DOI:
10.1039/C8SC05439H
(Edge Article)
Chem. Sci., 2019,
10, 4322-4327
Access to P-chiral phosphine oxides by enantioselective allylic alkylation of bisphenols†
Received
5th December 2018
, Accepted 12th March 2019
First published on 12th March 2019
Abstract
A novel biscinchona alkaloid-catalyzed highly enantioselective desymmetrization reaction of bisphenol compounds with achiral Morita–Baylis–Hillman carbonate agents was developed. Through the asymmetric allylic alkylation strategy, a broad range of optically active P-stereogenic phosphine oxides were generated with excellent to good yields (up to 99%) and high enantioselectivities (up to 98.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 e.r.). The reaction was further investigated by the linear free energy relationship (LFER) analysis. A possible transition state was proposed and furthered verified by theoretical calculations.
:1.5 e.r.). The reaction was further investigated by the linear free energy relationship (LFER) analysis. A possible transition state was proposed and furthered verified by theoretical calculations.
Introduction
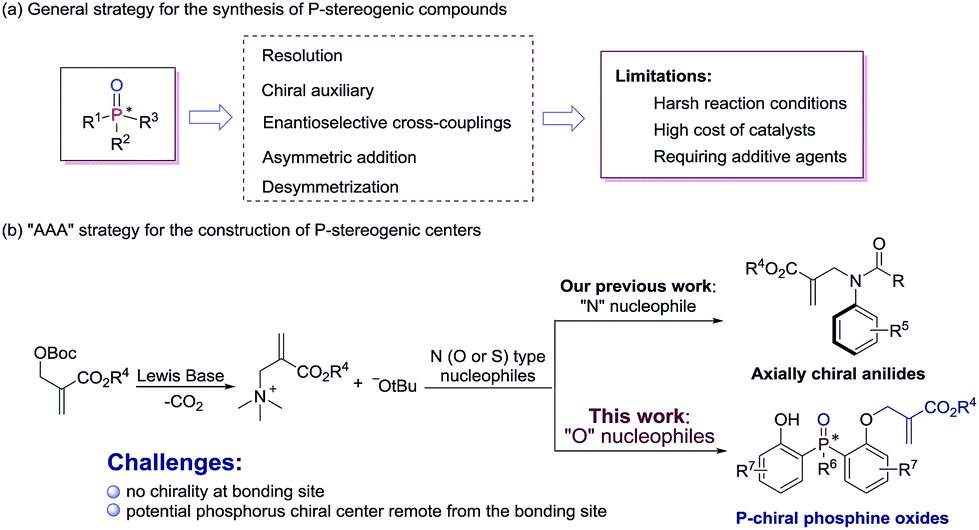
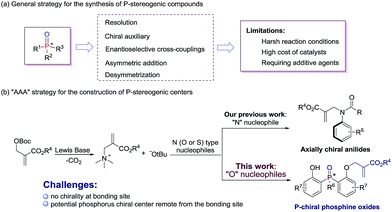
P-stereogenic compounds, in which the chirality is on the phosphorus atom, have been widely used as biologically active compounds,1 chiral ligands2 and useful building blocks.3 The significance of this privileged structural motif with P-stereogenic centers has led to a great demand for efficient synthetic methods. However, the methodologies used to synthesize such chiral structures were largely limited.4 Early reports on P-stereogenic center synthesis comprise the resolution of diastereoisomers,5 chiral auxiliary controlled asymmetric reactions,6 transition-metal-catalyzed enantioselective cross-coupling,7 asymmetric addition reactions of phosphorus nucleophiles,8 and desymmetrization of prochiral phosphorus derivatives (Scheme 1a).9 Among the above mentioned synthetic strategies of P-stereogenic centers, only two cases were achieved through organocatalysis.10 Therefore, it is currently desirable and challenging to develop an organocatalysis-based synthetic methodology for P-stereogenic centers, and to expand the facile and extensive substrate adaptability.
 |
| | Scheme 1 (a) Methods for the synthesis of P-stereogenic compounds and (b) our design. | |
Lewis base catalyzed asymmetric allylic alkylation (AAA) reaction with Morita–Baylis–Hillman (MBH) carbonates as electrophile precursors has been considered as one of the most attractive approaches to build stereogenic centers. However, the AAA strategy currently is restricted to the building of chiral carbon centers, because most frequently used allylation reagents are racemic MBH adducts.11 In fact, it is possible to expand the usage of the AAA strategy to building other non-carbon center chirality when the reactions proceed between achiral MBH adducts and N (O or S) type nucleophiles. Our group recently developed a general and efficient method for atroposelective construction of axially chiral anilides (Scheme 1b).12
In continuation of our ongoing interest in the AAA strategy, we speculated that the privileged phosphorus compounds with P-stereogenic centers could be generated via the AAA reaction between bis(2-hydroxyphenyl)phosphinates and achiral MBH carbonates (Scheme 1b). Thus, two challenges need to be solved in this scenario: (i) the difficulty in controlling stereoselectivities due to the lack of chirality at the bonding site; (ii) the difficulty in designing a proper chiral catalyst to induce enantiocontrol during the course of desymmetrization, because the targeted phosphorus center is remote from the enantiotopic site. Herein, we report a biscinchona alkaloid-catalyzed enantioselective desymmetrization of bis(2-hydroxyphenyl)phosphine oxides. Utilizing the AAA strategy based method, a number of phosphine oxides possessing chiral phosphorus(V) atoms were synthesized with good enantioselectivities.
Results and discussion
Reaction optimization
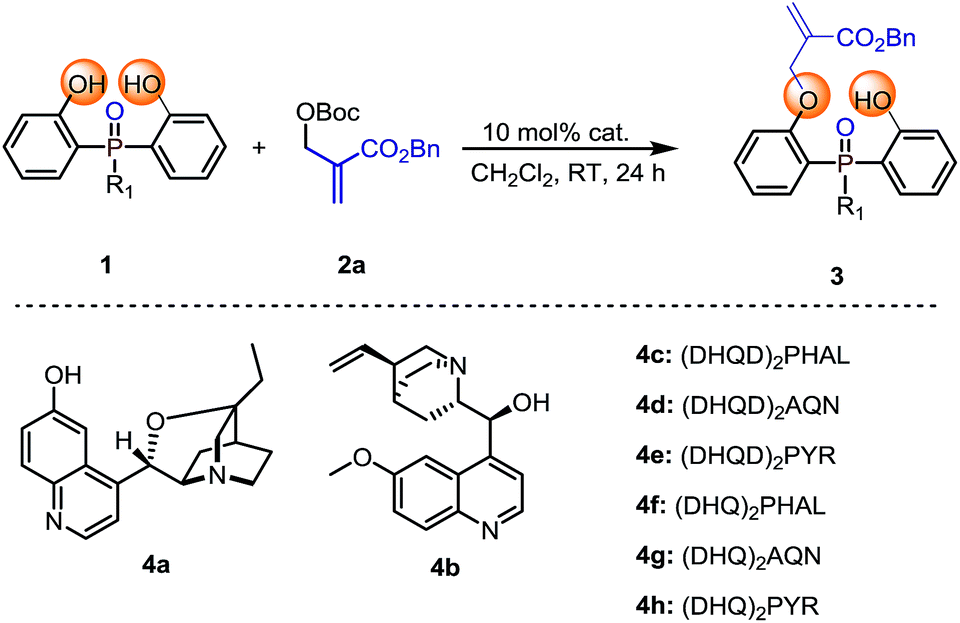
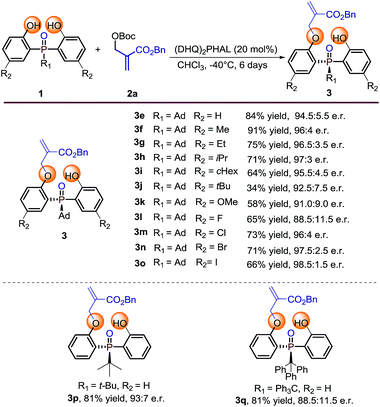
Our initial investigation was carried out with bis(2-hydroxyphenyl)phosphinate 1a and Boc protected MBH product 2a as the model substrates, 10 mol% of cinchona alkaloid 4a as the catalyst in CH2Cl2 at room temperature. The reaction gave the desymmetrization product 3a in 96% yield, albeit with a racemic result (Table 1, entry 1). To improve the enantioselectivity, we next evaluated different types of cinchona alkaloid catalysts. As shown in Table 1, the catalysts' backbone demonstrated remarkable impacts towards the outcome of the reaction (Table 1, entries 2–8), in which biscinchona alkaloid catalyst 4f gave the best 66:34 e.r (Table 1, entry 6). To further improve the enantioselectivity, we then screened different bisphenol substrates. To our delight, substrates 1b–1d, which had large sized ester groups, showed improved enantioselectivities (Table 1, entries 9–11). This result indicated that the enantioselectivity of the studied desymmetrization reaction may be closely related to the steric hindrance of the substituent linked to the pre-stereogenic P-center. This hypothesis was furthermore supported by the reaction with substrate phosphine oxide 1e, in which the enantioselectivity was obtained as 84:16 e.r (Table 1, entry 12). A series of subsequent screenings (for example, temperature, solvent, and substrate ratio) showed enhancement of the enantioselectivity. Lowering the reaction temperature to −40 °C can further increase the e.r. value to 94:6, while increasing the catalyst loading and prolonging the reaction time were necessary to ensure the conversion of the reaction (Table 1, entry 13). The evaluation of the solvents showed that CHCl3 was the best reaction medium in terms of reactivity and enantioselectivity (Table 1, entries 13–17). Increasing amount of substrate 2a obtained an improved yield of 84% with retention of 94.5:5.5 e.r (Table 1, entry 18).
Table 1 Reaction optimizationa
Substrate evaluation
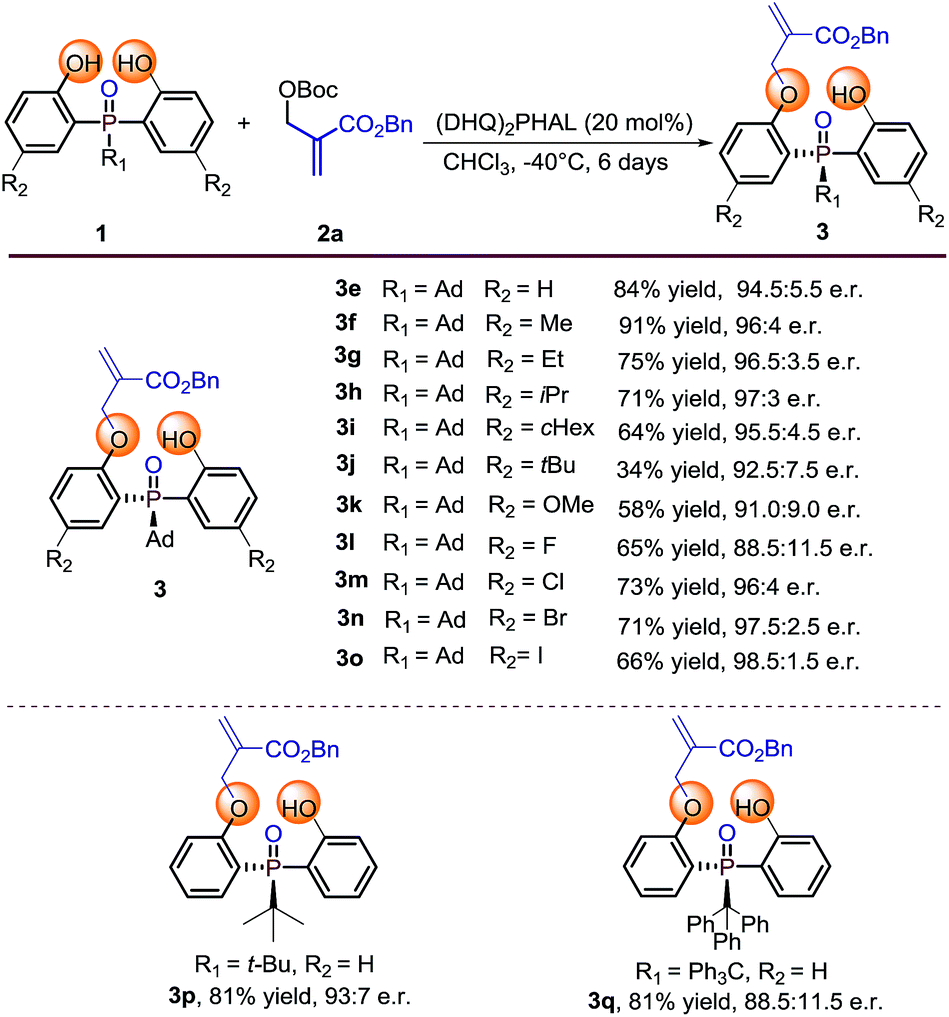
With the optimal reaction conditions in hand, we set out to explore the substrate generality of this desymmetrization strategy. Firstly, different substituted phosphine oxides were investigated. As shown in Table 2, substrates with electron-donating groups and electron-withdrawing groups on the phenyl ring were well tolerated in this reaction. The corresponding products 3e–3k and 3m–3o were afforded in 34–91% yields and 91.0:9.0–98.5:1.5 e.r. values. When the substituted atom was F, the enantioselectivity decreased to a moderate level (3l, 65% yield and 88.5:11.5 e.r.). It is valuable to note that the steric hindrance of the substituent seems to also influence on the reactivity. For example, the substrate with R2 as the tert-butyl group only provided the corresponding product 3j with 34% yield under the optimized reaction conditions. Two other substrates with large steric hindrance phosphine oxides, in which the groups linked to the P-center were tert-butyl and triphenyl methyl, were also examined. As a result, 3p was obtained in 81% yield with 93:7 e.r. and 3q was obtained in 81% yield with 88.5:11.5 e.r.
Table 2 Substrate scope of phosphine oxide substratesa
|
Reaction conditions: phosphine oxides 1 (0.1 mmol), MBH carbonate 2a (0.2 mmol), catalyst (20 mol%), 1 mL CHCl3. Isolated yields. e.r. values were determined by chiral HPLC analysis.
|
|
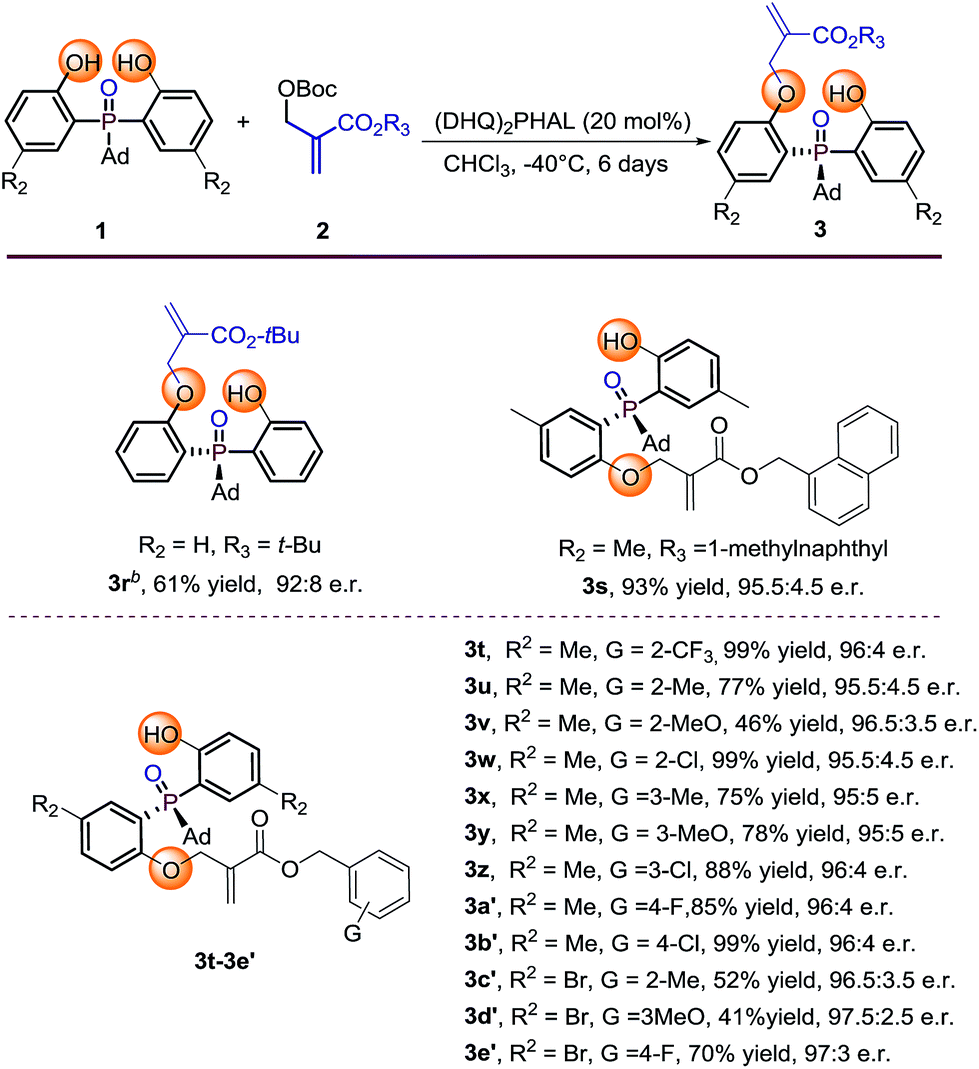
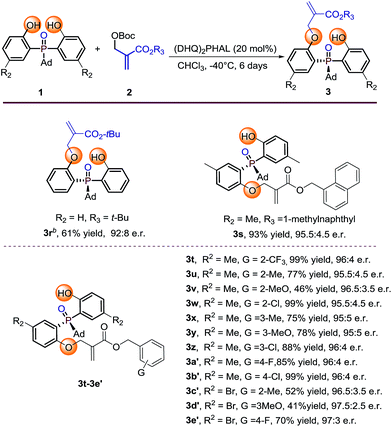
Further investigation of the substrate scope was focused on the Boc protected MBH carbonate substrates 2 (Table 3). The electronic nature or position of the substituent on the benzyl ring does not appear to affect the results, all the benzyl-substituted MBH carbonates afforded the target products 3t–3e′ in moderate to excellent yields (41–99%) with excellent enantioselectivities (95.5:4.5 to 97.5:2.5 e.r.). The 1-methylnaphthyl type MBH carbonate tolerated well under the optimal condition, gave 3s in 93% yield with 95.5:4.5 e.r. The tert-butyl substituted MBH carbonate is also transformed to the desired product 3r in 61% yield and 92:8 e.r. by running the reaction at 0 °C albeit with large steric hindrance. The absolute configuration of 3r was determined by X-ray diffraction analysis and those of other products were assigned by analogy.13
Table 3 Substrate scope of MBH carbonate substratesa
|
Reaction conditions: phosphine oxides 1 (0.1 mmol), MBH carbonates 2 (0.2 mmol), catalyst (20 mol%), 1 mL CHCl3. Isolated yields. e.r. values were determined by chiral HPLC analysis.
The reaction was conducted at 0 °C.
|
|
LFER research studies
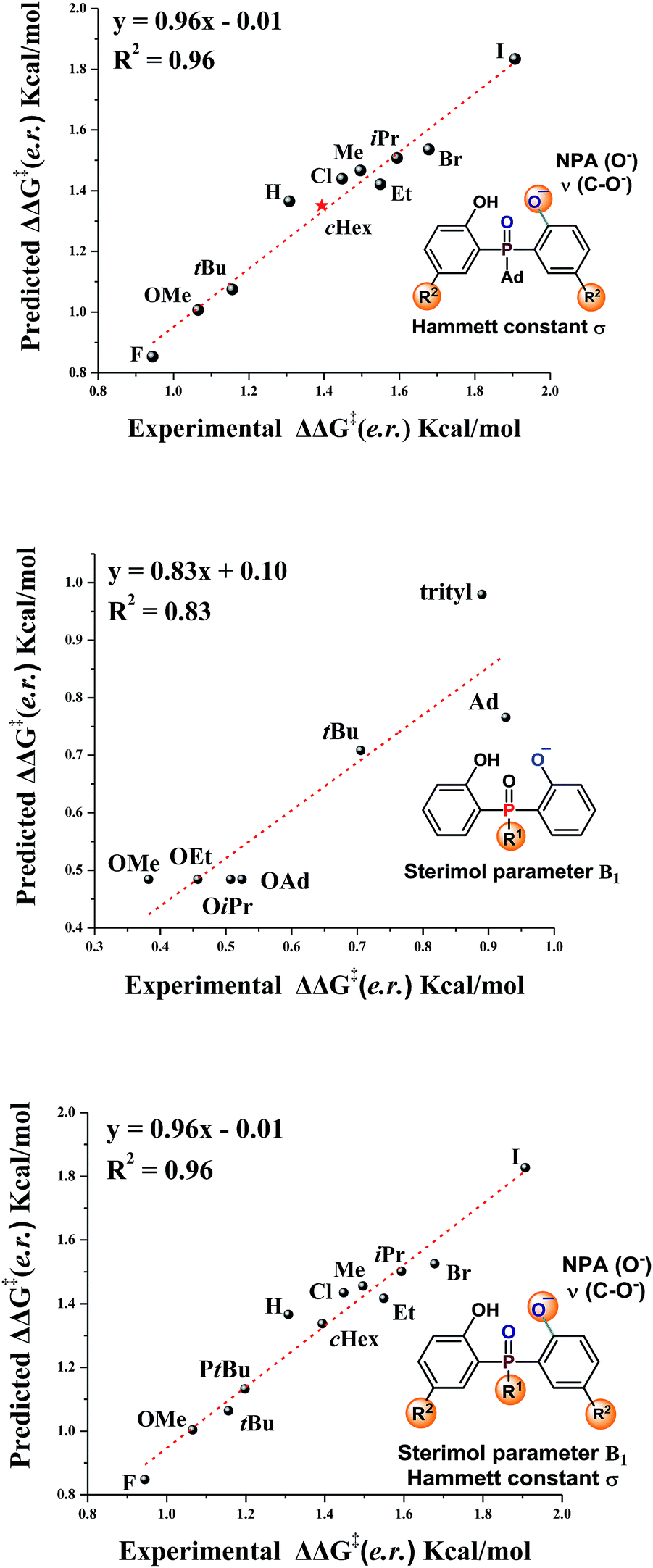
The substrate scope study reveals that, in general, products with large sized substituents on the aromatic ring of phosphine have better enantioselectivity. To investigate the effect of the steric factor on the enantioselectivity, we plotted the enantioselectivities against Charton values.14 Preliminary analysis revealed that substrates having large steric substituents R2 gave better enantioselectivities (For R2 = H, Me, Et, iPr, see Fig. S1 in the ESI†). This rule could also be expanded to substrates containing halogen substituents (R2 = F, Cl, Br, I, see Fig. S2 in the ESI†). It seemed that the steric factor may play a role in enantioselectivity control. However, the steric correlation could not explain the stereoselectivity for the substrates containing other large hindrance substituents, such as R2 = tBu or cHex. Thus, other factors, such as the electronic effect, should also be considered.
Recently, Sigman and coworkers utilized multivariate linear regression (MLR) models to effectively predict the experimental reaction outcome based on both experimentally derived and calculated physical organic molecular descriptors.15 Inspired by their work and our previous related study,12 we explored the factors that govern the stereoselectivity. The regression analysis was made with ten data sets and tested with one data set. After evaluating various parameters, the Hammett constant (σ), NPA charge (O−) and ν (C–O−),15b,c which can be used to embody the steric and electronic effects, were found to accurately predict the stereoselectivity (eqn (1), Fig. 1, slope = 0.96, intercept = −0.01, R2 = 0.96). Besides, the effects of substituents linked to the pre-stereogenic P-center were evaluated. The stereoselectivity is in good correlation with Sterimol parameter B1 (eqn (2)).16 The steric factor of substituents linked to the phosphorus atom significantly affected the enantioselectivity. Finally, based on eqn (2), eqn (1) could be expanded to substrates that have different substituents linked to the pre-stereogenic P-center (eqn (3), slope = 0.96, intercept = −0.01, R2 = 0.96).
| | | ΔΔG‡ (e.r.) = −3.04σ + 78.6NPA(O−) − 0.014ν(C–O−) + 88.2 | (1) |
| | | ΔΔG‡ (e.r.) = 0.16B1 + 0.24 | (2) |
| | | ΔΔG‡ (e.r.) = −3.03σ + 78.1NPA(O−) − 0.0144ν(C–O−) + 0.828B1 + 85.7 | (3) |
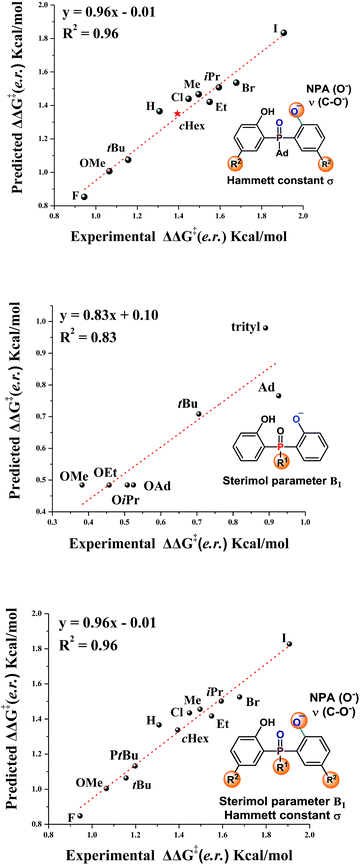 |
| | Fig. 1 Correlation of enantioselectivity with substrate parameters. | |
Theoretical calculations
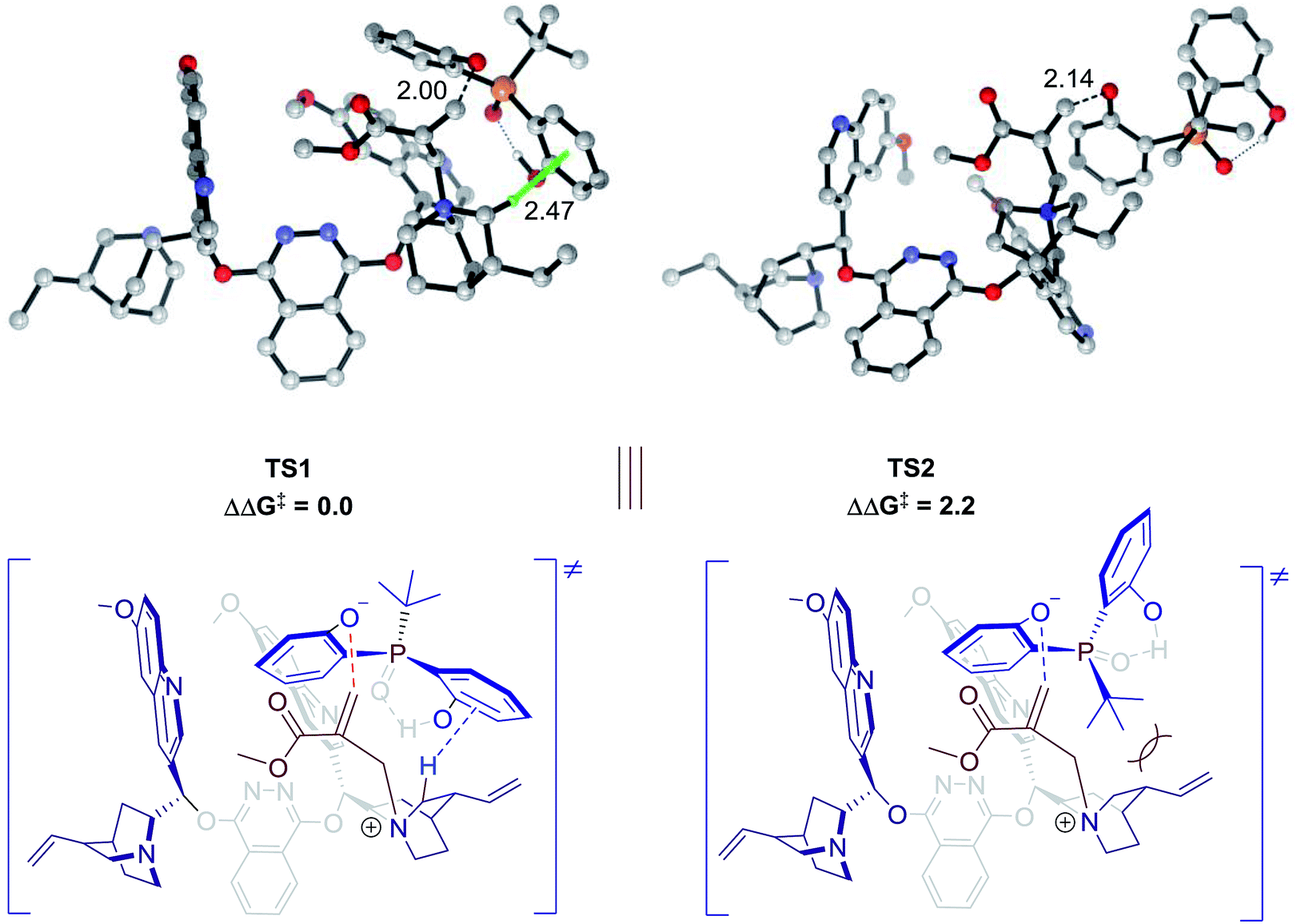
Theoretical calculations were conducted to support the above analysis.17,18 Extensive explorations of a variety of catalytic arrangements show that the two most stable transition state structures are TS1 and TS2 (Fig. 2). The free energy difference between TS1 and TS2 is 2.2 kcal mol−1, which agrees with experimental data, 85:15 e.r. As shown in Fig. 2, TS1 is stabilized by a C–H⋯π interaction between the methylene of the quinuclidine ring and the aromatic ring of phenol. However, this interaction is missing in TS2, which would be a key factor that contributes to the energy difference between TS1 and TS2. Previous studies of C–H⋯π interactions showed that the interaction energies correlated with Hammett constant (σm),19 which supported our LFER analysis. The steric factor of substituents linked to the pre-stereogenic P-center could also be explained by transition states. If the tert-butyl group linked to the phosphorus atom is replaced by an adamantyl group, the steric effect between the catalyst and substrate will destabilize TS2, which is crucial for excellent enantioselectivity control.
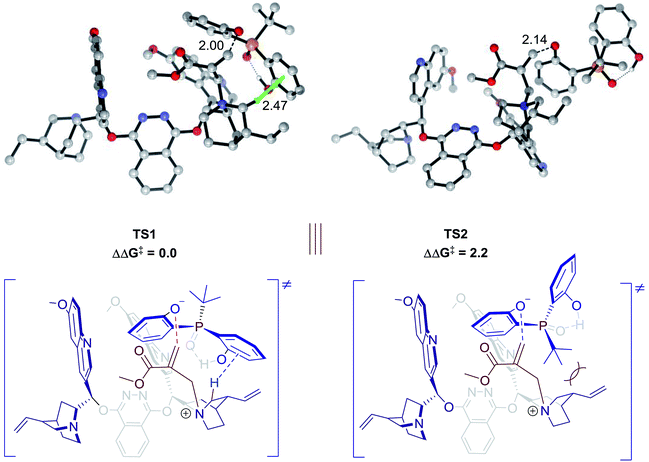 |
| | Fig. 2 Transition state structures and relative free energies (in kcal mol−1) for desymmetrization catalyzed by 4f. Noncritical hydrogen atoms have been omitted for clarity. The C–H⋯π interaction in TS1 is highlighted. | |
Large scale reaction and product transformation
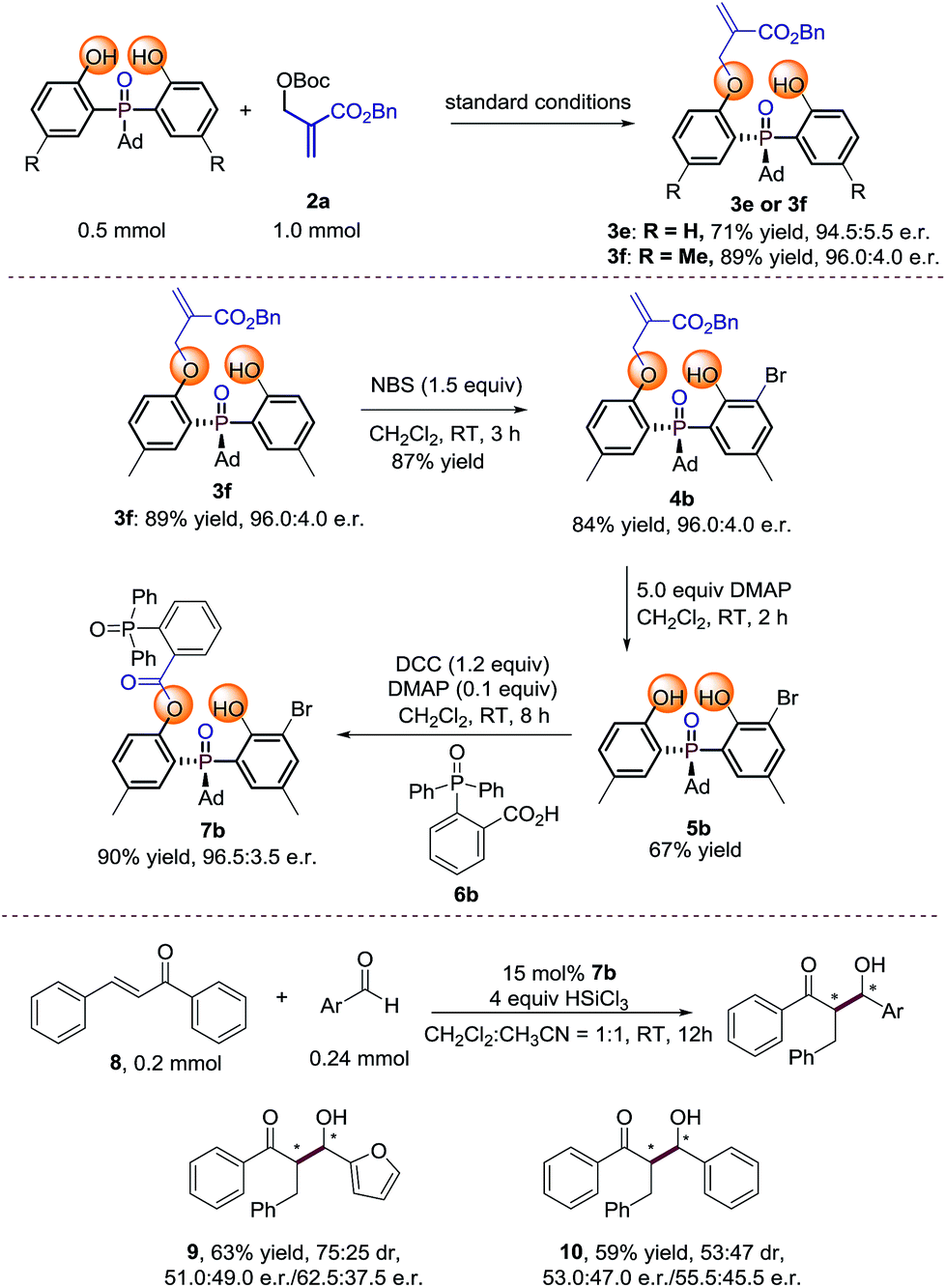
To probe the efficiency of the current studied desymmetrization strategy in preparative synthesis, large scale reactions were investigated under the optimal conditions. To our delight, the desired products 3e and 3f were obtained without any loss of enantioselectivity (Scheme 2). Further bromination of 3f by 1.5 equiv. NBS afforded 4b. Treating 4b with 5 equivalents of DMAP afforded 5b in moderate yield with retention of enantioselectivity. Finally, product 7b can be obtained in 90% yield with 96.5:3.5 e.r. under the conditions shown in Scheme 2. The preliminary application of the synthesized bidentate chiral phosphine oxide indicated that 7b could be used as a catalyst in asymmetric reactions between enones and aldehydes (Scheme 2).20
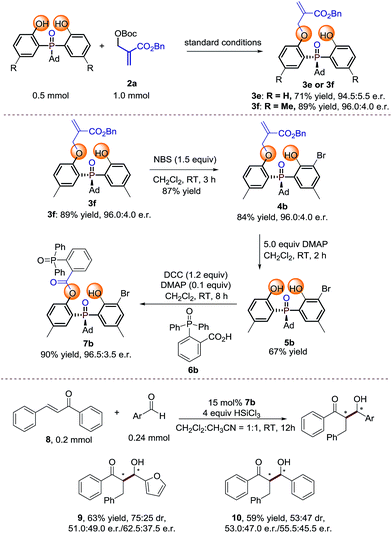 |
| | Scheme 2 Large scale reaction and synthetic transformations of the product.21 | |
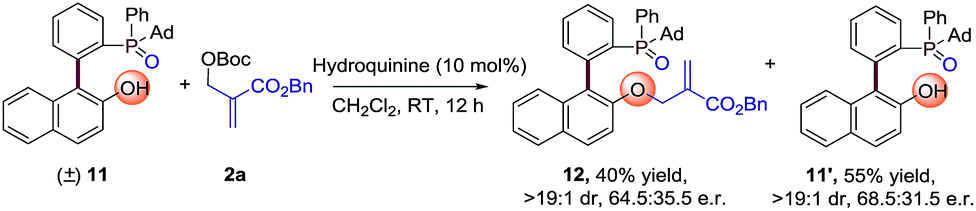
Moreover, we also made another kinetic resolution experiment with substrate (±)-11, which contains both axial and phosphorus prochirality (Scheme 3). The reaction of (±)-11 with 2a proceeds smoothly in the presence of 10 mol% hydroquinine under the standard conditions, resulting in 12 in 40% yield with 64.5:35.5 e.r. and 11’ in 55% yield with 68.5:31.5 e.r. Further optimization of reaction conditions may lead to better enantioselectivities. This result again proves the universality of the strategy for the synthesis of P-stereogenic compounds.
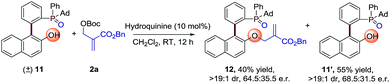 |
| | Scheme 3 Kinetic resolution experiment using the AAA strategy. | |
Conclusions
In summary, we have developed a catalytic enantioselective desymmetrization of bisphenols that hold pre-stereogenic P-centers, using a biscinchona alkaloid catalyst. This AAA strategy based method provides a novel and highly efficient way to the synthesis of P-stereogenic compounds, affording the desired functionalized phosphine oxides in good yields (up to 99%) and high enantioselectivities (up to 98.5:1.5 e.r.). A range of functional groups were tolerated under the mild reaction conditions. A possible transition state was proposed based on the linear free energy relationship analysis, which was further verified by theoretical calculations.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the NNSFC (grant no. 21390400) for financial support.
Notes and references
-
(a)
The Role of Phosphonates in Living Systemsk, ed. R. L. Hildebrand, CRC Press, Boca Raton, Fla, 1983 Search PubMed;
(b) G. Lavielle, P. Hautefaye, C. Schaeffer, J. A. Boutin, C. A. Cudennec and A. Pierre, J. Med. Chem., 1991, 34, 1998 CrossRef CAS PubMed;
(c) F. R. Atherton, C. H. Hassall and R. W. Lambert, J. Med. Chem., 1986, 29, 29 CrossRef CAS PubMed;
(d) K. Lin, S. Zhou, C. Xu and W. P. Liu, J. Agric. Food Chem., 2006, 54, 8134 CrossRef CAS PubMed.
-
(a)
Phosphorus Ligands in Asymmetric: Catalysis synthesis and applications, ed. A. Borner, Wiley-VCH, Weinheim, 2008 Search PubMed;
(b)
Phosphorus (III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, Wiley: Chichester, U.K., 2012. p. 3029 Search PubMed;
(c) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed;
(d) J. H. Xie and Q. L. Zhou, Acc. Chem. Res., 2008, 41, 581 CrossRef CAS PubMed.
-
(a) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS PubMed;
(b) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS PubMed;
(c) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS;
(d) B. J. Cowen and S. J. Miller, Chem. Soc. Rev., 2009, 38, 3102 RSC;
(e) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS PubMed;
(f) M. Sampath and T. P. Loh, Chem. Sci., 2010, 1, 739 RSC;
(g) A. Marinetti and A. Voituriez, Synlett, 2010, 174 CrossRef CAS;
(h) Q. Y. Zhao, Z. Lian, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 1724 RSC;
(i) C. E. Henry, Q. Xu, Y. C. Fan, T. J. Martin, L. Belding, T. Dudding and O. Kwon, J. Am. Chem. Soc., 2014, 136, 11890 CrossRef CAS PubMed;
(j) Z. Wang, X. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927 RSC;
(k) M. Gicquel, Y. Zhang, P. Aillard, P. Retailleau, A. Voituriez and A. Marinetti, Angew. Chem., Int. Ed., 2015, 54, 5470 CrossRef CAS PubMed.
-
(a) O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2012, 23, 1 CrossRef CAS;
(b) L. A. Wozniak and A. Okruszek, Chem. Soc. Rev., 2003, 32, 158 RSC;
(c) H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119 CrossRef PubMed;
(d) M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771 RSC.
-
(a) S. B. Wild, Coord. Chem. Rev., 1997, 166, 291 CrossRef CAS;
(b) D. S. Glueck, Chem.–Eur. J., 2008, 14, 7108 CrossRef CAS PubMed;
(c) V. S. Chan, M. Chiu, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6021 CrossRef CAS PubMed;
(d) J. S. Harvey and V. Gouverneur, Chem. Commun., 2010, 46, 7477 RSC;
(e) F. A. Kortmann, M. C. Chang, E. Otten, E. P. A. Couzijn, M. Lutz and A. J. Minnaard, Chem. Sci., 2014, 5, 1322 RSC.
-
(a) E. Bergin, C. T. O'Connor, S. B. Robinson, E. M. McGarrigle, C. P. O'Mahony and D. G. Gilheany, J. Am. Chem. Soc., 2007, 129, 9566 CrossRef CAS PubMed;
(b) D. Gatineau, L. Giordano and G. Buono, J. Am. Chem. Soc., 2011, 133, 10728 CrossRef CAS PubMed;
(c) Z. S. Han, N. Goyal, M. A. Herbage, J. D. Sieber, B. Qu, Y. Xu, Z. Li, J. T. Reeves, J. N. Desrosiers, S. Ma, N. Grinberg, H. Lee, H. P. R. Mangunuru, Y. Zhang, D. Krishnamurthy, B. Z. Lu, J. J. Song, G. Wang and C. H. Senanayake, J. Am. Chem. Soc., 2013, 135, 2474 CrossRef CAS PubMed;
(d) O. Berger and J. L. Montchamp, Angew. Chem., Int. Ed., 2013, 52, 11377 CrossRef CAS PubMed;
(e) S. Rast, B. Mohar and M. Stephan, Org. Lett., 2014, 16, 2688 CrossRef CAS PubMed;
(f) K. Nikitin, K. V. Rajendran, H. Müller-Bunz and D. G. Gilheany, Angew. Chem., Int. Ed., 2014, 53, 1906 CrossRef CAS PubMed;
(g) J. D. Sieber, D. Chennamadhavuni, K. R. Fandrick, B. Qu, Z. S. Han, J. Savoie, S. Ma, L. P. Samankumara, N. Grinberg, H. Lee, J. J. Song and C. H. Senanayake, Org. Lett., 2014, 16, 5494 CrossRef CAS PubMed.
-
(a) R. M. Alexander, R. C. Kevin and A. E. David, J. Am. Chem. Soc., 1995, 117, 9075 CrossRef;
(b) G. Cédric, S. J. Canipa, P. O'Brien and S. Taylor, J. Am. Chem. Soc., 2006, 128, 9336 CrossRef PubMed;
(c) C. Scriban and D. S. Glueck, J. Am. Chem. Soc., 2006, 128, 2788 CrossRef CAS PubMed;
(d) V. S. Chan, I. C. Stewart, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 2786 CrossRef CAS PubMed;
(e) Z. Q. Lin, W. Z. Wang, S. B. Yan and W. L. Duan, Angew. Chem., Int. Ed., 2015, 54, 6265 CrossRef CAS PubMed;
(f) R. Beaud, R. J. Phipps and M. J. Gaunt, J. Am. Chem. Soc., 2016, 138, 13183 CrossRef CAS PubMed;
(g) B. Pérez-Saavedra, N. Vázquez-Galiñanes, C. Saá and M. Fañanás-Mastral, ACS Catal., 2017, 7, 6104 CrossRef;
(h) Y. S. Jang, Ł. Woźniak, J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 12901 CrossRef CAS PubMed.
-
(a) C. Scriban, I. Kovacik and D. S. Glueck, Organometallics, 2005, 24, 4871 CrossRef CAS;
(b) D. P. Zhao and R. Wang, Chem. Soc. Rev., 2012, 41, 2095 RSC;
(c) Y. Huang, Y. Li, P. Leung and T. Hayashi, J. Am. Chem. Soc., 2014, 136, 4865 CrossRef CAS PubMed;
(d) C. Li, Q. Bian, S. Xu and W. Duan, Org. Chem. Front., 2014, 1, 541 RSC.
-
(a) D. Wiktelius, M. J. Johansson, K. Luthman and N. Kann, Org. Lett., 2005, 7, 4991 CrossRef CAS PubMed;
(b) J. S. Harvey, S. J. Malcolmson, K. S. Dunne, S. J. Meek, A. L. Thompson, R. R. Schrock, A. H. Hoveyda and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 762 CrossRef CAS PubMed;
(c) Z. J. Du, J. Guan, G. J. Wu, P. Xu, L. X. Gao and F. S. Han, J. Am. Chem. Soc., 2015, 137, 632 CrossRef CAS PubMed;
(d) Y. S. Jang, M. Dieckmann and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 15088 CrossRef CAS PubMed;
(e) Y. Zheng, L. N. Guo and W. W. Zi, Org. Lett., 2018, 20, 7039 CrossRef CAS PubMed.
-
(a) Z. J. Huang, X. Huang, B. S. Li, C. L. Mou, S. Yang, B. A. Song and Y. G. Robin Chi, J. Am. Chem. Soc., 2016, 138, 7524 CrossRef CAS PubMed;
(b) Y. Toda, M. Pink and J. N. Johnston, J. Am. Chem. Soc., 2014, 136, 14734 CrossRef CAS PubMed.
-
(a) O. Belda and C. Moberg, Acc. Chem. Res., 2004, 37, 159 CrossRef CAS PubMed;
(b) B. M. Trost, M. R. Machacek and A. Aponick, Acc. Chem. Res., 2006, 39, 747 CrossRef CAS PubMed;
(c) C. W. Cho and M. J. Krische, Angew. Chem., Int. Ed., 2004, 43, 6689 CrossRef CAS PubMed;
(d) Y. Q. Jiang, Y. L. Shi and M. Shi, J. Am. Chem. Soc., 2008, 130, 7202 CrossRef CAS PubMed;
(e) H. L. Cui, X. Feng, J. Peng, J. Lei, K. Jiang and Y. C. Chen, Angew. Chem., Int. Ed., 2009, 48, 5737 CrossRef CAS PubMed;
(f) X. Wang, F. Meng, Y. Wang, Z. Han, Y. J. Chen, L. Liu, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 9276 CrossRef CAS PubMed;
(g) X. Wang, P. Guo, Z. Han, X. Wang, Z. Wang and K. Ding, J. Am. Chem. Soc., 2014, 136, 405 CrossRef CAS PubMed.
- S. L. Li, C. Yang, Q. Wu, H. L. Zheng, X. Li and J. P. Cheng, J. Am. Chem. Soc., 2018, 140, 12836 CrossRef CAS PubMed.
- CCDC: 1579041, see ESI† for more details.
-
(a) M. Charton, J. Am. Chem. Soc., 1969, 91, 615 CrossRef CAS;
(b) M. Charton, J. Am. Chem. Soc., 1975, 97, 1552 CrossRef CAS;
(c) M. Charton, J. Am. Chem. Soc., 1975, 97, 3691 CrossRef CAS;
(d) M. Charton, J. Am. Chem. Soc., 1975, 97, 3694 CrossRef CAS.
-
(a) For selected review: M. S. Sigman, K. C. Harper, E. N. Bess and A. Milo, Acc. Chem. Res., 2016, 49, 1292 CrossRef CAS PubMed;
(b) C. B. Santiago, J. Y. Guo and M. S. Sigman, Chem. Sci., 2018, 9, 2398 RSC. For selected examples:
(c) A. Milo, E. N. Bess and M. S. Sigman, Nature, 2014, 507, 210 CrossRef CAS PubMed;
(d) C. Zhang, C. B. Santiago, L. Kou and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 7290 CrossRef CAS PubMed;
(e) M. Orlandi, M. J. Hilton, E. Yamamoto, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 12688 CrossRef CAS PubMed;
(f) R. T. Thornbury, V. Saini, T. d. A. Fernandes, C. B. Santiago, E. P. A. Talbot, M. S. Sigman, J. M. McKenna and F. D. Toste, Chem. Sci., 2017, 8, 2890 RSC;
(g) J. M. Crawford, E. A. Stone, A. Metrano, S. J. Miller and M. S. Sigman, J. Am. Chem. Soc., 2018, 140, 868 CrossRef CAS PubMed;
(h) S. B. Zhao, T. Gensch, B. Murray, Z. L. Niemeyer, M. S. Sigman and M. R. Biscoe, Science, 2018, 362, 670 CrossRef CAS PubMed;
(i) J. A. S. Coelho, A. Matsumoto, M. Orlandi, M. J. Hilton, M. S. Sigman and F. D. Toste, Chem. Sci., 2018, 9, 7153 RSC;
(j) S. Biswas, K. Kubota, M. Orlandi, M. Turberg, D. H. Miles, M. S. Sigman and F. D. Toste, Angew. Chem., Int. Ed., 2018, 57, 589 CrossRef CAS PubMed.
-
(a)
A. Verloop, Drug Design, Academic Press, New York, 1976 Search PubMed;
(b)
A. Verloop and J. Tipker, Biological Activity and Chemical Structure, Elsevier, Amsterdam, 1977 Search PubMed;
(c)
A. Verloop, IUPAC Pesticide Chemistry, Pergamon, Oxford, 1983 Search PubMed.
- To reduce the computational cost, a methyl group substituted MBH substrate and tert-butyl substituted phosphine oxide substrate were used. The experimental e.r. value of the product was 85:15, which was obtained under identical conditions.
-
M. J. Frisch, et al., Gaussian 09, revision D.01, Gaussian, Inc., 2013. See ESI† for full citation and calculation details Search PubMed.
-
(a) S. E. Wheeler and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10854 CrossRef CAS PubMed;
(b) B. W. Gung, B. U. Emenike, M. Lewis and K. Kirschbaum, Chem.–Eur. J., 2010, 16, 12357 CrossRef CAS PubMed;
(c) J. W. G. Bloom, R. K. Raju and S. E. Wheeler, J. Chem. Theory Comput., 2012, 8, 3167 CrossRef CAS PubMed.
-
(a) M. Sugiura, N. Sato, Y. Sonoda, S. Kotani and M. Nakajima, Chem. - Asian J., 2010, 5, 478 CrossRef CAS PubMed;
(b) M. Sugiura, N. Sato, S. Kotani and M. Nakajima, Chem. Commun., 2008, 44, 4309 RSC.
- For details of derivatization, see ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and computational details. CCDC 1579041. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05439h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  * and
Jin-Pei
Cheng
* and
Jin-Pei
Cheng