 9ab have been performed to briefly demonstrate the synthetic utility of the coupling system. First, a gram-scale synthesis of 9ab has been realized in 59% yield under reduced catalyst loading (Scheme 6a). Reduction of 9ab by Zn delivered the corresponding isoindole 12 in 59% yield (Scheme 6b). Furthermore, hydrogenation of conjugated diene using Pd/C as a catalyst gave 13 in 95% yield with retention of the N–O group (Scheme 6b). Diels–Alder reaction of 1,3-diene 9ab and N-phenylmaleimide in toluene at 80 °C proceeded smoothly to give adduct 14 in 77% yield with >95:5 endo:exo selectivity. In addition, 9ab underwent smooth oxidative cleavage when treated with NaIO4 to deliver α,β-unsaturated ketone 15 in 45% yield (Scheme 6b).
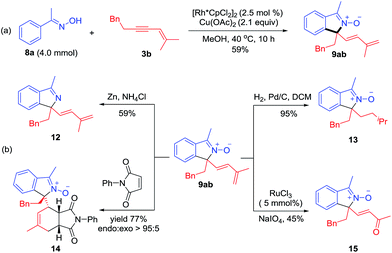 |
| | Scheme 6 Applications of the coupling reaction. (a) Reaction on a gram scale. (b) Derivations of a coupled product through chemoselective reduction, Diels–Alder reaction and oxidative cleavage. | |
Mechanistic investigations
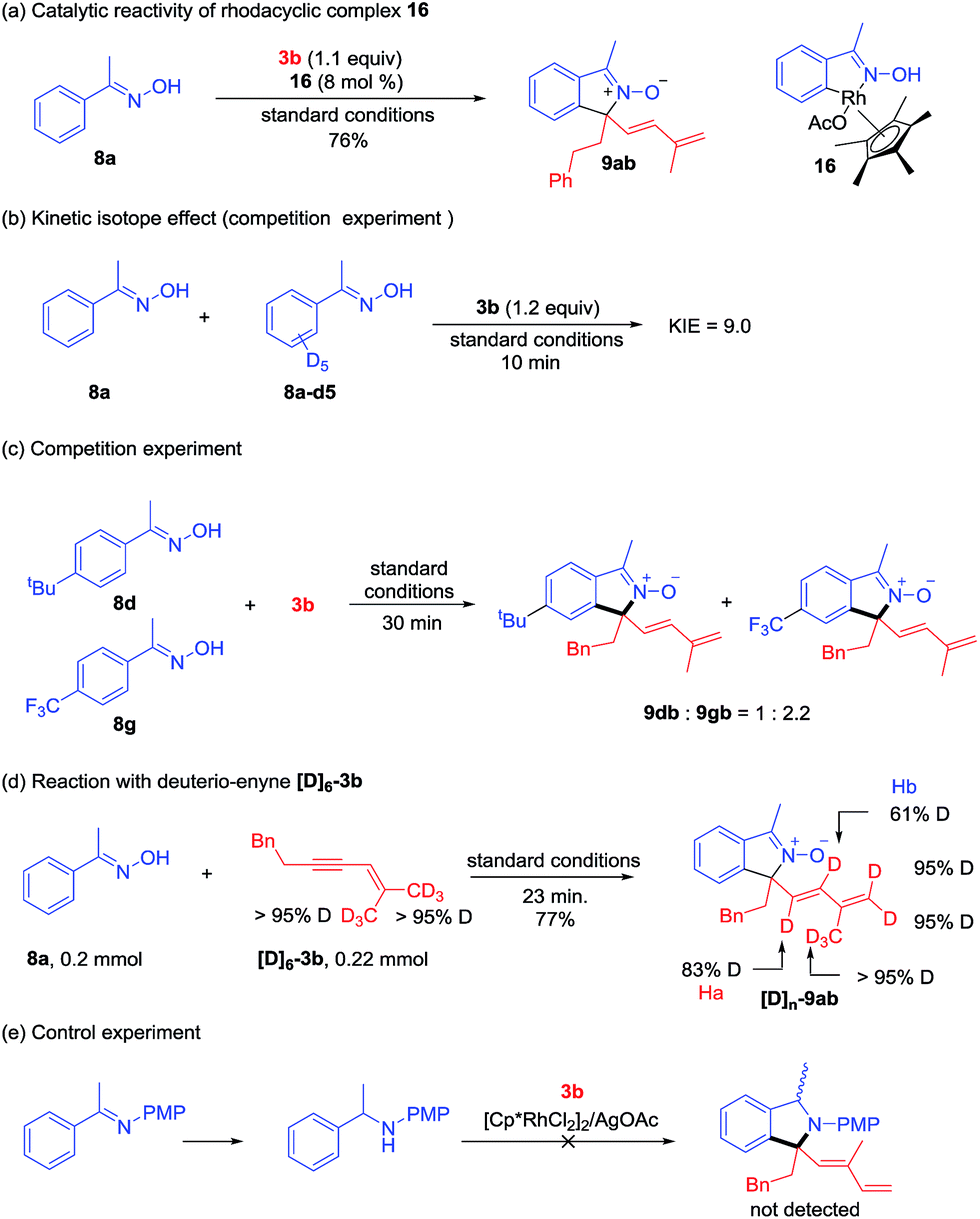
A series of experiments have been conducted to probe the reaction mechanism. Rhodacyclic complex 16 has been prepared from cyclometalation of oxime 8a, and it proved to be an active catalyst for the coupling of 8a and 3b (Scheme 7a), indicating relevancy of C–H activation. A kinetic isotope effect (KIE) value of 9.0 was then obtained in the competitive coupling of 8a and 8a-d5 with 3b under the standard conditions at a low conversion (Scheme 7b). A large KIE value was also obtained in the competitive coupling between 6a and 6a-d3 with 3a (see ESI Fig. 5†). These results indicated that cleavage of the C–H bond is likely involved in the turnover-limiting step of all the above coupling systems. When an equimolar mixture of 8d and 8g was allowed to competitively couple with 3b, products 8dn and 8gb were obtained in a 1:2.2 ratio, indicating that an electron-poor oxime reacted at a slightly higher rate (Scheme 7c).
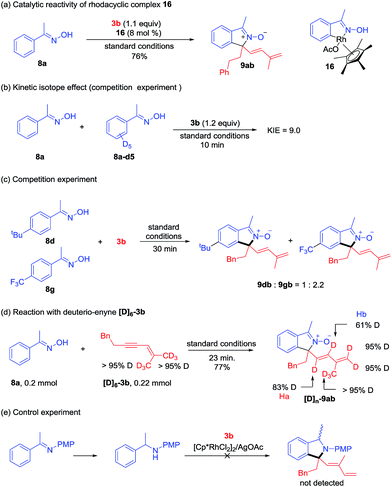 |
| | Scheme 7 Mechanistic studies. (a) Catalytic reactivity of a rhodacyclic intermediate. (b) Kinetic isotope effect. (c) Competition experiment. (d) Reaction with deuterio-enyne [D]6-3b. (e) Control experiment. | |
To understand the 1,4-Rh migration process, a reaction of 8a and hexadeuterated 1,3-enyne 3b-d6 was then conducted (Scheme 7d). Observation of partial deuteration (83% D) at the alkenyl carbon close to the quaternary center and slight loss of deuterium at the alkene terminus positions (95% D) suggested relevancy of reversible 1,4-Rh migration and this migration may occur under acetate-assistance (Ha), and this result is consistent with those in Lam's work.5,6 The partial deuteration (61% D) of Hb may suggest reversible protonolysis at the allylic position in our system (see ESI Fig. 10† for another proposed mechanism).13 Interestingly, the total number of deuterium atoms in [D]n-9ab is greater than that in 3b, and this may result from additional H/D exchange with the [D]6-3b. Additionally, a control experiment (Scheme 7e) revealed that an NH benzyl amine failed to react under similar conditions, indicating that the reaction had to be performed by a two-step process.
On the basis of mechanistic studies and previous reports,14 a plausible catalytic cycle is proposed in Scheme 8 for the representative coupling of 8a and 3b. Starting from an active [Cp*Rh(OAc)2] species (A), cyclorhodation of oxime 8a gives a five-membered rhodacycle B. Coordination of the incoming 1,3-enyne 3b and regioselective migratory insertion of Rh–C(aryl) afford a Rh(III) alkenyl intermediate C, and the regioselective insertion of this step is largely controlled by electronic effects of the biased alkyne.4m Subsequently, reversible 1,4-Rh(III) migration occurs under acetate-assisted concerted metalation–deprotonation to generate intermediate D, and subsequent protonolysis may produce E. Allyl-to-allyl rearrangement of F generates a π-allylrhodium species G, and nucleophilic attack of the directing group at π-allyl carbon provides the conjugate acid of the [4 + 1] annulation product, deprotonation of which delivers the final product 9ab. In the case of 2-phenylpyridine and 8-methylquinoline, the resulting ionic product is stable enough for isolation. The Rh(I) intermediate generated from the nucleophilic attack is reoxidized by Cu(II)/O2 to regenerate the active rhodium(III) species for the next catalytic cycle.
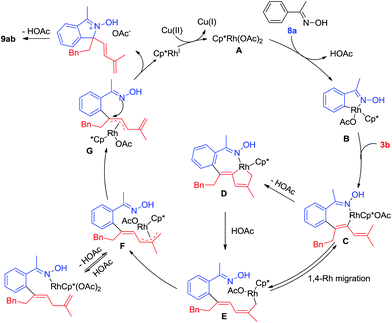 |
| | Scheme 8 Proposed mechanism. | |
Conclusions
In summary, we have realized Rh(III)-catalyzed diversified oxidative annulation of 2-arylpyridines, 8-methylquinolines, oximes, and ketimines with 1,3-enynes through alkenyl-to-allyl 1,4-rhodium(III) migration. The C–N coupling/annulation occurred via the nucleophilic attack of the sp2-hybridized N-nucleophiles. Various ionic, zwitterionic, and neutral N-containing five-membered azacycles bearing a quaternary carbon center have been efficiently synthesized. Mechanistic studies including KIE and competition experiments have been performed and supported the proposed mechanism. Further studies on the C–H activation of other arenes that highlight the unique role of 1,3-enynes are currently underway in our laboratories.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
The NSFC (No. 21525208 and 21801067), research fund from Henan Normal University (5101034011009), the Education Department of Henan Province Natural Science Research Program (18A150010), and start-up fund from Henan Normal University (qd17108) are gratefully acknowledged.
Notes and references
-
(a) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731–1770 CrossRef CAS PubMed;
(b) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed;
(c) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., 2009, 48, 5094–5115 CrossRef CAS PubMed;
(d) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed;
(e) T. W. Lyons and M. S. Sanford, Chem. Rev., 2011, 110, 1147–1169 CrossRef PubMed;
(f) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed;
(g) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC;
(h) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed;
(i) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC;
(j) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC;
(k) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC;
(l) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed;
(m) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2014, 41, 5588–5598 RSC;
(n) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173–6214 RSC;
(o) X.-X. Guo, D.-W. Gu, Z.-X. Wu and W.-B. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed;
(p) L. Yang and H. M. Huang, Chem. Rev., 2015, 115, 3468–3517 CrossRef CAS;
(q) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
-
(a) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed;
(b) N. Kuhl, N. Schroder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443–1460 CrossRef CAS;
(c) G.-Y. Song and X.-W. Li, Acc. Chem. Res., 2015, 48, 1007–1020 CrossRef CAS PubMed.
-
(a) Y. Yang, K. Li, Y. Cheng, D. Wan, M. Li and J. You, Chem. Commun., 2016, 52, 2872–2884 RSC;
(b) T. Piou and T. Rovis, Acc. Chem. Res., 2018, 51, 170–180 CrossRef CAS PubMed;
(c) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908–6909 CrossRef CAS PubMed;
(d) F. W. Patureau, T. Besset, N. Kuhl and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2154–2156 CrossRef CAS PubMed;
(e) B. J. Li, H. Y. Wang, Q. L. Zhu and Z. J. Shi, Angew. Chem., Int. Ed., 2012, 51, 3948–3952 CrossRef CAS PubMed;
(f) M.-B. Zhou, R. Pi, M. Hu, Y. Yang, R. J. Song, Y. Z. Xia and J. H. Li, Angew. Chem., Int. Ed., 2014, 53, 11338–11341 CrossRef CAS PubMed;
(g) T. Zhou, Y. W. Wang, B. Li and B. Q. Wang, Org. Lett., 2016, 18, 5066–5069 CrossRef CAS PubMed;
(h) R. T. Stemmler and C. Bolm, Adv. Synth. Catal., 2007, 349, 1185–1198 CrossRef CAS.
-
(a) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed;
(b) T. Satoh and M. Miura, Org. Lett., 2007, 9, 1407–1409 CrossRef PubMed;
(c) D. A. Colby, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 3645–3651 CrossRef CAS PubMed;
(d) L. Li, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2008, 130, 12414–12419 CrossRef CAS PubMed;
(e) D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474–16475 CrossRef CAS PubMed;
(f) N. Umeda, H. Tsurugi, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2008, 47, 4019–4022 CrossRef CAS PubMed;
(g) N. Guimond and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 12050–12051 CrossRef CAS PubMed;
(h) P. C. Too, Y.-F. Wang and S. Chiba, Org. Lett., 2010, 12, 5688–5691 CrossRef CAS PubMed;
(i) D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326–18339 CrossRef CAS PubMed;
(j) K. Muralirajan, K. Parthasarathy and C.-H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 4169–4172 CrossRef CAS PubMed;
(k) N. Umeda, K. Hirano, T. Satoh, N. Shibata, H. Sato and M. Miura, J. Org. Chem., 2011, 76, 13–24 CrossRef CAS PubMed;
(l) H. Yan, H.-L. Wang, X.-C. Li, X.-Y. Xin, C.-X. Wang and B.-S. Wan, Angew. Chem., Int. Ed., 2015, 54, 10613–10617 CrossRef CAS PubMed;
(m) M. P. Huestis, L. Chan, D. R. Stuart and K. Fagnou, Angew. Chem., Int. Ed., 2011, 50, 1338–1341 CrossRef CAS PubMed;
(n) D. Ghoraia and J. Choudhury, Chem. Commun., 2014, 50, 15159–15162 RSC;
(o) D. Ghorai and J. Choudhury, ACS Catal., 2015, 5, 2692–2696 CrossRef CAS;
(p) D. L. Davies, C. E. Ellul, S. A. Macgregor, C. L. McMullin and K. Singh, J. Am. Chem. Soc., 2015, 137, 9659–9669 CrossRef CAS PubMed;
(q) P. Gandeepan and C. -H. Cheng, Chem.–Asian J., 2016, 11, 448–546 CrossRef CAS PubMed;
(r) J. L. Mascareñas, Angew. Chem., Int. Ed., 2016, 55, 11000–11019 CrossRef PubMed;
(s) C.-Q. Wang, L. Ye, C. Feng and T.-P. Loh, J. Am. Chem. Soc., 2017, 139, 1762–1765 CrossRef CAS PubMed.
-
(a) D. J. Burns and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 9931–9935 CrossRef CAS PubMed;
(b) D. J. Burns, D. Best, M. D. Wieczysty and H. W. Lam, Angew. Chem., Int. Ed., 2015, 54, 9958–9962 CrossRef CAS PubMed;
(c) J. D. Dooley and H. W. Lam, Chem.–Eur. J., 2018, 24, 4050–4054 CrossRef CAS PubMed;
(d) S. E. Korkis, D. J. Burns and H. W. Lam, J. Am. Chem. Soc., 2016, 138, 12252–12257 CrossRef CAS PubMed.
-
(a) H. B. Hepburn and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 11605–11610 CrossRef CAS PubMed;
(b) M. Callingham, B. M. Partridge, W. Lewis and H. W. Lam, Angew. Chem., Int. Ed., 2017, 56, 16352–16356 CrossRef CAS PubMed;
(c) B. M. Partridge, M. Callingham, W. Lewis and H. W. Lam, Angew. Chem., Int. Ed., 2017, 56, 7227–7232 CrossRef CAS PubMed.
-
(a) J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed;
(b) J. Fortage, C. Peltier, F. Nastasi, F. Puntoriero, F. Tuyèras, S. Griveau, F. Bedioui, C. Adamo, I. Ciofini and S. Campagna, J. Am. Chem. Soc., 2010, 132, 16700–16713 CrossRef CAS PubMed.
-
(a) A. Núñez, A. M. Cuadro, J. Alvarez-Builla and J. J. Vaquero, Org. Lett., 2009, 9, 2977–2980 CrossRef PubMed;
(b) T. Fukutani, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Commun., 2009, 5141–5143 RSC;
(c) K. S. Stowers, C. Kevin, K. C. Fortner and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 6541–6544 CrossRef CAS PubMed.
-
(a) J. Jayakumar, K. Parthasarathy and C. H. Cheng, Angew. Chem., Int. Ed., 2012, 51, 197–220 CrossRef CAS PubMed;
(b) D. Zhao, Q. Wu, X. Huang, F. Song, T. Lv and J. You, Chem.–Eur. J., 2013, 19, 6239–6244 CrossRef CAS PubMed;
(c) N. Senthilkumar, P. Gandeepan, J. Jayakumar and C. H. Cheng, Chem. Commun., 2014, 50, 3106–3108 RSC;
(d) N. S. Upadhyay, J. Jayakumar and C. H. Cheng, Chem. Commun., 2017, 53, 2491–2494 RSC;
(e) Y. R. Han, S.-H. Shim, D.-S. Kim and C.-H. Jun, Org. Lett., 2018, 20, 264–267 CrossRef CAS PubMed;
(f) J. Jayakumar and C.-H. Cheng, Chem.–Eur. J., 2016, 22, 1800–1804 CrossRef CAS;
(g) D. Sucunza, A. M. Cuadro, J. Alvarez-Builla and J. J. Vaquero, J. Org. Chem., 2016, 81, 10126–10135 CrossRef CAS PubMed;
(h) Y. Zhao, S. Li, X. Zheng, J. Tang, Z. She, G. Gao and J. You, Angew. Chem., Int. Ed., 2017, 56, 4286–4289 CrossRef CAS PubMed;
(i) G.-Y. Zhang, L. Yang, Y.-Y. Wang, Y.-J. Xie and H.-M. Huang, J. Am. Chem. Soc., 2013, 135, 8850–8853 CrossRef CAS.
- X. Yang, S. Liu, S. Yu, L. Kong, Y. Lan and X. Li, Org. Lett., 2018, 20, 2698–2701 CrossRef CAS PubMed.
-
(a) Z. Shi, D. C. Koester, M. Boultadakis-Arapinis and F. Glorius, J. Am. Chem. Soc., 2013, 135, 12204–12207 CrossRef CAS PubMed;
(b) Q. Wang, F. Wang, X. Yang, X. Zhou and X. Li, Org. Lett., 2016, 18, 6144–6147 CrossRef CAS PubMed.
- W. Zhang, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2017, 56, 5336–5340 CrossRef CAS PubMed.
-
(a) A. Lerchen, T. Knecht, M. Koy, J. B. Ernst, K. Bergander, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 15248–15252 CrossRef CAS PubMed;
(b) T. A. F. Nelson and S. B. Blakey, Angew. Chem., Int. Ed., 2018, 57, 14911–14915 CrossRef CAS PubMed.
-
(a) S. Ma and Z. Gu, Angew. Chem., Int. Ed., 2005, 44, 7512–7517 CrossRef CAS PubMed;
(b) Y. Ikeda, K. Takano, S. Kodama and Y. Ishii, Chem. Commun., 2013, 49, 11104–11106 RSC;
(c) Y. Ikeda, K. Takano, M. Waragai, S. Kodama, N. Tsuchida, K. Takano and Y. Ishii, Organometallics, 2014, 33, 2142–2145 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1861504 and 1457168. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00545e |
| ‡ D. B. and J. X. contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jintao
Xia‡
bd,
Fangfang
Song
a,
Xueyan
Li
a,
Bingxian
Liu
a,
Jintao
Xia‡
bd,
Fangfang
Song
a,
Xueyan
Li
a,
Bingxian
Liu