
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnnihilator dimers enhance triplet fusion upconversion†
Andrew B.
Pun
 a,
Samuel N.
Sanders
b,
Matthew Y.
Sfeir
cd,
Luis M.
Campos
*a and
Daniel N.
Congreve
*b
a,
Samuel N.
Sanders
b,
Matthew Y.
Sfeir
cd,
Luis M.
Campos
*a and
Daniel N.
Congreve
*b
aDepartment of Chemistry, Columbia University, New York, New York 10027, USA. E-mail: lcampos@columbia.edu
bRowland Institute at Harvard University, Cambridge, Massachusetts 02142, USA. E-mail: congreve@rowland.harvard.edu
cPhotonics Initiative, Advanced Science Research Center, City University of New York, New York, New York 10031, USA
dDepartment of Physics, Graduate Center, City University of New York, New York, New York 10016, USA
First published on 7th March 2019
Abstract
Optical upconversion is a net process by which two low energy photons are converted into one higher energy photon. There is vast potential to exploit upconversion in applications ranging from solar energy and biological imaging to data storage and photocatalysis. Here, we link two upconverting chromophores together to synthesize a series of novel tetracene dimers for use as annihilators. When compared with the monomer annihilator, TIPS–tetracene, the dimers yield a strong enhancement in the triplet fusion process, also known as triplet–triplet annihilation, as demonstrated via a large increase in upconversion efficiency and an order of magnitude reduction of the threshold power for maximum yield. Along with the ongoing rapid improvements to sensitizer materials, the dimerization improvements demonstrated here open the way to a wide variety of emerging upconversion applications.
Introduction
The ability to design and synthesize materials that convert infrared light into visible light is remarkably important due to widespread potential applications, including solar energy capture,1 photocatalysis,2 data storage,3 night vision,4 and biological imaging.5 In this vein, the process by which low energy photons are converted to a single high energy photon is known as optical upconversion.6,7 In recent years this process has received a surge of interest for its potential in solar energy applications.8 As a result, much work has been done to optimize upconversion in highly ordered and condensed systems, where there are large numbers of annihilators in very close proximity, thus leading to high upconversion efficiencies.9 Considering that upconversion can also greatly benefit other areas, such as imaging10 and photochemistry,2,11 where lower concentrations are required, a major challenge remains to find efficient upconverting materials that are robust with respect to concentration level. Thus, it is imperative to investigate how structural modifications to annihilators can be exploited to tune the process of upconversion.One promising approach to exhibit efficient upconversion is using mixed conjugated organic systems operating via triplet fusion (TF) upconversion, also known as triplet–triplet annihilation upconversion (Fig. 1A).12,13 In such systems, an organometallic sensitizer absorbs long wavelength light, exciting the sensitizer to a singlet state, which then rapidly undergoes intersystem crossing to a long lived triplet state, shown in Fig. 1B. This triplet can then be transferred from the sensitizer to an organic annihilator via collisional triplet energy transfer (TET), provided the triplet energy level of the annihilator is lower than that of the sensitizer. Two annihilators in their excited triplet states (T1) can then come together and undergo intermolecular triplet fusion (xTF), populating the excited singlet (S1) state of one of the annihilators. This S1 then decays to the ground state singlet (S0) via fluorescence, giving off a photon of higher energy than the incident light. In order for the xTF process to occur, it must conserve energy, such that the energy of the singlet state is less than twice the energy of the triplet state, i.e. E(S1) < 2 × E(T1). A schematic of the xTF process can be seen in Fig. 1C.
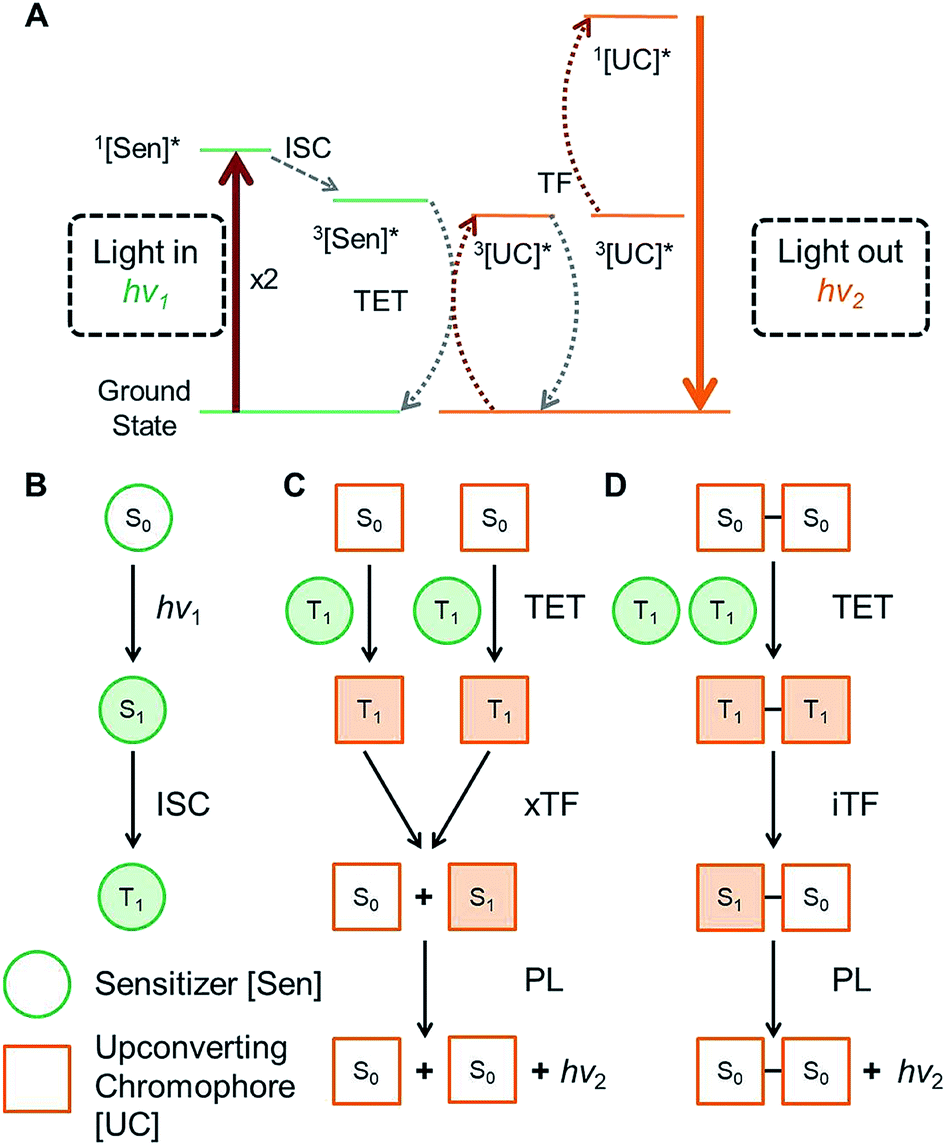 | ||
| Fig. 1 (A) Qualitative energy level diagram of the full triplet fusion upconversion mechanism. *Denotes first excited state ([Sen] = sensitizer, [UC] = upconverting chromophore) (B) mechanism by which triplet states are populated in a molecular sensitizer by photoexcitation (hν1), followed by intersystem crossing (ISC). (C) Mechanism of conventional intermolecular triplet fusion upconversion (xTF) giving off a high energy photon (hν2). (D) Proposed mechanism for intramolecular triplet fusion upconversion (iTF). Unfilled and filled shapes represent ground state and excited state molecules, respectively. S and T denote singlet and triplet states respectively. | ||
In order for TF to be a technically viable process, it is necessary to absorb low energy photons (such as IR and near infrared, NIR) that are inaccessible to many optoelectronic materials. Thus, a major focus in the field of TF upconversion has been on finding sensitizers which can absorb sufficiently low energy. Great strides have been made in recent years to this end, including the use of lead and cadmium chalcogenide nanocrystals as sensitizers to complement existing molecular annihilators.14–17 While much focus has been given to improving the sensitizer, comparatively little work has been done on the annihilator.18,19 Novel and exciting methods focusing on the structure of the materials to assemble the annihilators have been explored by the Simon and Kimizuka groups.20–22 But to date, there has been very little work to modify the annihilator itself to intrinsically enhance upconversion, especially operating at low concentrations in solution. Thus far, most work in the field of optical upconversion has employed common acene derivatives, such as diphenyl anthracene and rubrene, or perylene as the organic upconverter.23–25 In this work, we investigate how covalently coupling two upconverting chromophores to form an annihilator dimer affects the efficiency of upconversion. In contrast to previously studied polymeric systems, which focus on large assemblies,26–28 we sought to investigate interactions between two individual chromophores. Thus, we synthesized a series of tetracene dimers linked by 0, 1, 2, and 4 p-phenylene spacers (BTn; n = 0, 1, 2, 4), which were designed to undergo intramolecular triplet fusion upconversion (iTF, Fig. 1D), in addition to the typical xTF caused by collisions between chromophores.
Experimental
Synthesis of BT0,1,2 and 4
Full synthetic details for BT0, BT1, BT2 and BT4 including characterization data is provided in the ESI.†Optical characterization
Solutions of BT0,1,2,4 or TIPS–tetracene (TIPS–Tc) with PdPc(OBu)8 were prepared from anhydrous toluene in a nitrogen glovebox. Solutions were made in 1 cm × 1 cm cuvettes from Spectrocell and were degassed by sparging with nitrogen for 30 seconds then sealed before removing from glovebox for measurement. Solutions were excited with a 730 nm laser diode purchased from Thorlabs, focused to a beam diameter of 0.15 mm. Unless otherwise noted, the excitation density in all experiments was 113 W cm−2. All upconverted PL spectra were measured with a QEPro spectrometer purchased from Ocean Optics through a 700 nm shortpass filter. Power intensity dependence measurements were taken by varying the beam intensity using neutral density filters.PLQY
PLQY measurements were made using an integrating sphere purchased from Labsphere following de Mello et al.29 Briefly, the 10 mm cuvette was placed inside the sphere and excited with either 730 nm or 445 nm light focused from a laser diode. The upconversion quantum yield was determined by comparing the quantum yields when excited at 730 nm and 445 nm, with the upconversion yield defined such that it is the number of excitations leading to a singlet exciton on the annihilator. The cited yields are for annihilator concentrations of 5 × 10−4 M. The sphere and all optical equipment were calibrated against a calibrated silicon photodetector from Newport Corp.Results and discussion
Our approach in designing annihilators for upconversion is rooted in our fundamental understanding of how the platform of acene dimers operates in intramolecular singlet fission (iSF) – the reverse process of iTF.30 In all reported acene dimers, the spatial proximity of the generated triplet pairs leads to extremely rapid recombination.31 Even in the case of acene chromophores spaced by up to two p-phenylene spacers, the lifetime of a pair of triplets on one acene dimer is no more than 270 ns,30,32 ensuring us that iTF can occur with a relatively long bridge between the chromophores. However, it should be noted that when the energetic criteria for iSF is not met, the dimeric system is ideal to undergo iTF, because of the aforementioned rapid triplet recombination. Considering that the energy of the singlet state in tetracene is roughly less than twice the energy of the triplet state – an observation that is not favorable for iSF but good for iTF – we investigate the upconversion process in a series of tetracene dimers shown in Fig. 2A. The tetracene dimer BT0 does not contain a bridge, but BT1 and BT2 contain a phenylene and biphenylene bridge, respectively. These bridges effectively separate the triplets after sensitization, but maintain electronic communication between them. We also synthesized a tetracene dimer with a quaterphenylene bridge, BT4, to observe the effects of dimerization in a system where the two tetracene chromophores are too distant to be in good electronic communication. BT4 was chosen as it is the longest p-phenylene spaced dimer we could readily synthesize. These compounds were chosen due to the presence of the triisopropylsilylacetylene (TIPS) groups which impart stability and solubility, as well as facilitating iterative synthesis.33 These compounds were compared to a monomeric TIPS–Tc annihilator. PdPc(OBu)8 was chosen as the sensitizer, as it has been previously shown that this compound can efficiently donate triplets to tetracene derivatives upon absorption of near IR light.34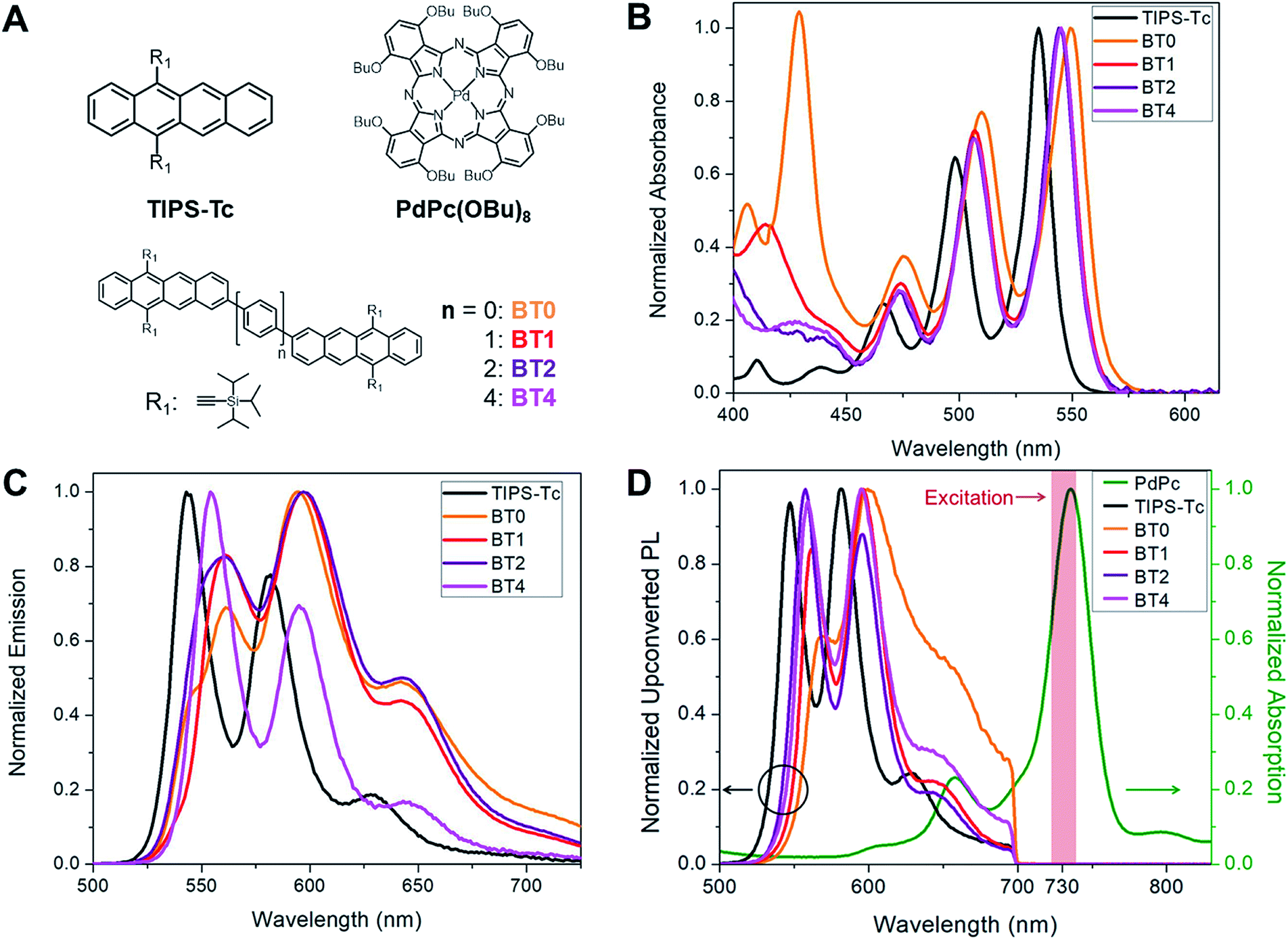 | ||
| Fig. 2 (A) Structures of the compounds used in this work. (B) Normalized steady state absorbance spectra of the annihilators used. (C) Normalized photoluminescence spectra of the annihilators used, excited at 490 nm. (D) Normalized upconverted photoluminescence spectra at 5 × 10−4 M annihilator and 2.5 × 10−5 M PdPc(OBu)8 (700 nm short pass filter used). Normalized steady state absorbance of PdPc(OBu)8 shown in green. The shaded red region represents excitation wavelength from 730 nm diode. | ||
The absorption and emission spectra of the annihilators used in this work can be seen in Fig. 2B and C, respectively. The absorption of the tetracene dimers exhibits a modest redshift in the onset of absorption relative to TIPS–Tc. This is characteristic of the extension of a π system typically seen in conjugated organic molecules. The emission spectra of the upconverting chromophores are similar to one another, with TIPS–Tc and BT4 exhibiting a more pronounced peak at 550 nm relative to the peak at 600 nm, and vice versa for BT0–BT2. The normalized absorption spectra of PdPc(OBu)8 can be seen in green in Fig. 2D, exhibiting strong absorption in the near IR. Upon excitation with a 730 nm laser diode (shaded red Fig. 2D), optical upconversion was observed for TIPS–Tc as well as all four tetracene dimers. The normalized upconversion photoluminescence (UCPL) of these compounds can be seen in Fig. 2D.
To confirm that iTF was possible, we performed DFT calculations of the T1 states of BT0, BT1, BT2, and BT4.35Fig. 3 shows one of the doubly degenerate T1 orbitals of BT0, BT1, BT2, and BT4. In the case of closely linked dimers such as BT0, BT1, and BT2, the T1 state is generally localized on one of the tetracene chromophores, but spills onto the second chromophore. This mixing of triplet states across multiple chromophores facilitates iTF. In the case of BT4, the two tetracene chromophores are too far apart to allow the T1 state to be shared. As a result, BT4 should not be able to undergo iTF, and instead we would expect it to behave similarly to TIPS–Tc as an upconversion annihilator.
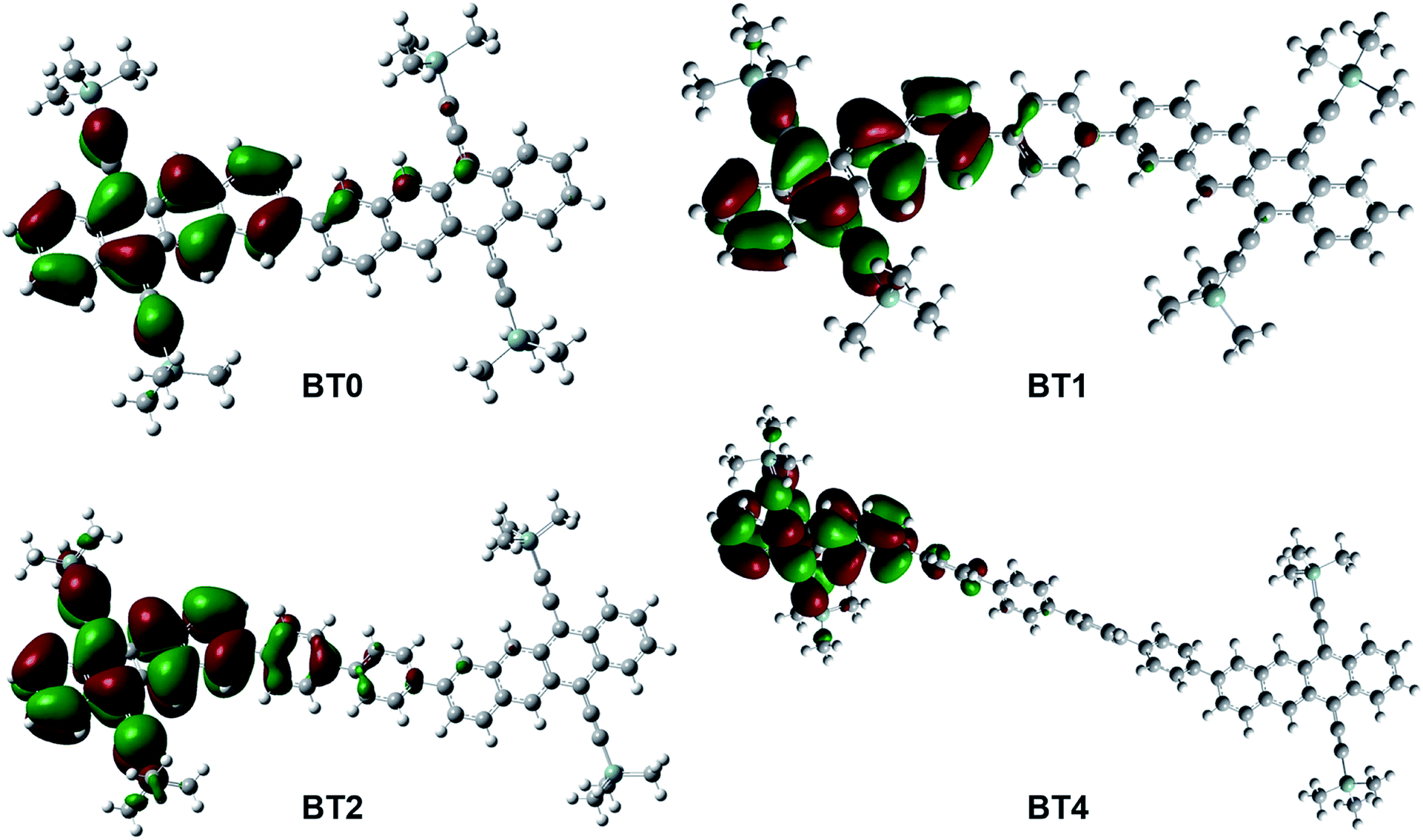 | ||
| Fig. 3 T1 orbitals of tetracene dimers studied generated via DFT calculations. | ||
After establishing the presence of upconversion when using tetracene dimers as annihilators, we explored the dependence of upconversion photoluminescence intensity on the concentration of these materials. We began with a sensitizer concentration of 2.5 × 10−5 M, and an annihilator concentration of 5 × 10−4 M, these being typical of solution state measurements of upconversion.19,34 We then lowered the concentration of the annihilator down to 3.75 × 10−5 M while holding the amount of sensitizer constant in order to study the effects of iTF on concentration. The dependence of upconversion PL as a function of concentration of the annihilator can be seen in Fig. 4A. BT4 decays nearly identically to the monomeric TIPS–Tc, as we would expect in a system where the two tetracene chromophores are electronically independent of one another and lack the iTF upconversion pathway. BT0 exhibits the lowest upconversion PL across all concentrations, which is likely due to its low photoluminescence quantum yield (PLQY) as a result of singlet fission, a competing pathway for singlet decay.36,37 In stark contrast, BT1 and BT2 both exhibit higher upconversion PL compared to TIPS–Tc, across all concentrations. This is even in spite of the significantly lower PLQYs of BT1 and BT2, a result of the fact that these compounds undergo singlet fission as well. In fact, the overall upconversion yield (defined as the percentage of absorbed photons that become a singlet on the annihilator) is significantly higher in BT1 than in TIPS–Tc, at 4.2% vs. 0.70%, respectively. This upconversion yield is equivalent to that of a previously reported tetracene dimer19 but we note that we are using a phosphor that absorbs lower energy light and lower concentrations of sensitizer. Calculations show that the T1 energies of the annihilators studied in this work vary by only 7 meV (Table S1†), ruling out changes in T1 energy as the source of our enhanced upconversion performance in these materials. Indeed, the robustness of the BT1 upconverting chromophore to these non-ideal conditions proves its usefulness in a wide variety of demanding upconversion applications where lower concentrations are required.
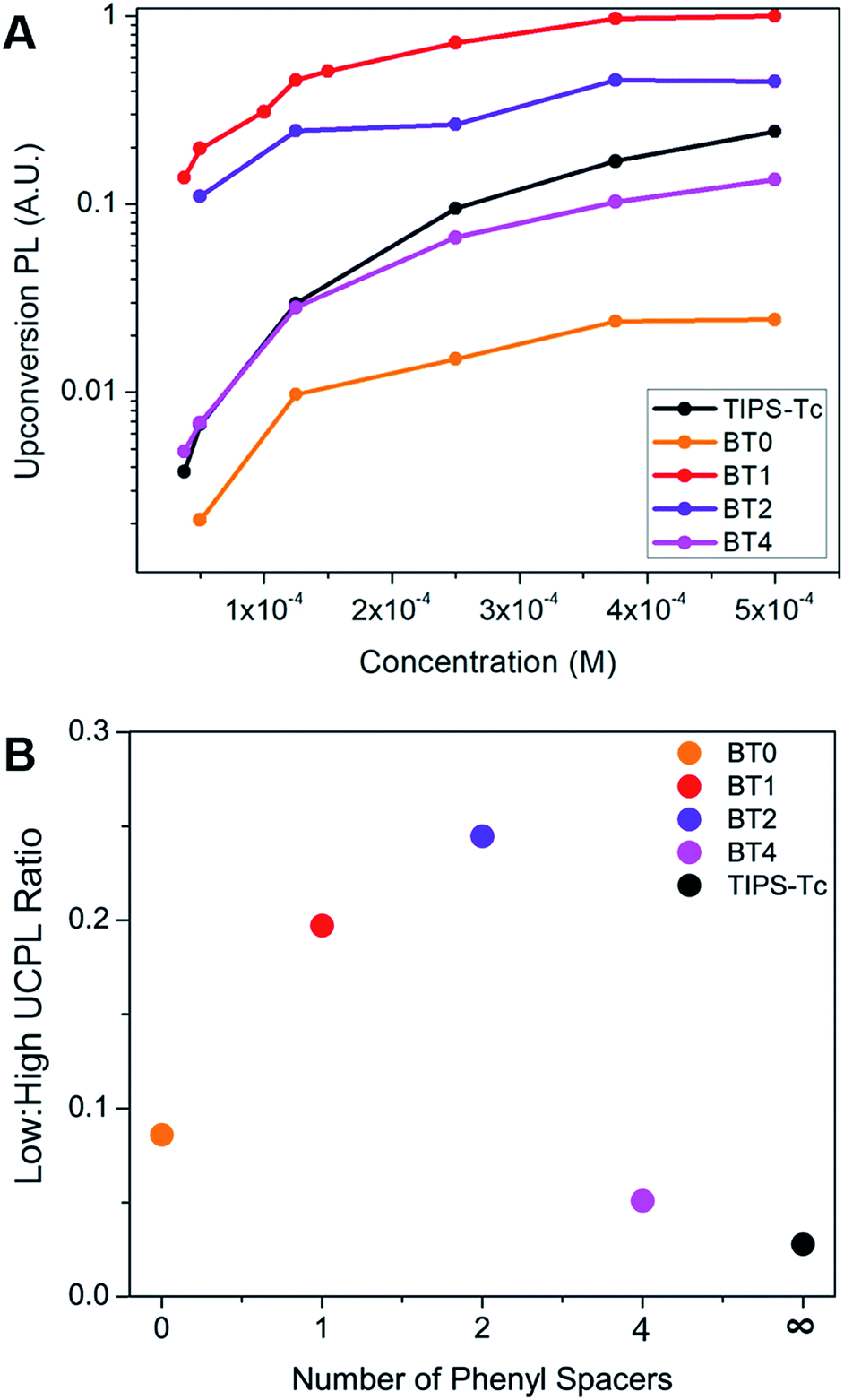 | ||
| Fig. 4 (A) Change in upconversion PL of compounds studied as a function of annihilator concentration at 2.5 × 10−5 M PdPc(OBu)8. (B) UCPL at 5 × 10−4 M divided by UCPL at 5 × 10−5 M annihilator concentration plotted as a function of the number of phenyl spacers in the annihilator used. | ||
We then performed Stern–Volmer quenching experiments on our system to measure the rate of triplet energy transfer (kTET) from sensitizer to annihilator. This was done to rule out an increase in kTET as the source of enhanced UCPL in our tetracene dimers (Fig. S9, Table S2†). These measurements showed a slight enhancement in kTET of the dimers relative to TIPS–Tc. The change in kTET is small relative to the overall enhancement in UCPL for these materials, but in order to provide further evidence of iTF, we constructed a kinetic model of our system. The details of this model can be found in the ESI (Fig. S11†). Using this model, we are able to fit the rate of triplet fusion (kTF) to our experimental data in Fig. 4A. Using the values of kTET extracted from our Stern–Volmer measurements, we find the only way to fit our data using the model is with an enhanced rate of kTF (Table S6†). TIPS–Tc has a kTF of 2.3 × 106 (M−1 s−1), compared to 250 × 106 (M−1 s−1) for BT1. We believe this enhancement in kTF is additional supporting evidence of iTF, which leads to enhanced UPCL yields of our BT dimers.
In the case of classical xTF, a high concentration of annihilator molecules has been considered crucial for efficient upconversion, despite the fact that this high concentration can lead to fluorescence quenching via aggregation.7,38,39 At high annihilator concentrations, we expect xTF to dominate (that is, excited annihilator molecules are much more likely to collide with one another than with a sensitizer). This is demonstrated by the fact that TIPS–Tc is competitive with both BT1 and BT2 at higher concentrations. At low concentrations, however, excited annihilators are as likely to collide with sensitizers as they are with another annihilator, and thus the ability to hold two triplets is greatly advantageous, and the dimeric materials dominate, suggesting that iTF is occurring. This is exhibited by the greater than an order of magnitude reduction of UCPL for TIPS–Tc, while BT1 and BT2 only see a modest decrease in UCPL at low concentrations (Fig. 4A). For this reason, most demonstrations of upconversion in solution do so with an annihilator concentration of ∼1 mM, whereas we can see bright upconversion even at annihilator concentrations as low as 3.75 × 10−5 M. The benefit of the extra iTF pathway can clearly be seen in its high upconversion PL seen at very low concentrations. This is further demonstrated in Fig. 4B, where we compare the UCPL of these annihilators and low and high concentration. This opens the door for the use of iTF capable upconversion materials in photochemical or imaging applications, where high concentrations of extrinsic materials are undesirable. For example, it is known that a high concentration of annihilators will reduce the propagation of the upconverted PL in solution.40 This is disadvantageous for imaging applications where it is crucial that the upconverted PL be detectable throughout the species. Photochemical reactions where the photocatalyst is excited by upconverted PL11 would also benefit from a low concentration of annihilator, because a high concentration of these extra species could easily lead to undesirable side reactions.
Because it requires two initial species, upconversion PL has a quadratic dependence on light intensity at low fluence. This dependence on light intensity then exhibits a change from quadratic to linear once TF becomes the dominant recombination mechanism.41Fig. 5 shows the dependence of upconversion PL intensity on incident power density for TIPS–Tc and BT1 (BT0, BT2, and BT4 shown in Fig. S1, S2, and S3† respectively). The crossover point of BT1, 4.3 W cm−2 is an order of magnitude lower than that of TIPS–Tc at 44.5 W cm−2. This crossover point reduction is a crucial benefit for BT1, as it ensures that upconversion is occurring at maximum efficiency even at low photon flux.
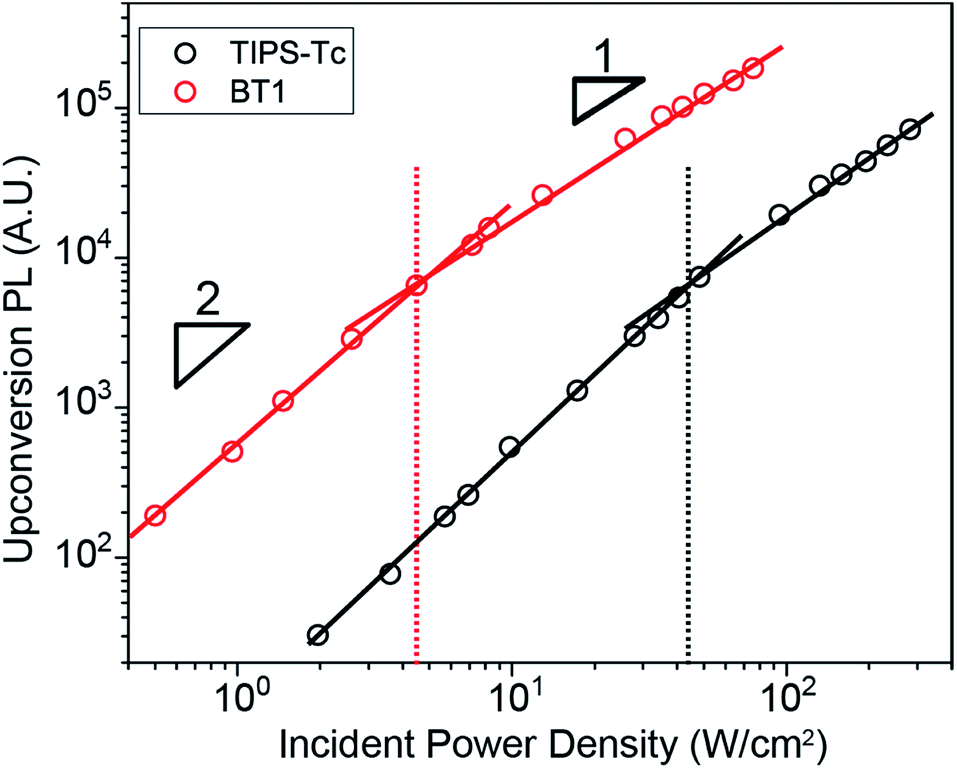 | ||
| Fig. 5 Dependence of upconverted PL on incident light intensity at 2.5 × 10−4 M TIPS–Tc and BT1 concentration. The transition between quadratic and linear dependences occurs at 44.5 and 4.3 W cm−2 for TIPS–Tc and BT1 respectively. | ||
Conclusions
In conclusion, we have synthesized a new series of tetracene dimers that suggest that intramolecular triplet fusion is occurring in these materials. Dimerization is an attractive strategy towards enhanced upconversion because it offers intrinsic benefits to upconversion, rather than relying on complex processing or assembly techniques. This should ease the transition of these materials into non-ideal media where they could be widely applicable, such as in biological systems or photochemical reactions, where a greater number of exogenous species can reduce the efficiency of xTF. As compared to monomeric TIPS–Tc, a tetracene dimer exhibits greater upconversion yield, less sensitivity to concentration, and reduced power thresholds. These improvements pave the way towards upconversion as a viable candidate for a host of applications, especially when a high concentration of outside species is either unfavorable or untenable.Conflicts of interest
The authors declare the following competing financial interest(s): Columbia University is filing a patent based on this work.Acknowledgements
A. B. P. thanks the NSF for GRFP (DGE 16-44869). L. M. C. acknowledges support from the Cottrell Scholar Award and the National Science Foundation (NSF) CAREER Program (DMR 1351293). We are grateful to Jonathan Owen for use of the fluorimeter. D. N. C. acknowledges the support of the Rowland Fellowship Program of the Rowland Institute at Harvard University. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number TG-DMR150018. This research used resources of the Center for Functional Nanomaterials, which is a US DOE Office of Science Facility at Brookhaven National Laboratory under contract No. DE-SC0012704.Notes and references
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 RSC.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef PubMed.
- Y. Zhou, S.-T. Han, X. Chen, F. Wang, Y.-B. Tang and V. A. L. Roy, Nat. Commun., 2014, 5, 4720 CrossRef CAS PubMed.
- S.-W. Liu, C.-C. Lee, C.-H. Yuan, W.-C. Su, S.-Y. Lin, W.-C. Chang, B.-Y. Huang, C.-F. Lin, Y.-Z. Lee, T.-H. Su and K.-T. Chen, Adv. Mater., 2015, 27, 1217–1222 CrossRef CAS PubMed.
- Y. Il Park, K. T. Lee, Y. D. Suh and T. Hyeon, Chem. Soc. Rev., 2015, 44, 1302–1317 RSC.
- T. W. Schmidt and F. N. Castellano, J. Phys. Chem. Lett., 2014, 5, 4062–4072 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- J. C. Goldschmidt and S. Fischer, Adv. Opt. Mater., 2015, 3, 510–535 CrossRef CAS.
- P. Bharmoria, S. Hisamitsu, H. Nagatomi, T. Ogawa, M. Morikawa, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2018, 140, 10848–10855 CrossRef CAS PubMed.
- U. Agrawal, D. T. Reilly and C. M. Schroeder, Curr. Opin. Biotechnol., 2013, 24, 646–653 CrossRef CAS PubMed.
- S. H. C. Askes, A. Bahreman and S. Bonnet, Angew. Chem., Int. Ed., 2014, 53, 1029–1033 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Phys. Rev. Lett., 2006, 97, 143903 CrossRef CAS PubMed.
- M. Wu, D. N. Congreve, M. W. B. Wilson, J. Jean, N. Geva, M. Welborn, T. Van Voorhis, V. Bulović, M. G. Bawendi and M. A. Baldo, Nat. Photonics, 2016, 10, 31–34 CrossRef CAS.
- Z. Huang, X. Li, M. Mahboub, K. M. Hanson, V. M. Nichols, H. Le, M. L. Tang and C. J. Bardeen, Nano Lett., 2015, 15, 5552–5557 CrossRef CAS PubMed.
- Z. Huang, X. Li, B. D. Yip, J. M. Rubalcava, C. J. Bardeen and M. L. Tang, Chem. Mater., 2015, 27, 7503–7507 CrossRef CAS.
- V. Gray, P. Xia, Z. Huang, E. Moses, A. Fast, D. A. Fishman, V. I. Vullev, M. Abrahamsson, K. Moth-Poulsen and M. Lee Tang, Chem. Sci., 2017, 8, 5488–5496 RSC.
- D. Dzebo, K. Börjesson, V. Gray, K. Moth-Poulsen and B. Albinsson, J. Phys. Chem. C, 2016, 120, 23397–23406 CrossRef CAS.
- S. Baluschev, V. Yakutkin, T. Miteva, Y. Avlasevich, S. Chernov, S. Aleshchenkov, G. Nelles, A. Cheprakov, A. Yasuda, K. Müllen and G. Wegner, Angew. Chem., Int. Ed., 2007, 46, 7693–7696 CrossRef CAS PubMed.
- D. C. Thévenaz, S. H. Lee, F. Guignard, S. Balog, M. Lattuada, C. Weder and Y. C. Simon, Macromol. Rapid Commun., 2016, 37, 826–832 CrossRef PubMed.
- P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887–1894 CrossRef CAS PubMed.
- K. Mase, K. Okumura, N. Yanai and N. Kimizuka, Chem. Commun., 2017, 53, 8261–8264 RSC.
- D. Di, L. Yang, J. M. Richter, L. Meraldi, R. M. Altamimi, A. Y. Alyamani, D. Credgington, K. P. Musselman, J. L. MacManus-Driscoll and R. H. Friend, Adv. Mater., 2017, 29, 1605987 CrossRef PubMed.
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC.
- Y. Y. Cheng, B. Fückel, T. Khoury, R. G. C. R. Clady, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, J. Phys. Chem. A, 2011, 115, 1047–1053 CrossRef CAS PubMed.
- P. C. Boutin, K. P. Ghiggino, T. L. Kelly and R. P. Steer, J. Phys. Chem. Lett., 2013, 4, 4113–4118 CrossRef CAS.
- S. H. Lee, D. C. Thévenaz, C. Weder and Y. C. Simon, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1629–1639 CrossRef CAS.
- V. Gray, K. Moth-Poulsen, B. Albinsson and M. Abrahamsson, Coord. Chem. Rev., 2018, 362, 54–71 CrossRef CAS.
- J. C. de Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. T. Trinh, B. Choi, J. Xia, E. J. Taffet, J. Z. Low, J. R. Miller, X. Roy, X.-Y. Zhu, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, J. Am. Chem. Soc., 2015, 137, 8965–8972 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, Angew. Chem., Int. Ed., 2016, 55, 3373–3377 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, K. Appavoo, M. L. Steigerwald, L. M. Campos and M. Y. Sfeir, J. Am. Chem. Soc., 2016, 138, 7289–7297 CrossRef CAS PubMed.
- E. Kumarasamy, S. N. Sanders, A. B. Pun, S. A. Vaselabadi, J. Z. Low, M. Y. Sfeir, M. L. Steigerwald, G. E. Stein and L. M. Campos, Macromolecules, 2016, 49, 1279–1285 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. A, 2008, 112, 3550–3556 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revis. D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- N. V. Korovina, S. Das, Z. Nett, X. Feng, J. Joy, R. Haiges, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2016, 138, 617–627 CrossRef CAS PubMed.
- A. M. Müller, Y. S. Avlasevich, W. W. Schoeller, K. Müllen and C. J. Bardeen, J. Am. Chem. Soc., 2007, 129, 14240–14250 CrossRef PubMed.
- C. E. McCusker and F. N. Castellano, Inorg. Chem., 2015, 54, 6035–6042 CrossRef CAS PubMed.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- Y. Y. Cheng, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 66–71 RSC.
- A. Haefele, J. Blumhoff, R. S. Khnayzer and F. N. Castellano, J. Phys. Chem. Lett., 2012, 3, 299–303 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03725f |
| This journal is © The Royal Society of Chemistry 2019 |