
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile access to functionalized chiral secondary benzylic boronic esters via catalytic asymmetric hydroboration†
Suman
Chakrabarty
 a,
Hector
Palencia
b,
Martha D.
Morton
ac,
Ryan O.
Carr
a and
James M.
Takacs
*ac
a,
Hector
Palencia
b,
Martha D.
Morton
ac,
Ryan O.
Carr
a and
James M.
Takacs
*ac
aDepartment of Chemistry, University of Nebraska-Lincoln, Lincoln, Nebraska 68588-0304, USA. E-mail: jtakacs1@unl.edu
bDepartment of Chemistry, University of Nebraska-Kearney, Kearney, Nebraska 68849, USA
cNebraska Center for Integrated Biomolecular Communication (NCIBC), University of Nebraska-Lincoln, Lincoln, Nebraska 68588-0304, USA
First published on 25th March 2019
Abstract
Allylic and homoallylic phosphonates bearing an aryl or heteroaryl substituent at the γ- or δ-position undergo rhodium-catalyzed asymmetric hydroboration by pinacolborane to give functionalized chiral secondary benzylic boronic esters in yields up to 86% and enantiomer ratios up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Compared to minimally-functionalized terminal and 1,1-disubstituted vinyl arenes, there are relatively few reports of efficient catalytic asymmetric hydroboration (CAHB) of more highly functionalized internal alkenes. Phosphonate substrates bearing a variety of common heterocyclic ring systems, including furan, indole, pyrrole and thiophene derivatives, as well as those bearing basic nitrogen substituents (e.g., morpholine and pyrazine) are tolerated, although donor substituents positioned in close proximity of the alkene can influence the course of the reaction. Stereoisomeric (E)- and (Z)-substrates afford the same major enantiomer of the borated product. Deuterium-labelling studies reveal that rapid (Z)- to (E)-alkene isomerization accounts for the observed (E/Z)-stereoconvergence during CAHB. The synthetic utility of the chiral boronic ester products is illustrated by stereospecific C–B bond transformations including stereoretentive electrophile promoted 1,2-B-to-C migrations, stereoinvertive SE2 reactions of boron-ate complexes with electrophiles, and stereoretentive palladium- and rhodium-catalyzed cross-coupling protocols.
:1. Compared to minimally-functionalized terminal and 1,1-disubstituted vinyl arenes, there are relatively few reports of efficient catalytic asymmetric hydroboration (CAHB) of more highly functionalized internal alkenes. Phosphonate substrates bearing a variety of common heterocyclic ring systems, including furan, indole, pyrrole and thiophene derivatives, as well as those bearing basic nitrogen substituents (e.g., morpholine and pyrazine) are tolerated, although donor substituents positioned in close proximity of the alkene can influence the course of the reaction. Stereoisomeric (E)- and (Z)-substrates afford the same major enantiomer of the borated product. Deuterium-labelling studies reveal that rapid (Z)- to (E)-alkene isomerization accounts for the observed (E/Z)-stereoconvergence during CAHB. The synthetic utility of the chiral boronic ester products is illustrated by stereospecific C–B bond transformations including stereoretentive electrophile promoted 1,2-B-to-C migrations, stereoinvertive SE2 reactions of boron-ate complexes with electrophiles, and stereoretentive palladium- and rhodium-catalyzed cross-coupling protocols.
Introduction
Chiral alkyl boronic esters possess a unique blend of benchtop stability and the potential to undergo a variety of C–B bond transformations via stereospecific 1,2-migration from an in situ generated boron “ate-complex” rendering them especially versatile intermediates for asymmetric synthesis.1,2 Metal-catalyzed asymmetric protoboration3 and hydroboration4,5 of alkenes are among the most common approaches for the preparation of chiral alkyl boronic esters. While catalytic asymmetric hydroboration (CAHB) of minimally-functionalized terminal and 1,1-disubstituted vinyl arenes (e.g. simple substituted styrene derivatives) have been extensively investigated,6 there are relatively few reports using more highly functionalized di- or trisubstituted internal alkenes.7 The latter have been a focus of our research into CAHB. Herein, we disclose that 1,2-disubstituted allyl phosphonates bearing an aryl substituent at the γ-position are efficient substrates and provide facile access to functionalized chiral, secondary benzylic and related α-aryl boronic esters.8We previously reported that methylidene substrates bearing β-phenyl substitution undergo efficient rhodium-catalyzed CAHB with pinacolborane (pinBH) to give chiral tertiary boronic esters via regioselective β-boration. For example, 1 affords (R)-2 (81%) in a 97:3 enantiomer ratio (er); the β:γ regioisomer ratio (rr) is 4:1 or greater under the conditions described in Fig. 1.5a Similarly, the trisubstituted alkene (E)-3, bearing phenyl substituents at both the β- and γ-positions, undergoes preferential β-boration (3:1 rr) to afford the chiral, tertiary boronic ester product (R)-4 (60%, 97:3 er).5a
 | ||
| Fig. 1 Directed-CAHB of allylic phosphonate-functionalized vinyl arenes varying in substitution pattern. Standard CAHB conditions: 1 mol% Rh(nbd)2BF4 or 0.5 mol% [Rh(cod)Cl]2/1 mol% AgBF4, 1 mol% (R,R)-T1 or (R)-B1, 1.1 eq. pinacolborane (pinBH), 3 h, rt. Oxidation conditions: NaBO3·4H2O/H2O. | ||
We now report that 5a and related substrates bearing a phenyl, aryl or heteroaryl substituent at the γ-position undergo preferential γ-boration to afford new chiral, secondary benzylic boronic esters. The benzylic regiochemistry presumably arises from the favorable formation of a rhodium π-benzyl complex of the substrate in the course of reaction.9 We were surprised to find that the alkene stereochemistry does not impact the overall regio- or stereochemical course of the reaction. Both (E)- and (Z)-5a afford the same γ-borated product 6a in greater than 20:1 rr using (R)-B1; C–B bond oxidation using NaBO3·4H2O affords the known chiral alcohol10 shown (82%, 96:4 er).
Results and discussions
Two alternative rhodium catalyst precursors are used in this study. A 1:1 combination of [Rh(nbd)2BF4/(R)-B1] can be replaced by an in situ generated 1:1 [Rh(cod)BF4/(R)-B1] catalyst formed by treating [Rh(cod)Cl]2 with AgBF4 and the chiral ligand. The catalysts afford near identical results, but the latter protocol is more economical. While pinBH is known to undergo relatively facile rhodium-catalyzed degradation,11 thus often necessitating its use in excess, a stoichiometric amount of pinBH is usually enough for complete CAHB of these allylic phosphonates.
The choice of chiral ligand is, of course, critical to the success of CAHB.12 As indicated in Fig. 1, rhodium catalyst systems incorporating the TADDOL-derived chiral cyclic monophosphite (R,R)-T1 give efficient enantioinduction for β-aryl methylidene substrate 1 and the β,γ-bisaryl trisubstituted alkene (E)-3. Its use with 5a gives (S)-6a in good yield (81%, after oxidation), but with only a low level of enantioinduction (64:36 er). In contrast, the BINOL-derived phosphoramidite (R)-B1 affords high levels of regiocontrol and enantioselectivity.13
Scope of aryl and heteroaryl substrates
Fig. 2 summarizes results obtained for a series of substrates 5b–t in which the nature of the aromatic ring appended at the γ-position varies. In addition to the major γ-boration product 6 and the minor regioisomeric β-boration product 7, alkene reduction (i.e., 8) typically comprises the remaining 5–15% of the product mixture. The latter reaction mode arises from a competing catalytic cycle and is the subject of ongoing studies;14 it will not be discussed further here. Herein, we focus on the regio- and enantioselectivity of the rhodium-catalyzed hydroboration mode.15 | ||
| Fig. 2 Substrate scope for CAHB of γ-aryl allylic phosphonates. Note: atwo equivalents of pinacolborane were used. bReaction carried out for 12 hours. | ||
A range of donor and acceptor substituents are well-tolerated with relatively minor fluctuations in regio- and/or enantioselectivity. For example, the 4-methylphenyl derivative 5b and the 4-trifluoromethylphenyl derivative 5c undergo γ-boration with high regioselectivity (>20:1 rr) yielding 9b (84%, 97:3 er) and 9c (78%, 97:3 er) after oxidation of the corresponding secondary benzylic boronic esters (i.e., 6b and 6c). The 4-fluorophenyl (5d) and 4-methyoxyphenyl (5e) derivatives exhibit somewhat lower levels of γ:β regioselectivity (6d–e, 10–11:1 rr) and enantioselectivity affording 9d (71%, 94:6 er) and 9e (77%, 94:6 er), respectively. In contrast, the 3-methoxyphenyl derivative 5f and 3,4-methylenedioxyphenyl derivative 5g again exhibit higher regioselectivity (6f–g: >20:1 rr) leading to 9f (83%, 98:2 er) and 9g (70%, 94:6 er), after oxidation. The 2-methoxyphenyl derivative 5h exhibits reduced regioselectivity (6h: 9:1 rr), but otherwise good conversion to yield 9h (72%, 94:6 er).16 The 4-dimethylamino, morpholine, and pyrazine derivatives 5i–k demonstrate the viability of substrates bearing basic nitrogen functionality; 9i–k are obtained in moderate to good yields (55–68%) and up to 94:6 er.
In addition to demonstrating tolerance for basic nitrogen in several of the substrates described in Fig. 2, it is pleasing to see that substrates incorporating some heteroaromatic ring systems also undergo efficient CAHB, albeit with some unusual variation in regio- and/or enantioselectivity. For example, the Boc-protected indole derivative 5l exhibits high γ-regioselectivity (6l, >20:1 rr) with good enantioinduction after oxidation to 9l (78%, 96:4 er). Similarly, the 3-substituted N-Boc-protected pyrrole derivative 5m is both highly regioselective (6m, >20:1 rr) and highly enantioselective; the er obtained for 9m (86%, 99:1 er) is the highest obtained among the substrates tested. However, the results obtained for CAHB of the 2-substituted N-Boc-protected pyrrole derivative 5n differ markedly. The regioselectivity of 6n (>20:1 rr) is excellent. However, the level of enantioselectivity for 9n (85%, 80:20 er) is not only much lower but results from hydroboration with the opposite sense of π-facial selectivity compared to most other substrates (vide infra); (R)-9n is the major product.
The 3- and 2-substituted thiophene substrates (i.e., 5o and 5p) exhibit high regioselectivity leading to 6o and 6p (>20:1 rr), respectively. However, the 3-substituted thiophene 5o affords 9o (83%, 85:15 er) with only modest levels of stereocontrol, while the 2-substituted thiophene 5p gives 9p (80%, 95:5 er) with good stereocontrol. The 2-substituted benzothiophene 5q gives both lower levels of regiocontrol (6q, 5:1 rr) and enantioselectivity for 9q (65%, 89:11 er). The corresponding furan derivatives 5r–t give similar results.
Stereochemical assignments
The variable regio- and stereochemical results obtained with heteroaryl derivatives illustrate a caveat for substrates bearing multiple donor groups in proximity to the alkene or bear relatively bulky aromatic ring systems. Using the Birman benzotetramisole (BTM)17 chiral acylation catalyst, a kinetic resolution (KR) strategy was employed to confirm the absolute configuration assignments for several of the previously unreported chiral secondary benzylic alcohols 9 prepared via CAHB. As shown in Fig. 3A, the (S)-BTM-catalyzed acylation of a racemic sample of (±)-9a with 0.5 equivalents of isobutyric anhydride results in the rapid acylation of (S)-9a to (S)-10a and recovery of the known (R)-9a unreacted alcohol. | ||
| Fig. 3 Stereochemical assignments based on kinetic resolution: (A) General scheme for kinetic resolution via acylation using benzotetramisole (BTM); (B) 31P NMR analysis for assignment of absolute configuration via KR; (C) absolute configuration assignments of benzyl alcohol products containing heterocyclic ring systems via KR. | ||
Alcohol 9 and ester 10 are readily differentiated by 31P NMR spectroscopy providing a convenient protocol for the rapid determination of absolute configuration of these previously unreported chemical entities (Fig. 3B). As demonstrated by the 31P NMR stack-plot, (S)-9a (96:4 er) is more rapidly acylated using (S)-BTM than with (R)-BTM. Using this method, we assigned the absolute configurations for the α-hydroxy heteroaryl products obtained via CAHB/oxidation (Fig. 3C). For example, the 3-substituted pyrrole derivative 9m (99:1 er), synthesized via CAHB/oxidation using (R)-B1, undergoes more rapid (S)-BTM-catalyzed acylation to 10m (27% conversion in 12 h) compared to (R)-BTM-catalyzed acylation (2% conversion in 12 h). The relative rates are consistent with predominant (S)-configuration of 9m. In contrast, the 2-pyrrole derivative 9n (80:20 er), also prepared via CAHB/oxidation, undergoes relatively sluggish (S)-BTM-catalyzed acylation to 10n compared to (R)-BTM-catalyzed acylation. The results indicate that predominantly (R)-9n is formed from the 2-substituted pyrrole by CAHB with (R)-B1. We speculate that the N-Boc moiety in the latter acts as an alternative directing group in the rhodium-catalyzed hydroboration thereby switching the sense of alkene π-facial selectivity. Although one might reasonably expect that 2-subsituted thiophene and furan derivatives behave similarly, the data summarized in Fig. 3c indicate that (S)-9o–p and (S)-9r–s are the major stereoisomers produced via CAHB/oxidation.
In our prior studies of phosphonate-directed CAHB, we illustrated the synthetic utility of chiral bifunctional tertiary organoboron derivatives through thiophosphonate olefination5b chemistry and α-oxophosphonate active ester5a chemistry as well as through a number of stereospecific C–B bond transformations. Here, we focus on the use of (S)-6a (96:4 er) in stereoretentive and stereoinvertive C–B bond transformations via intermediate boron-ate complexes (Fig. 4). Stereoretentive cross-coupling with carbanions derived from electron rich vinyl and aromatic derivatives via electrophile-promoted 1,2-B-to-C migration of a boron–ate complex is generally facile under conditions reported by Aggarwal.2b,2e For example, treatment of (S)-6a with excess vinyl magnesium bromide followed by I2 and sodium methoxide affords the vinyl derivative (R)-11 (79%, 95:5 er) in high levels of stereoretention.2e Similarly, reaction of (S)-6a with 2-lithiobenzofuran followed by NBS affords the gem-bisaryl product (S)-12 (68%, 96:4 er) with essentially complete enantiospecificity.2b
 | ||
Fig. 4 Product utility illustrated by stereoretentive and stereoinvertive C–B bond transformations. Reagents and conditions: (a) (i) excess CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHMgBr, THF, −78 °C to rt; (ii) I2/MeOH, −78 °C; (iii) NaOMe/MeOH; (iv) Na2S2O3 (aq.); (b) (i) benzofuran-2-yllithium, THF, −78 °C; (ii) NBS, THF, −78 °C; (iii) Na2S2SO3 (aq.); (c) 2-iodobenzofuran, Ag2O, Pd2(dba)3/PPh3, THF, 60 °C; (d) 4-iodoanisole, Ag2O, Pd2(dba)3/PPh3, THF, 60 °C; (e) (i) 4-nitrobenzaldehyde, [Rh(cod)Cl]2, KHF2, dioxane/H2O; (ii) TEMPO, TCCA, CH2Cl2; (f) (3,5-bis(trifluoromethyl)phenyl)lithium, THF, −78 °C to −40 °C; (ii) cycloheptatrienyl tetrafluoroborate, rt.; (iii) NaHCO3 (aq.). (g) (i) (3,5-bis(trifluoromethyl)phenyl)lithium, THF, −78 °C to −40 °C; (ii) 4-Methoxybenzenediazonium tetrafluoroborate, 0 °C (iii) NaHCO3 (aq.). CHMgBr, THF, −78 °C to rt; (ii) I2/MeOH, −78 °C; (iii) NaOMe/MeOH; (iv) Na2S2O3 (aq.); (b) (i) benzofuran-2-yllithium, THF, −78 °C; (ii) NBS, THF, −78 °C; (iii) Na2S2SO3 (aq.); (c) 2-iodobenzofuran, Ag2O, Pd2(dba)3/PPh3, THF, 60 °C; (d) 4-iodoanisole, Ag2O, Pd2(dba)3/PPh3, THF, 60 °C; (e) (i) 4-nitrobenzaldehyde, [Rh(cod)Cl]2, KHF2, dioxane/H2O; (ii) TEMPO, TCCA, CH2Cl2; (f) (3,5-bis(trifluoromethyl)phenyl)lithium, THF, −78 °C to −40 °C; (ii) cycloheptatrienyl tetrafluoroborate, rt.; (iii) NaHCO3 (aq.). (g) (i) (3,5-bis(trifluoromethyl)phenyl)lithium, THF, −78 °C to −40 °C; (ii) 4-Methoxybenzenediazonium tetrafluoroborate, 0 °C (iii) NaHCO3 (aq.). | ||
The palladium-catalyzed cross-coupling of chiral boronic esters has attracted recent interest both due to its synthetic utility and the interesting mechanistic issue of stereoretention18 or stereoinversion;19 the outcome is often dependent upon participation or non-participation of polar substituents in the substrate.20,21 Benzylic boronic esters are rather unique in the context of palladium-catalyzed cross-coupling. Only the protocol recently introduced by Crudden22 is reportedly effective. Its efficiency varies, but in favorable constructs, the latter proceeds with 84–94% stereoretention (sr). Thus, the outcome for the bifunctional substrate (S)-6a (96:4 er) was uncertain. In our hands, cross-coupling with 2-iodobenzofuran under the [Pd2(dba)3/PPh3/Ag2O] conditions reported by Crudden gives (S)-12 (55%, 70:30 er, 73% sr); cross-coupling proceeds with predominant stereoretention, albeit with significant erosion of enantiopurity. Using the same conditions, cross-coupling of (S)-6a with 4-iodoanisole yields (S)-13 (60%, 77:23 er, 80% sr). The latter is useful, since we were unable to prepare 13via the 1,2-B-to-C migration protocol. An alternative rhodium-catalyzed cross-coupling procedure reported by Aggarwal23 effects the stereoretentive addition of (S)-6a to 4-nitrobenzaldehyde to afford aryl ketone (S)-14 (74%, 94:6 er) after oxidation; the latter cross-coupling proceeds with only slight erosion of enantiopurity over two steps.
Aggarwal reported that ate-complexes of secondary boronic esters react with strong electrophiles via a stereoinvertive SE2 mechanism.2f We find that the intermediate boron–ate complex formed by addition of (3,5-bis(trifluoromethyl)phenyl) lithium to (S)-6a (96:4 er) readily reacts with cycloheptatrienyl tetrafluoroborate to effect the net C–B to C–C bond substitution to give (S)-15 (95%, 95:5 er) in excellent yield. Similarly, the net stereoinvertive C–B to C–N bond substitution is accomplished by treating the in situ generated boron ate-complex with 4-methoxybenzenediazonium tetrafluoroborate to give the diazo compound (R)-16 (55%, 94:6 er).
Additional substrate scope and some key mechanistic insights
The question naturally arises as to whether the phosphonate directing group is unique in promoting γ-boration with these vinyl arene substrates. That appears not to be the case (Fig. 5A); the corresponding benzylamide substrate (E)-17 undergoes regioselective (7:1 rr) amide-directed CAHB using (R)-B1 to afford the analogous γ-borated benzylic ester (S)-18 (78%, 94:6 er).24
 | ||
| Fig. 5 The influence of other structural variations on the directed CAHB of 1,2-disubstituted alkenes: (A) analogous amide substrate undergoes efficient CAHB using B1; (B) one-carbon homologous substrates perform with similar efficiency: Same major enantiomer obtained from (E)- and (Z)-substrates. Note: Yield and er are determined after oxidation to the corresponding alcohols. aZ:E ratio = 4:1. | ||
The one-carbon homolog of (E)-5a, that is, the γ,δ-unsaturated phosphonate substrate (E)-19, reacts under the standard conditions to afford predominantly (>20:1 rr) δ-borated benzylic boronic ester (S)-20 (79%, 94:6 er) after oxidation (Fig. 5B). Furthermore, as was noted for (E)- and (Z)-5a, the same major enantiomer of 20 is formed independent of the substrate alkene geometry; (Z)-19 also affords (S)-20 (81%, 94:6 er). The ability to start with pure (E)-, pure (Z)- or an (E/Z)-mixture and arrive at the same product is a practical advantage but raises mechanistic questions. We previously reported that the amide-directed CAHBs of (E)- and (Z)-trisubstituted alkenes proceed with the same sense of π-facial selectivity and therefore lead to diastereomers.5f Those results would have suggested that (E)- and (Z)-19 (and similarly (E)- and (Z)-5a) should lead to enantiomers not the same product.
The origin of (E/Z)-isomer stereoconvergence is resolved based on the results of CAHB of (Z)-5a using a limiting amount of pinBH (Fig. 6A) and deuterium labelling via CAHB of (E)- and (Z)-5a with pinBD (Fig. 6B). With respect to the first test, a sample enriched in (Z)-isomer of 5a (90:10 Z:E) is subjected to the otherwise standard CAHB conditions, but using a limiting amount of pinBH (0.4 equiv.), leading to partial boration and recovered alkene. The 1H NMR spectral windows for the starting and recovered E/Z-mixtures of 5a shown in Fig. 6A indicate that (Z)-5a is essentially completely converted to the (E)-isomer under the reaction conditions, thus providing a mechanism by which the two isomers lead to the same product.
 | ||
| Fig. 6 Mechanistic studies indicating the origin of (E/Z)-stereoconvergent CAHB arising via Z to E isomerization under the reaction conditions: (A) (Z)- to (E)-substrate isomerization is observed under standard CAHB conditions; (B) different distributions of deuterated products are obtained when isomeric (E)- or (Z)- substrates react with pinBD under standard CAHB conditions. | ||
Fig. 6B shows the results of deuterium labelling. CAHB of (E)- and (Z)-5a using pinBD affords different distributions of non-, isomeric mono- and di-deuterated products. CAHB/oxidation of (E)-5a using pinBD yields essentially a single monodeuterated product 2-d-(2S,3S)-9a (83%) accompanied by the non-deuterated (3S)-9a (17%) with no apparent di-deuteration as determined by mass spectral analysis. In contrast, CAHB/oxidation of (Z)-5a using pinBD affords: 30% of non-deuterated (3S)-9a; 47% of a mixture of diastereomeric monodeuterated products 2-d-(2S,3S)-9a; and 2-d-(2R,3S)-9a and 21% of the dideuterated product 2,2-d2-(3S)-9a. The dideuterated product presumably arises by the reaction of 2-d-(E)-5a with pinBD. The latter, when generated in situ via isomerization of (Z)-5a as described above, also generates an equivalent amount of pinBH that can react competitively to give the observed diastereomeric monodeuterated product and an increased amount of the non-deuterated product as is observed. See the ESI† for more detailed mechanistic schemes.
Some current limitations
The attempted CAHB of several related phosphonates reveal some current limitations of the [Rh(nbd)2BF4/B1] catalyst system (Fig. 7). (i) The vinyl (i.e., α,β-unsaturated) phosphonate (E)-21 is largely recovered unchanged when subjected to the standard CAHB conditions. (ii) The trisubstituted variants (E)-22 and (E)-23 react only sluggishly under the standard conditions. (iii) In contrast to the vinylarene substrate (E)-5a, similar internal alkenes bearing an alkyl rather than aryl/heteroaryl γ-substituent (e.g., (E)-24), undergo predominantly β-boration, albeit with modest regio- and enantioselectivity with the standard catalyst system. (iv) The δ,ε-unsaturated vinyl arene (E)-25 affords the corresponding benzylic boronic ester upon CAHB but again with only modest regioselectivity, yield, and enantiopurity. It should be noted, however, that these results can only be said to reflect limitations of the [Rh(nbd)2BF4/B1] catalyst system; systematic catalyst optimizations have not been carried out for these substrates.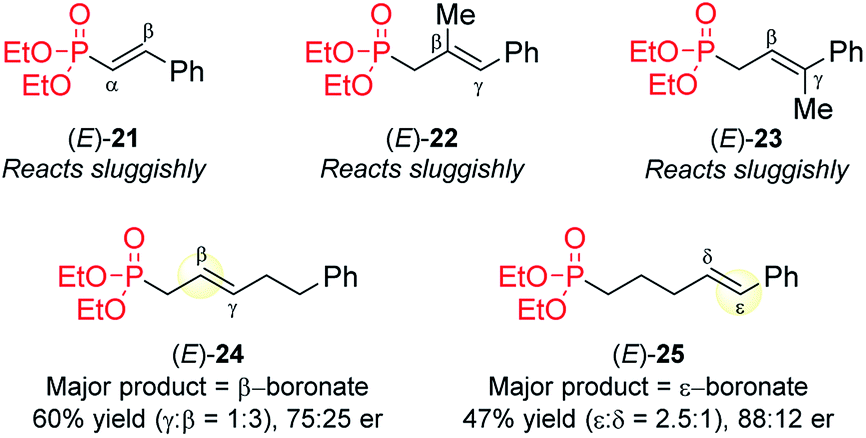 | ||
| Fig. 7 Current limitations of the methodology. | ||
Conclusions
While the regioselective, CAHBs of minimally-functionalized terminal and 1,1-disubstituted vinyl arenes (e.g. simple substituted styrene derivatives) have been investigated rather extensively and quite successfully, there are relatively few reports using more highly functionalized di- or trisubstituted internal alkenes. We find that the rhodium-catalyzed CAHBs of allylic and homoallylic phosphonates derived from internal vinyl arenes give facile access to functionalized chiral secondary benzylic boronic esters. A range of substrates including some bearing heteroaromatic ring systems of interest in medicinal chemistry, such as furan, indole, morpholine, pyrazine, pyrrole and thiophene derivatives, can be accommodated. The absolute configurations of selected chiral secondary benzylic boronic ester products were confirmed or assigned via kinetic acylation of the corresponding benzylic alcohols using the BTM acylation catalyst highlighting the potential for other donor substituents in proximity of the alkene to influence the stereochemical course of the reaction. Stereoretentive and stereoinvertive C–B bond transformation protocols highlight the versatility of this methodology. We find that palladium-catalyzed cross-coupling under the conditions reported by Crudden proceeds mostly with stereoretention. Diastereomeric substrates, for example, (E)- and (Z)-5a and (E)- and (Z)-16, give the same borated product stereochemistry. Mechanistic studies reveal that (Z)- to (E)-alkene isomerization occurs rapidly under the reaction conditions. This observation along with deuterium incorporation data provides a reasonable explanation for the origin of (E/Z)-stereoconvergence during CAHB.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Funding from the NIH National Institutes of General Medical Sciences (R01 GM100101) is gratefully acknowledged. We thank Dr Jian Zhang, Dr Jingshi Lu and Mr Nicholas Armada for assistance in preparing (Z)-5a and (Z)-19via photoisomerization.25Notes and references
- For leading examples, see (a) J. Wu, P. Lorenzo, S. Zhong, M. Ali, C. P. Butts, E. L. Myers and V. K. Aggarwal, Nature, 2017, 547, 436 CrossRef CAS PubMed; (b) B. S. L. Collins, C. M. Wilson, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 11700 CrossRef CAS PubMed; (c) J. Schmidt, J. Choi, A. T. Liu, M. Slusarczyk and G. C. Fu, Science, 2016, 354, 1265 CrossRef CAS PubMed; (d) C. M. Crudden, C. Ziebenhaus, J. P. G. Rygus, K. Ghozati, P. J. Unsworth, M. Nambo, S. Voth, M. Hutchinson, V. S. Laberge, Y. Maekawa and D. Imao, Nat. Commun., 2016, 7, 11065 CrossRef PubMed; (e) S. C. Matthew, B. W. Glasspoole, P. Eisenberger and C. M. Crudden, J. Am. Chem. Soc., 2014, 136, 5828 CrossRef CAS PubMed.
- For selected examples, see: (a) L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70 CrossRef CAS PubMed; (b) A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584 CrossRef CAS PubMed; (c) M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 513, 183 CrossRef CAS PubMed; (d) C. G. Watson, A. Balanta, T. G. Elford, S. Essafi, J. N. Harvey and V. K. Aggarwal, J. Am. Chem. Soc., 2014, 136, 17370 CrossRef CAS PubMed; (e) R. P. Sonawane, V. Jheengut, C. Rabalakos, R. Larouche-Gauthier, H. K. Scott and V. K. Aggarwal, Angew. Chem., Int. Ed., 2011, 50, 3760 CrossRef CAS PubMed; (f) R. Larouche-Gauthier, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2011, 133, 16794 CrossRef CAS PubMed. For reviews, see: (g) C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481 RSC; (h) R. J. Armstrong and V. K. Aggarwal, Synthesis, 2017, 49, 3323 CrossRef CAS; (i) D. Leonori and V. K. Aggarwal, Angew. Chem., Int. Ed., 2015, 54, 1082 CrossRef CAS PubMed.
- For key examples, see (a) B. Liu, H.-H. Wu and J. Zhang, ACS Catal., 2018, 8, 8318 CrossRef CAS; (b) Y. Cai, X.-T. Yang, S.-Q. Zhang, F. Li, Y.-Q. Li, L.-X. Ruan, X. Hong and S.-L. Shi, Angew. Chem., Int. Ed., 2018, 57, 1376 CrossRef CAS PubMed; (c) L. Chen, X. Zou, H. Zhao and S. Xu, Org. Lett., 2017, 19, 3676 CrossRef CAS PubMed; (d) C. Jarava-Barrera, A. Parra, A. Lopez, F. Cruz-Acosta, D. Collado-Sanz, D. J. Cardenas and M. Tortosa, ACS Catal., 2016, 6, 442 CrossRef CAS PubMed; (e) H. Lee, B. Y. Lee and J. Yun, Org. Lett., 2015, 17, 764 CrossRef CAS PubMed; (f) Z. T. He, Y. S. Zhao, P. Tian, C. C. Wang, H. Q. Dong and G. Q. Lin, Org. Lett., 2014, 16, 1426 CrossRef CAS PubMed; (g) A. Parra, L. Amenos, M. Guisan-Ceinos, A. Lopez, J. L. G. Ruano and M. Tortosa, J. Am. Chem. Soc., 2014, 136, 15833 CrossRef CAS PubMed; (h) J. C. H. Lee, R. McDonald and D. G. Hall, Nat. Chem., 2011, 3, 894 CrossRef CAS PubMed; (i) Y. Sasaki, C. Zhong, M. Sawamura and H. Ito, J. Am. Chem. Soc., 2010, 132, 1226 CrossRef CAS PubMed; (j) Y. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 3160 CrossRef CAS PubMed; (k) J. M. O'Brien, K. S. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 10630 CrossRef PubMed; (l) I.-H. Chen, L. Yin, W. Itano, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 11664 CrossRef CAS PubMed.
- For selected examples, see: (a) W. J. Jang, S. M. Song, J. H. Moon, J. Y. Lee and J. Yun, J. Am. Chem. Soc., 2017, 139, 13660 CrossRef CAS PubMed; (b) J. R. Smith, B. S. L. Collins, M. J. Hesse, M. A. Graham, E. L. Myers and V. K. Aggarwal, J. Am. Chem. Soc., 2017, 139, 9148 CrossRef CAS PubMed; (c) Y. Xi and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 6703 CrossRef CAS PubMed; (d) N. Hu, G. Zhao, Y. Zhang, X. Liu, G. Li and W. Tang, J. Am. Chem. Soc., 2015, 137, 6746 CrossRef CAS PubMed.
- (a) S. Chakrabarty and J. M. Takacs, ACS Catal., 2018, 8, 10530 CrossRef CAS; (b) S. Chakrabarty and J. M. Takacs, J. Am. Chem. Soc., 2017, 139, 6066 CrossRef CAS PubMed; (c) G. L. Hoang and J. M. Takacs, Chem. Sci., 2017, 8, 4511 RSC; (d) V. M. Shoba, N. C. Thacker, A. J. Bochat and J. M. Takacs, Angew. Chem., Int. Ed., 2016, 55, 1465 CrossRef CAS PubMed; (e) G. L. Hoang, Z. D. Yang, S. M. Smith, R. Pal, J. L. Miska, D. E. Perez, L. S. W. Pelter, X. C. Zeng and J. M. Takacs, Org. Lett., 2015, 17, 940 CrossRef CAS PubMed; (f) S. M. Smith and J. M. Takacs, J. Am. Chem. Soc., 2010, 132, 1740 CrossRef CAS PubMed; (g) S. M. Smith, N. C. Thacker and J. M. Takacs, J. Am. Chem. Soc., 2008, 130, 3734 CrossRef CAS PubMed.
- Selected examples of CAHB of 1,1-disubstituted vinyl arenes: (a) Z. Wang, Z. He, R. Zhang, G. Zhang, G. Xu, Q. Zhang, T. Xiong and Q. Zhang, Org. Lett., 2017, 19, 3067 CrossRef CAS PubMed; (b) H. Zhang and Z. Lu, ACS Catal., 2016, 6, 6596 CrossRef CAS; (c) L. Zhang, Z. Zuo, X. Wan and Z. J. Huang, J. Am. Chem. Soc., 2014, 136, 15501 CrossRef CAS PubMed; (d) R. Corberan, N. W. Mszar and A. H. Hoveyda, Angew. Chem., Int. Ed., 2011, 50, 7079 CrossRef CAS PubMed; (e) C. Mazet and D. Gerard, Chem. Commun., 2011, 47, 298 RSC.
- For CAHB of 1,2-disubstituted vinyl arenes, see: (a) G. L. Hoang, S. Zhang and J. M. Takacs, Chem. Commun., 2018, 54, 4838 RSC; (b) D. Noh, S. K. Yoon, J. Won, J. Y. Lee and J. Yun, Chem.–Asian J., 2011, 6, 1967 CrossRef CAS PubMed . For CAHB of trisubstituted internal alkenes (bearing alkyl/aryl substituents), see ref. 5a, b, d and f.
- Other recent methods for synthesizing chiral secondary benzylic/α-aryl boronic esters: (a) Z. Tao, K. A. Robb, J. L. Panger and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 15621 CrossRef CAS PubMed; (b) J. Guo, B. Cheng, X. Shen and Z. Lu, J. Am. Chem. Soc., 2017, 139, 15316 CrossRef CAS PubMed; (c) C. Margarita and P. G. Andersson, J. Am. Chem. Soc., 2017, 139, 1346 CrossRef CAS PubMed; (d) C. H. Basch, K. M. Cobb and M. P. Watson, Org. Lett., 2016, 18, 136 CrossRef CAS PubMed; (e) H. M. Nelson, B. D. Williams, J. Miró and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 3213 CrossRef CAS PubMed; (f) C. Sun, B. Potter and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 6534 CrossRef CAS PubMed; (g) X. Feng, H. Jeon and J. Yun, Angew. Chem., Int. Ed., 2013, 52, 3989 CrossRef CAS PubMed; (h) A. Guzman-Martinez and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 10634 CrossRef CAS PubMed.
- D. R. Edwards, Y. B. Hleba, C. J. Lata, L. A. Calhoun and C. M. Crudden, Angew. Chem., Int. Ed., 2007, 46, 7799 CrossRef CAS PubMed.
- Y. H. Zhang, C. F. Xu, J. F. Li and C. Y. Yuan, Chin. J. Chem., 2003, 21, 883 CrossRef CAS.
- S. A. Westcott, H. P. Blom and T. B. Marder, Inorg. Chem., 1993, 32, 2175 CrossRef CAS.
- Our recent studies on rhodium-catalyzed CAHB of functionalized alkene substrates focus on using the simple and readily available TADDOL- and BINOL-derived chiral phosphites and phosphoramidites. Our studies consistently find that these two privileged ligand scaffolds tend to be complementary in their effectiveness; in part, the choice of TADDOL- or BINOL-derived ligands hinges on the alkene substitution pattern.
- The parent BINOL-derived ligand B2 gives a similar yield and regioselectivity but lower enantioselectivity for (S)-9a (90:10 er); see the ESI† for ligand screening data.
- (a) V. M. Shoba and J. M. Takacs, J. Am. Chem. Soc., 2017, 139, 5740 CrossRef CAS PubMed; (b) N. G. Leonard and P. J. Chirik, ACS Catal., 2018, 8, 342 CrossRef CAS ; see also ref. 8b.
- The regioisomeric γ- and β-boronic esters 6 and 7 often proved difficult to separate by chromatography on silica gel. Thus, the 6:7 ratio is determined via 31P NMR analysis of crude reaction mixture. The isolated yield of the major γ-regioisomer and its er are obtained after oxidation.
- It is worth noting that unlike our previous work on 1,1-disubstituted alkenes (ref. 5a) in which ortho-substitution of the arene leads to a change in regioselectivity, substrates bearing 2-substituted arenes in this series of 1,2-disubstituted internal vinylarenes do not significantly impact the regioselectivity. For example, in addition to 5h, the 2-methylphenyl derived substrate 5 (Ar = 2-methylphenyl) also undergoes regioselective γ-boration (13:1 rr) to afford 6 (Ar = 2-methylphenyl) but with a relatively modest level of enantioselectivity determined after oxidation (74%, 91:9 er); see the ESI.†.
- V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351 CrossRef CAS PubMed.
- For examples of stereoretentive cross-coupling of chiral secondary organoboron derivatives, see ref. 5c, e and (a) T. P. Blaisdell and J. P. Morken, J. Am. Chem. Soc., 2015, 137, 8712 CrossRef CAS PubMed; (b) M. Daini and M. Suginome, J. Am. Chem. Soc., 2011, 133, 475 CrossRef PubMed.
- For examples of stereoinvertive cross-coupling of chiral secondary organoboron derivatives, see: (a) J. C. H. Lee, H.-Y. Sun and D. G. Hall, J. Org. Chem., 2015, 80, 7134 CrossRef CAS PubMed; (b) L. Li, S. Zhao, A. Joshi-Pangu, M. Diane and M. R. Biscoe, J. Am. Chem. Soc., 2014, 136, 14027 CrossRef CAS PubMed; (c) J. C. Lee, R. McDonald and D. G. Hall, Nat. Chem., 2011, 3, 894 CrossRef CAS PubMed; (d) T. Awano, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2011, 133, 20738 CrossRef CAS PubMed; (e) D. L. Sandrock, L. Jean-Gerard, C. Chen, S. D. Dreher and G. A. Molander, J. Am. Chem. Soc., 2010, 132, 17108 CrossRef CAS PubMed; (f) T. Ohmura, T. Awano and M. Suginome, J. Am. Chem. Soc., 2010, 132, 13191 CrossRef CAS PubMed.
- For ligand-controlled stereoretentive and stereoinvertive cross-coupling of chiral secondary organoboron derivatives, see: S. Zhao, T. Gensch, B. Murray, Z. L. Niemeyer, M. S. Sigman and M. R. Biscoe, Science, 2018, 362, 670 CrossRef CAS PubMed.
- For a perspective on enantiospecific and iterative Suzuki-Miyaura cross-couplings, see: J. P. G. Rygus and C. M. Crudden, J. Am. Chem. Soc., 2017, 139, 18124 CrossRef CAS PubMed.
- D. Imao, B. W. Glasspoole, V. S. Laberge and C. M. Crudden, J. Am. Chem. Soc., 2009, 131, 5024 CrossRef CAS PubMed.
- A. Ros and V. K. Aggarwal, Angew. Chem., Int. Ed., 2009, 48, 6289 CrossRef CAS PubMed.
- The 3,3′-bisphenyl substituents in ligand B1 play a significant role in determining regioselectivity. B2, the analogous BINOL-derived phosphoramidite derived from the parent BIONOL ring system, gives a slight preference for β-boration (ca. 2:1 rr) with (E)-14. Using B2, γ,δ-unsaturated amides afford δ-borated benzylic boronic esters in excellent yields and enantioselectivities; see: ref. 7a.
- J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang and J. Zhang, J. Am. Chem. Soc., 2018, 140, 13719 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data & dpectral data. See DOI: 10.1039/c8sc05613g |
| This journal is © The Royal Society of Chemistry 2019 |