
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDe novo synthesis of mesoporous photoactive titanium(IV)–organic frameworks with MIL-100 topology†
Javier
Castells-Gil
 a,
Natalia
M. Padial
ab,
Neyvis
Almora-Barrios
a,
Ivan
da Silva
c,
Diego
Mateo
d,
Josep
Albero
d,
Hermenegildo
García
d and
Carlos
Martí-Gastaldo
*a
a,
Natalia
M. Padial
ab,
Neyvis
Almora-Barrios
a,
Ivan
da Silva
c,
Diego
Mateo
d,
Josep
Albero
d,
Hermenegildo
García
d and
Carlos
Martí-Gastaldo
*a
aUniversidad de Valencia (ICMol), Catedrático José Beltrán-2, 46980, Paterna, Spain. E-mail: carlos.marti@uv.es
bDepartment of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA
cISIS Facility, Rutherford Appleton Laboratory, Chilton, Didcot, Oxfordshire OX11 0QX, UK
dInstituto Universitario de Tecnología Química CSIC-UPV, Universitat Politècnica de València, Av. De Los Naranjos s/n, 46022, Valencia, Spain
First published on 8th March 2019
Abstract
Most developments in the chemistry and applications of metal–organic frameworks (MOFs) have been made possible thanks to the value of reticular chemistry in guiding the unlimited combination of organic connectors and secondary building units (SBUs) into targeted architectures. However, the development of new titanium-frameworks still remains limited by the difficulties in controlling the formation of persistent Ti-SBUs with predetermined directionality amenable to the isoreticular approach. Here we report the synthesis of a mesoporous Ti-MOF displaying a MIL-100 topology. MIL-100(Ti) combines excellent chemical stability and mesoporosity, intrinsic to this archetypical family of porous materials, with photoactive Ti3(μ3-O) metal-oxo clusters. By using high-throughput synthetic methodologies, we have confirmed that the formation of this SBU is thermodynamically favored as it is not strictly dependent on the metal precursor of choice and can be regarded as an adequate building block to control the design of new Ti-MOF architectures. We are confident that the addition of a mesoporous solid to the small number of crystalline, porous titanium-frameworks available will be a valuable asset to accelerate the development of new porous photocatalysts without the pore size limitations currently imposed by the microporous materials available.
Introduction
Metal–organic frameworks (MOFs) are crystalline, porous materials built from the interlinking of metal-oxo clusters with organic connectors.1 Their internal porosity can be engineered in size, shape and chemical function to provide a family of crystalline solids with structural and functional diversities beyond comparison. These intrinsic characteristics make MOFs appealing materials for modern storage, separation, delivery and catalysis technologies.2–6 The problems linked to the development of these applications have stressed the need for producing MOFs with superior chemical stability.7 Combination of hydrolytic stability with high surface area was first reported for sodalite-type frameworks built from imidazolate (ZIF)8 or azolate9 connectors due to hydrophobic shielding and strong metal–nitrogen bonds. Control over the strength of the coordination bond is also key to the robustness of Zr(IV) and Hf(IV) MOFs.Compared to these metals, titanium offers several advantages. It is less toxic, naturally more abundant and more adequate to endow these porous materials with photoactivity. Still, the synthesis of crystalline, porous titanium–organic frameworks remains comparatively underexplored.10 Though the discovery of the UiO family, based on Zr6O4(OH)4(RCO2)12 clusters,11 boosted the synthesis of multiple MOFs from isostructural Zr and Hf metal-oxo clusters,12–14 the controllable synthesis of Ti-MOFs remains quite challenging yet. This is due to the higher reactivity of Ti precursors, more prone to hydrolysis, that facilitates the formation of amorphous titanium oxide under solvothermal conditions. The formation of titanium-oxo-carboxylate clusters, capable of directing the formation of high symmetry, periodic architectures, demands stricter control over the hydrolysis-condensation equilibria. These synthetic difficulties are possibly the reason due to which almost all Ti-MOFs available are built from different secondary building units (SBUs). Fig. 1 summarizes the diversity of Ti-SBUs reported for the limited number of MOFs available: Ti12O15/Ti6O9 titanium-oxo clusters in MIL-177,15 Ti8O8 in MIL-125,16 Ti7O4 in PCN-22,17 Ti6O6 in MOF-901 (ref. 18) and MTM-1,19 Ti3O in COK-69,20 heterometallic clusters like Ti8Zr2O12 in PCN-415 (ref. 21) and Ti2M2O2 (M = Ca, Mn) in MUV-10,22 or simple titanium nodes in NTU-9 (ref. 23) and Ti-CAT-5,24 confirm the complexity for targeting the formation of specific MOF architectures due to the difficulties in controlling the formation of persistent SBUs.
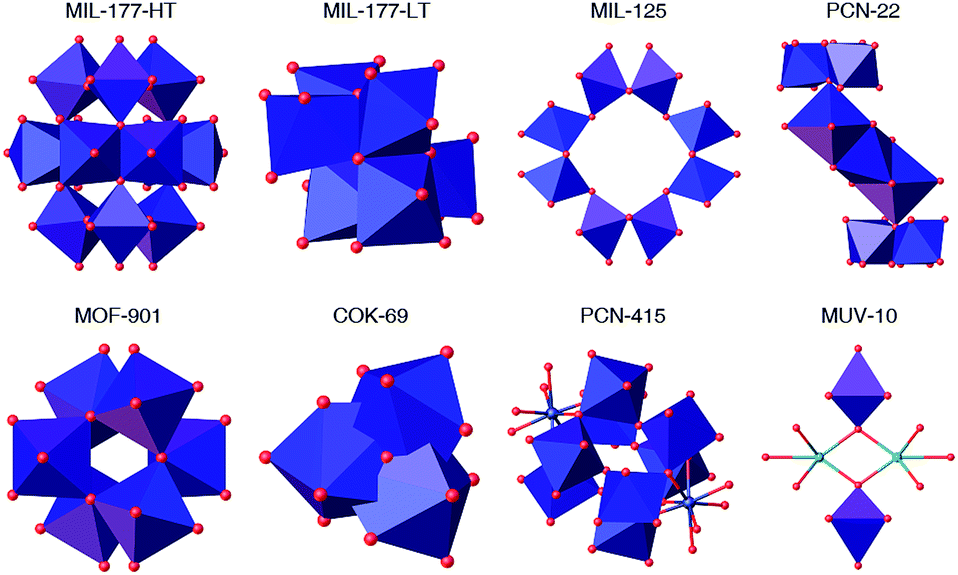 | ||
| Fig. 1 Summary of the SBUs in the structure of titanium–organic frameworks. | ||
This lack of chemical control over the final material is arguably limiting further development, highlighting the importance of finding alternative synthetic strategies that make Ti-MOFs amenable to the design principles of isoreticular chemistry. One strategy used to gain control is to produce daughter frameworks by incorporation of titanium(IV) into pre-formed materials by partial post-synthetic replacement of parent metals. This was originally demonstrated for the UiO-66(Zr)25 and MOF-5(Zn)26 families. More recent examples describe the use of titanium(III) precursors followed by mild oxidation for partial doping of MOF-74, MIL-100 and PCN-333 with Ti.27 However, these methodologies do not allow for complete metal replacement and also suffer from poor control over the positioning of titanium atoms in the structure,28 which can result in spurious deposition of metal oxide coatings.29
Compared to this post-synthetic strategy, here we report the formation of MIL-100(Ti) by direct synthesis. Systematic exploration of the chemical space, by using high-throughput (HT) methodologies, has enabled identification of the optimal synthetic conditions to produce highly crystalline solids from different titanium precursors. MIL-100(Ti) combines the chemical stability and mesoporosity, intrinsic to the MIL-100 family, with photoactive Ti3(μ3-O) metal-oxo clusters. Our work suggests that the formation of this SBU, classical in the chemistry of MOFs based on trivalent metals, is not strictly dependent on the metal source and might be used for the controlled design of new Ti-MOF architectures.
Results and discussion
Synthesis from pre-formed metal clusters
Our first approach to the direct synthesis of Ti-MOFs involved the use of titanium-oxo clusters as pre-formed SBUs. In principle, this was expected to favour the formation of crystalline solids under milder conditions than those conventionally used in MOF synthesis. This strategy has been successfully used to produce Zr(IV),30 Ce(IV),31 and Fe(III)-MOFs32 by direct reaction of metal-oxo clusters with polyaromatic carboxylic acids. Compared to simple precursors, we also expected to minimize the precipitation of amorphous oxides due to the higher stability of their cluster against hydrolysis. Accordingly, we reacted the as-made [Ti6O6(4-tbbz)6(OiPr)6] (tbbz = 4-tert-butylbenzoic acid) clusters (Fig. 2a) with trimesic acid (H3btc = benzene-1,3,5-tricarboxylic acid) between 80 and 160 °C for multiple combinations of solvents (DMF, DEF, NMP, MeOH, EtOH…), metal to linker ratios, concentration and addition of different modulators like acetic or benzoic acid. We used a FLEX SHAKE high-throughput workstation from Chemspeed© for robotic dispensing of solids and liquids in order to optimize the screening of a broad range of synthetic conditions whilst ensuring reproducibility. All attempts were unsuccessful as we could only isolate the unreacted cluster or amorphous solids for temperatures below or above 100 °C, respectively. We argued this was due to the poor solubility of the cluster under these conditions and decided to investigate an alternative mixture of solvents. After several tests, we observed that heating of a mixture of [Ti6O6(4-tbbz)6(OiPr)6] (Ti6) and H3btc in acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :tetrahydrofuran (ACN:THF, 3:1 v/v%) led to the formation of a white, microcrystalline solid. In order to optimize the crystallinity of the material we systematically varied the temperature of the reaction, the addition of acetic acid as the modulator, and the metal-to-linker ratio. We observed that, for a given Ti:btc ratio of 1:5, only the unreacted cluster was obtained at temperatures below 140 °C with no addition of the modulator (Fig. S1–S4†). However, a crystalline solid is observed in the presence of acetic acid as the modulator (180–550 equiv., 250–750 μL) and at temperatures above 100 °C. Note that a secondary crystalline phase is obtained as a side-product at low temperatures and high amounts of acetic acid (i.e. 550 equiv., 750 μL) as evidenced by powder X-ray diffraction (PXRD) (Fig. S2–S5†), whereas it is not observed for a lower concentration of the modulator. The formation of this secondary phase can be reduced by increasing the temperature of the reaction up to 160 °C (Fig. 2b–c, S6 and S7†). We next varied the Ti:btc ratio at 160 °C for a fixed amount of acetic acid (i.e. 180 equiv., 250 μL). PXRD of the solids confirms the formation of a highly crystalline MIL-100 phase for Ti:btc ratios of 1:2–5 (Fig. S8†). PXRD of the solids synthesized with increasing temperatures and concentration of the modulator confirms the critical effect of these parameters on the crystallinity of the material (Fig. 2b and c). The optimum conditions for the formation of highly crystalline MIL-100(Ti) corresponds to a Ti:btc ratio of 1:5 and addition of 250 μL of acetic acid (180 equiv.) and reaction at 160 °C (see ESI† for details). The use of more conventional solvents like DMF or DEF yielded amorphous or poorly crystalline materials under equivalent reaction conditions (Fig. S9 and S10†). Optical microscopy suggested the formation of small crystals, which was confirmed by scanning electron microscopy (SEM). As shown in Fig. 2e, the bulk is composed of single crystals with an octahedral habit, typical of cubic MIL-100 phases, and sizes oscillating below 5 μm. We also identified small fractions of amorphous material partially covering the surface of the crystals (Fig. S11†). The solid was recovered by centrifugation and washed with DMF and MeOH prior to drying in a vacuum at room temperature.
:tetrahydrofuran (ACN:THF, 3:1 v/v%) led to the formation of a white, microcrystalline solid. In order to optimize the crystallinity of the material we systematically varied the temperature of the reaction, the addition of acetic acid as the modulator, and the metal-to-linker ratio. We observed that, for a given Ti:btc ratio of 1:5, only the unreacted cluster was obtained at temperatures below 140 °C with no addition of the modulator (Fig. S1–S4†). However, a crystalline solid is observed in the presence of acetic acid as the modulator (180–550 equiv., 250–750 μL) and at temperatures above 100 °C. Note that a secondary crystalline phase is obtained as a side-product at low temperatures and high amounts of acetic acid (i.e. 550 equiv., 750 μL) as evidenced by powder X-ray diffraction (PXRD) (Fig. S2–S5†), whereas it is not observed for a lower concentration of the modulator. The formation of this secondary phase can be reduced by increasing the temperature of the reaction up to 160 °C (Fig. 2b–c, S6 and S7†). We next varied the Ti:btc ratio at 160 °C for a fixed amount of acetic acid (i.e. 180 equiv., 250 μL). PXRD of the solids confirms the formation of a highly crystalline MIL-100 phase for Ti:btc ratios of 1:2–5 (Fig. S8†). PXRD of the solids synthesized with increasing temperatures and concentration of the modulator confirms the critical effect of these parameters on the crystallinity of the material (Fig. 2b and c). The optimum conditions for the formation of highly crystalline MIL-100(Ti) corresponds to a Ti:btc ratio of 1:5 and addition of 250 μL of acetic acid (180 equiv.) and reaction at 160 °C (see ESI† for details). The use of more conventional solvents like DMF or DEF yielded amorphous or poorly crystalline materials under equivalent reaction conditions (Fig. S9 and S10†). Optical microscopy suggested the formation of small crystals, which was confirmed by scanning electron microscopy (SEM). As shown in Fig. 2e, the bulk is composed of single crystals with an octahedral habit, typical of cubic MIL-100 phases, and sizes oscillating below 5 μm. We also identified small fractions of amorphous material partially covering the surface of the crystals (Fig. S11†). The solid was recovered by centrifugation and washed with DMF and MeOH prior to drying in a vacuum at room temperature.
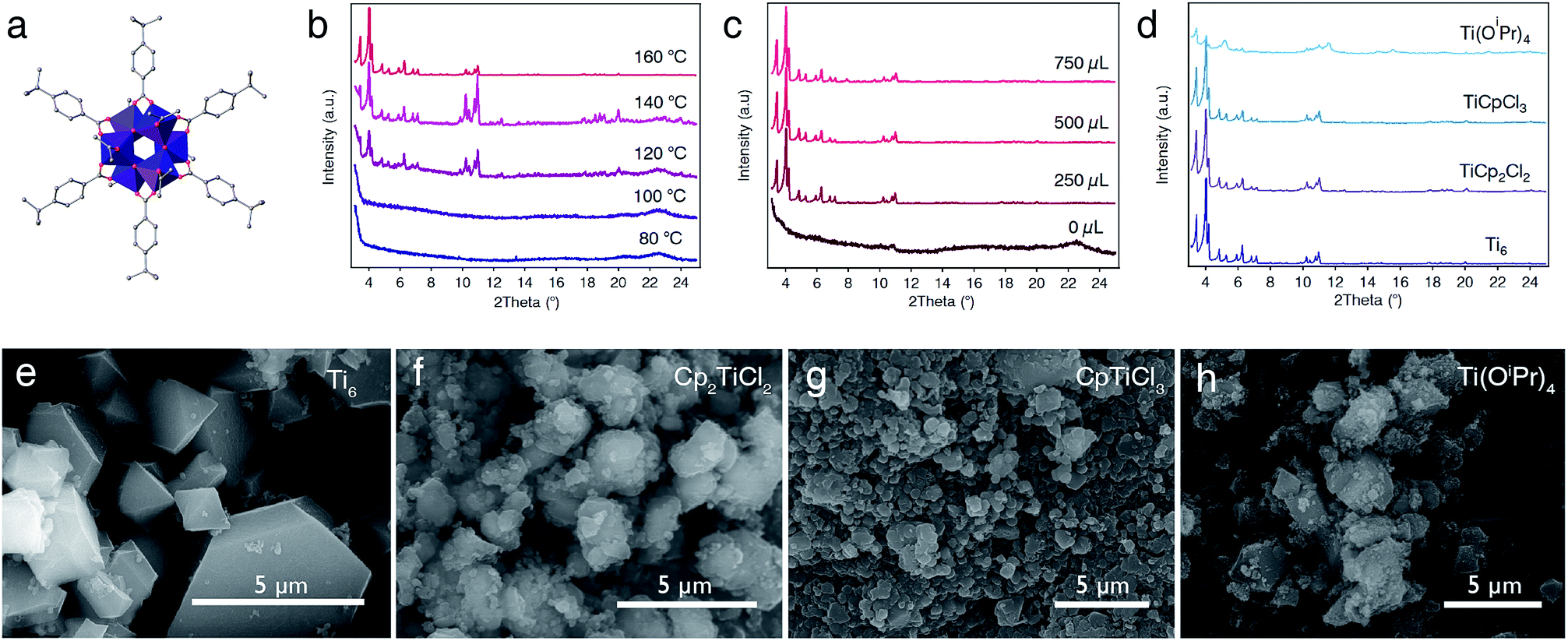 | ||
| Fig. 2 Optimization of reaction conditions for different titanium precursors to form MIL-100(Ti). (a) Structure of the titanium-oxo cluster [Ti6O6(4-tbbz)6(OiPr)6] (Ti6) used as the metal source. PXRD of the materials formed under different conditions showing the effect of (b) temperature, (c) amount of modulator at 160 °C and (d) Ti(IV) precursor used in the synthesis on the crystallinity of the formed phase. (e–h) SEM micrographs of the solid obtained with each Ti(IV) precursor showing the formation of micrometric, octahedral single crystals for Ti6. | ||
Structure and characterization
PXRD patterns of the solid collected at our laboratory revealed clear similarities to the reported phases of MIL-100(Cr, Fe) (Fig. S16†).33,34 For a clearer structural description, we collected high-resolution PXRD data that were refined by using an starting model built from the reported structure of MIL-100(Fe) by using Materials Studio. As shown in Fig. 3a, Rietveld refinement of the activated solid converged with excellent residual values (Rwp = 5.24%, Rexp = 1.93%) for a cubic Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group with cell parameter a = 73.5168(16) Å. The structure of MIL-100(Ti) is built from the interlinking of Ti3(μ3-O) clusters and btc linkers to conform a zeolitic mnt topology that features two types of mesoporous cages with pentagonal and hexagonal pore windows (Fig. 3b).
m space group with cell parameter a = 73.5168(16) Å. The structure of MIL-100(Ti) is built from the interlinking of Ti3(μ3-O) clusters and btc linkers to conform a zeolitic mnt topology that features two types of mesoporous cages with pentagonal and hexagonal pore windows (Fig. 3b).
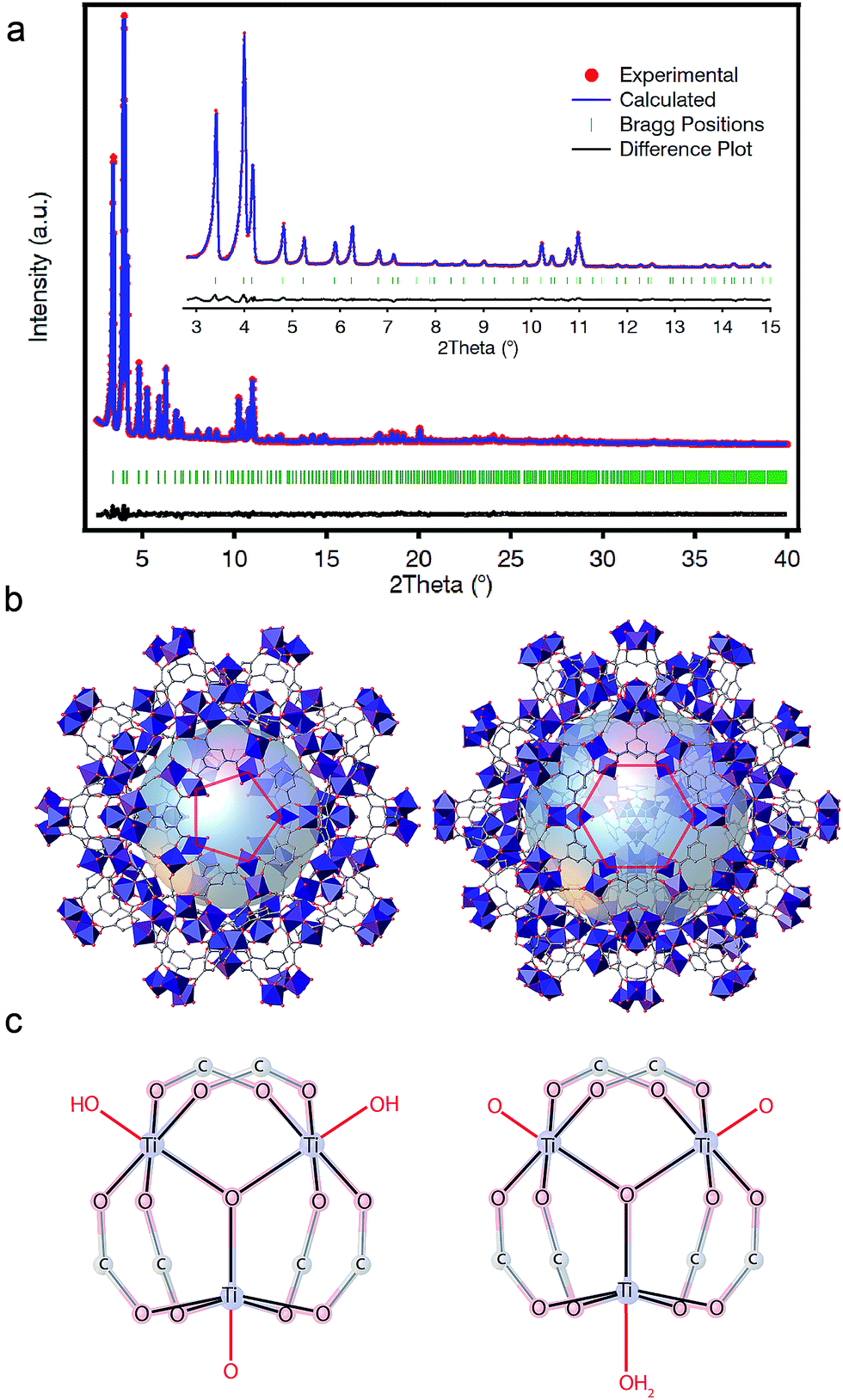 | ||
| Fig. 3 Structural characterization of MIL-100(Ti). (a) Rietveld refinement of MIL-100(Ti) (λCuKα1 = 1.540598 Å, λCuKα2 = 1.544426 Å). See SI3 for crystallographic data and full details. (b) Perspective of the structure along the [001] and [111] showing the presence of two types of mesoporous cages (2.4 and 2.9 nm) with pentagonal and hexagonal windows intrinsic to the MIL-100 family. (c) Proposed structure of the more favorable Ti-SBUs compatible with the formation of neutral MIL-100(Ti) featuring OH−/O2− (left) or O2−/H2O (right) capping ligands. | ||
The assembly of this structure from the starting Ti6O6 cluster used in the synthesis necessarily involves its degradation for the assembly of a thermodynamically more stable unit under the reaction conditions. This dynamic equilibrium between interconverting species is similar to that controlling the formation of heptameric clusters or Ti-oxo chains from Ti6O6(abz)6(OiPr)6 (abz = 4-aminobenzoate) during the synthesis of PCN-22 (ref. 17) or DGIST-1,35 respectively. By using comparatively milder conditions, the integrity of the starting homo or heterometallic polynuclear Ti(IV) clusters can also be preserved and they can be incorporated into the framework, as demonstrated for MOF-901,18 MNT-1 (ref. 19) or PCN-405.21 Compared to other precursors, these precedents seem to confirm the suitability of robust, molecular Ti clusters to either retain their nuclearity or control the formation of alternative SBUs more prone to induce the formation of specific MOF architectures under thermodynamic control. For example, M3(μ3-O) (M = Cr, Al, Fe) SBUs are very common in frameworks produced under relatively harsh conditions like MIL-96 (ref. 36) or MIL-101.37
The replacement of trivalent metals with Ti(IV) is not directly compatible with the local structure of MIL-100 materials, which features 6-connected [MIII3(μ3-O)(X)(H2O)2(btc)6] (X− = F, Cl, OH) nodes. In our case, the introduction of an excess of one positive charge per metal atom will be counterbalanced by additional anions. We hypothesized that these could be directly coordinated to the Ti sites or occupy the empty space available from the mesoporous cages for neutral or cationic frameworks, respectively. FT-IR spectra of the solid, before and after Soxhlet washing (Fig. S19†), 1H-NMR (Fig. S26†) and solid-state 13C-CP-MAS-NMR (Fig. S28†), all discard the presence of Ti6 clusters or free trimesate or acetate anions occupying the pores. Hence, we modelled computationally different neutral clusters by modification of the molecules coordinated to the axial position of the octahedral Ti(IV) centers whilst fixing the positions corresponding to the btc linkers and the μ3-O central bridge. We used the same methodology recently reported by De Vos and co-workers for rationalizing the most plausible formula of COK-69,20 based on analogous Ti(IV)-oxo trimers. We calculated the formation energies for the clusters that would generate a neutral structure by different combinations of O2−, OH− or H2O molecules acting as capping linkers. We optimized their structure with dispersion-corrected density functional theory (DFT-D3) as implemented in the Vienna ab initio simulation package (VASP) (see S5† for calculation details). Our results suggests that the formation of the cluster [Ti3(μ3-O)O(OH)2(btc)6] (Fig. 3c, left) is 0.21 eV more favourable than [Ti3(μ3-O)(O)2(H2O)(btc)6] (Fig. 3c, right). Nonetheless, like in the case of COK-69, the small difference between both clusters (c.f. 0.4 eV) is not sufficient to confirm unambiguously the formation of only one of them and both formulae might be reasonable.
The proposed formulae agree well with the percentage of residual oxide formed from the thermal decomposition of the solid (Fig. S21†). The experimental 32.2% of TiO2 is quite close to the calculated value of 33.9%. Permanent porosity was studied using N2 adsorption–desorption isotherms at 77 K after activation of the solid under dynamic vacuum (10−3 mbar) at 150 °C overnight. MIL-100(Ti) displays a non-hysteretic, type-I N2 adsorption with two secondary gas uptakes at P/P0 = 0.05 and 0.14 (Fig. 4a). The multi-point BET surface area was found to be 1321 m2 g−1 with a total pore volume of 0.66 cm3 g−1 (Fig. S22 and Table S3†). Pore size distribution (PSD) calculated using Non-Linear Density Functional Theory (NLDFT) models reveals two types of mesoporous pores between 1.6–2.4 and 2.6–3.6 nm that correlate well with the theoretical pore size values of 2.4 and 2.9 nm estimated from the structure of MIL-100(Ti). This surface area lies below the values reported for MIL-100(Cr, Fe) phases,33,34 suggesting ineffective activation or the partial blocking of the pores by molecular precursors with poor solubility. We used other activation protocols more adequate for highly porous materials like solvothermal treatment of the solid or Soxhlet washing with hot EtOH or MeOH. None of them led to a substantial increase in porosity. As commented before, FT-IR, 1H-NMR and 13C-CP-MAS-NMR of the activated solid rule out the presence of molecular components occluded in the pores of MIL-100(Ti). Thus, we ascribe the reduction of surface area to the formation of an amorphous fraction of the material that can be inferred from SEM (Fig. S11†), similar to previous reports for other mesoporous phases like MIL-101-NDC(Cr).38
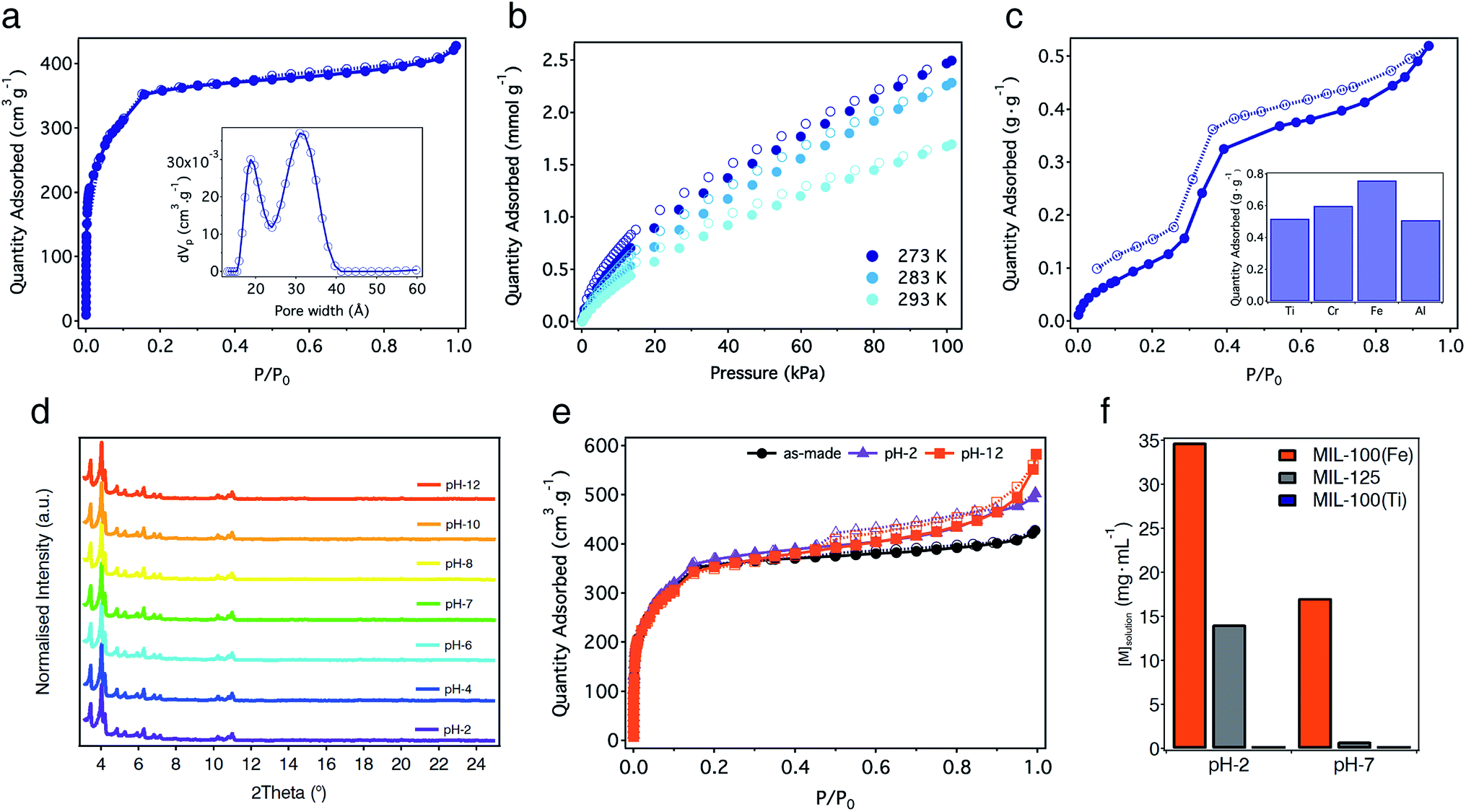 | ||
| Fig. 4 Sorption properties and chemical stability of MIL-100(Ti). (a) N2 isotherm at 77 K and PSD (inset). (b) CO2 isotherms obtained at 273, 283 and 293 K. (c) Water isotherm at 298 K and comparison of the total water adsorption at P/P0 = 0.9 with the isostructural Cr, Fe and Al MIL-100 phases (inset). (d) PXRD of the solid after soaked in water for 24 hours at variable pH. (e) Comparison of the N2 adsorption isotherms at 77 K of the as-synthesized MIL-100(Ti) and after water treatment at pH 2 and 12. (f) ICP-MS measurements for MIL-100(Fe), MIL-125 and MIL-100(Ti) at acidic and neutral pH. | ||
CO2 adsorption studies reveal a modest uptake of 1.70 mmol g−1 (7.5 wt%) at 293 K and 1 bar, with an isosteric heat of adsorption (Qst) of 34.8 kJ mol−1 (Fig. 4b and S24†). This value is very similar to the 30–35 kJ mol−1 reported for the Fe(III) phase39 and agrees well with the presence of vacant metal sites. Water adsorption also reveals clear similarities with other MIL-100 derivatives. As shown in Fig. 4c, the isotherm at 298 K shows an ‘S’-shaped profile with hysteretic behaviour, indicative of irreversible capillary condensation. Water adsorption at low relative pressures, which can be ascribed to the presence of open metal sites in the structure, is followed by a steep uptake at a relative humidity between 0.25 and 0.45% with an inflection point at P/P0 = 0.32, characteristic of hydrophilic materials.40 The maximum water adsorption capacity of MIL-100(Ti) calculated at P/P0 = 0.9 is 0.52 g g−1. This gravimetric adsorption is very similar to the value reported for MIL-100(Al) (0.51 g g−1) but smaller than those for the Cr and Fe phases ranging from 0.60 to 0.76 g g−1 (Fig. 4c and Table S4†).41,42
Thermal and chemical stability
One of the main features of MIL-100 materials is their stability in water. For Fe, Al and Sc phases, this stability is thermodynamic in origin and arises from strong coordination bonds with M–O bond dissociation energies of 444, 502 and 671 kJ mol−1.43 In turn, the kinetic inertness of Cr(III) ions in MIL-100(Cr) is responsible for its outstanding hydrolytic stability. Accordingly, the incorporation of highly charged Ti(IV) metals into MIL-100 topologies will result in high thermal and chemical stability due to thermodynamic parameters. Compared to the Cr and Fe phases that decompose between 300 and 350 °C,33,34 the thermogravimetric analysis (TGA) of MIL-100(Ti) confirms a significant increase in the thermal stability of the solid that remains stable up to 450 °C (Fig. S21†). This thermal stability is similar to other Ti-MOFs and agrees well with the higher dissociation energy of O–Ti(IV) bonds of 667 kJ mol−1.43 We next evaluated the hydrolytic stability of MIL-100(Ti) by soaking freshly made solids in water under acidic and basic conditions (pH 1–14). PXRD of the solids after incubation in water for 24 hours confirms that the structure of the solid remains unchanged between pH 2 and 12 (Fig. 4d and S32†). Stability was also confirmed by gas sorption. N2 adsorption measurements of the solids after water treatment show negligible changes in their surface area compared to the as-made material (Fig. 4e, S33 and Table S5†). These results confirm that MIL-100(Ti) displays excellent hydrolytic stability, comparable to other benchmark materials.7For further understanding on the role of Ti3(μ3-O) nodes over the chemical stability of the material we decided to carry out equivalent experiments for direct comparison with the isostructural MIL-100(Fe)44 and a representative Ti-MOF like MIL-125.16 PXRD and N2 sorption confirm that replacement of Fe(III) with Ti(IV) centers does not induce important changes under equivalent conditions. MIL-100(Fe) only shows minor degradation of the structure at pH 12 (Fig. S34†), whereas its surface area is maintained close to the original value under acidic and basic conditions (Fig. S35 and Table S6†). This behaviour is different for MIL-125 that retains its structural integrity and porous function almost intact at pH-12 but undergoes significant degradation at acidic pH (Fig. S36, S37 and Table S7†). We observe more important differences in the ICP-MS measurements of the supernatants after incubation in water. As shown in Fig. 4f, titanium leaching in MIL-100(Ti) is almost negligible for acidic and neutral conditions. In turn, the concentration of iron and titanium in solution increases with decreasing pH up to a maximum of 35 and 14 mg mL−1 at pH 2 for MIL-100(Fe) and MIL-125, respectively (see Table S8† for details). Concerning the isostructural series, we presume that the different degradation rates of the Ti and Fe phases might result in different drug delivery kinetics under simulated biological conditions. Although this study is beyond the scope of our work, we are confident that MIL-100(Ti) might be of interest to the MOF community for biomedical applications.
Optical response and photoactivity
The applications of Ti-MOFs as photocatalysts for solar fuel production or light-induced organic transformations are pivotal to this family of materials due to the promising combination of high surface areas, chemical stability and photoactivity.45 The optical band-gap of MIL-100(Ti) obtained by diffuse reflectance spectroscopy is 3.40 eV (Fig. S30†). This value lies between those reported for MIL-125 (3.68 eV)46 or MUV-10(Ca) (3.10 eV)22 and is compatible with UV light photoexcitation. This was confirmed by irradiating a suspension of the solid in dry THF with UV-B light (λ = 280–315 nm). After a few hours, the color of the solid changed from white to dark blue (Fig. S31†) and remained stable in the absence of oxygen. The electron paramagnetic resonance (EPR) spectra of the solid before/after irradiation confirms the occurrence of a broad signal at 350 mT, with g-fitted parameters g‖ = 1.927 and g⊥ = 1.952 characteristic of Ti(III) species, only for the irradiated sample (Fig. 5a and S31). This is accompanied by a weak, sharp peak at lower fields, which corresponds to the formation of btc radicals with g ≈ 2.00.22 This behaviour is consistent with a ligand-to-metal charge transfer (LMCT) mechanism, in which an electron is transferred from the photoexcited state of the linker to the metal sites (Fig. 5b).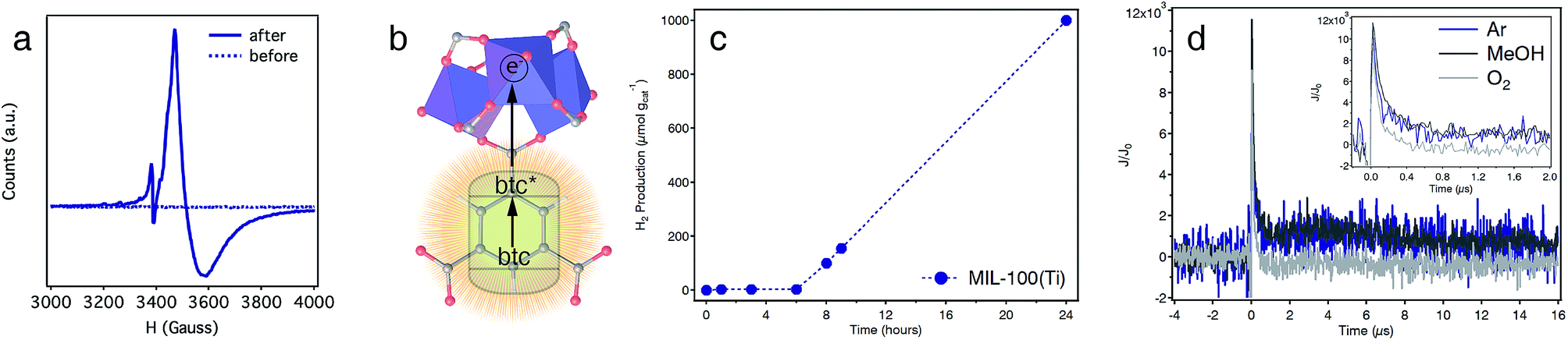 | ||
| Fig. 5 Photoactivity of MIL-100(Ti). (a) EPR of MIL-100(Ti) before and after irradiation with UV-light. (b) Schematic showing the ligand-to-metal charge transfer responsible for the photogeneration of Ti(III) species. (c) Photocatalytic H2 production of MIL-100(Ti) in a mixture of H2O:MeOH (4:1, v/v%). (d) Transient absorption kinetics of MIL-100(Ti) suspended in ACN under Ar(blue), in the presence of MeOH (dark grey) and in the presence of O2 (light grey). Laser excitation: 355 nm, monitored at 500 nm. The inset shows a zoom-in of the first 2 μs. | ||
We also tested the photocatalytic activity of MIL-100(Ti) by irradiating a suspension of the solid in a mixture of H2O:MeOH (4:1, v/v%) with a Xe lamp (300 W) and measuring the H2 produced at different time intervals. As shown in Fig. 5c, MIL-100(Ti) shows an induction period of ca. 6 h for a total H2 production of 1000 μmol gcat−1 after 24 hours (see S6† for experimental details). Stability was confirmed with PXRD and cyclability experiments for 5 consecutive cycles with minimum changes to the total H2 production measured (Fig. S40 and S41†).
The generation of photoinduced charge separation in MIL-100(Ti), a hallmark feature of semiconductors, was confirmed with transient absorption spectroscopy (TAS). The decay kinetics profile of the solid suspended in ACN, measured at 500 nm, shows two different processes. A first process that decays during the first 500 ns, and a second one that remains stable in Ar or in the presence of MeOH for approximately 20 μs after the light pulse (Fig. 5d). In addition, the longer-lived component is inhibited in the presence of O2 suggesting that it likely originates from photogenerated Ti(III) species in the framework. This is further confirmed by the TAS spectrum recorded at 60 ns after the light pulse, which shows an absorption band centred at ca. 640 nm, characteristic of Ti(III) species (Fig. S42†). The intensity of this band decreases after 6 μs showing a broad absorption between 400 and 600 nm, likely due to the decay of an important fraction of the photogenerated species.
Synthesis from other metal precursors
Previous studies confirm the importance of choosing suitable titanium precursors for directing the synthesis of crystalline titanium frameworks. We are confident that the use of high-throughput synthetic methodologies might help in studying this reactivity more systematically in order to enlarge the pool of precursors at hand. This pushed us to investigate the synthesis of MIL-100(Ti) by using other Ti salts with variable reactivity in water. We replaced Ti6 with precursors like Ti(OiPr)4, Cp2TiCl2 or CpTiCl3 and introduced minor changes to the synthesis optimized for the cluster. Our results confirm that MIL-100(Ti) can be also prepared from organometallic precursors. However, Ti(OiPr)4 only afforded poorly crystalline solids for all the conditions explored (Fig. 2d), PXRD LeBail refinement was used to confirm the phase purity of the solids formed with Cp2TiCl2 and CpTiCl3. Full-profile fits yield excellent agreement factors and profile differences for both solids (Fig. S17 and S18†). SEM analysis also confirms the formation of micrometric particles of sizes below 1 μm for Cp2TiCl2 and CpTiCl3 compared to the amorphous solid with ill-defined morphologies prepared with Ti(OiPr)4 (Fig. 2f–h, S12 and S13†). Our experiments suggest that organometallic precursors are more adequate than the isopropoxide to induce the formation of crystalline MIL-100(Ti), likely due to their higher resistance against hydrolysis.For a better understanding of the impact of the metal precursor over the properties of the MOF, we next analysed the porosity of MIL-100(Ti)-Cp2TiCl2 with N2 sorption experiments at 77 K. Compared to MIL-100(Ti)-Ti6, the solid displays a similar isotherm and pore size distribution – indicative of the formation of the same open framework – but a substantial decrease in the BET surface area and pore volume to 1130 m2 g−1 and 0.51 cm3 g−1 (Fig. S23 and Table S3†). We argued this could be linked to the presence of bulkier Cp units replacing OH− as capping ligands for a concomitant decrease in porosity. The ability of cyclopentadienyl ligands to induce the formation of Cp-capped titanium clusters was first anticipated for COK-69.20 To confirm this point we carried out FT-IR, 1H-NMR and 13C-CP-MAS-NMR studies of the solid after acid digestion (Fig. S20, S27 and S29†). All experiments discard the incorporation of Cp units into the material for the synthetic conditions used. To further confirm this point, we theoretically calculated the formation energy of the cluster [Ti3(μ3-O)O(Cp)2(btc)6] (SI5). As shown in Fig. S38,† our calculations suggest that the incorporation of Cp linkers in axial positions in the Ti sites is thermodynamically disfavoured, for a difference in energy of 1.1 or 1.3 eV over the formation of OH−/O2− or O2−/H2O capped clusters.
This systematic exploration of conditions and metal precursors suggests that the formation of trimeric Ti3(μ3-O) clusters units for the formation of MIL-100(Ti) under optimized reaction conditions is thermodynamically favoured not only for pre-formed clusters but also for organometallic complexes. However, the latter are less effective in preventing the formation of amorphous fractions of the material responsible for the observed reduction in porosity in MIL-100(Ti)-Cp2TiCl2. Overall, our experiments highlight the importance of controlling the reactivity of Ti precursors with water to enable the formation of crystalline, porous titanium-frameworks.
Conclusions
The design of new titanium-frameworks is arguably limited by the high reactivity of Ti(IV) with water and the subsequent difficulties in controlling the formation of persistent SBUs with the rigidity and directionality required to target specific architectures. We have shown how the use of high-throughput synthetic methodologies is useful to identify the conditions required to form Ti3(μ3-O) metal-oxo clusters from different titanium precursors. This classical SBU can be then reticulated into a MIL-100 topology by direct reaction with trimesic acid to produce a mesoporous material with excellent thermal and hydrolytic stability, linked to the incorporation of highly charged Ti(IV) atoms. MIL-100(Ti) also inherits the photoactivity of other titanium-frameworks for the generation of reactive Ti(III) species with light upon LMCT. In this regard, the combination of chemical stability and photoactivity into a mesoporous framework, also amenable to the generation of vacant sites, offers a promising route for the design of advanced porous photocatalysts without the metric limitations imposed by the microporosity of the materials currently available. Based on the preliminary results, we are confident that this MOF will also be compatible with linker functionalization or metal doping for engineering its optical response with visible light. In the context of biomedical applications, the low-toxicity of titanium and the different degradation rates compared to MIL-100(Ti) might also be relevant for approaching targeted release of drugs.Experimental section
Chemical and physical characterization
Infrared spectra were recorded on an Agilent Cary 630 FTIR spectrometer equipped with an ATR module. Thermogravimetric analysis was carried out with a Mettler Toledo TGA/SDTA 851e/SF/1100 between 25 and 800 °C at a rate of 5 °C min−1 with a flow of N2:O2 (4:1). PXRD patterns were collected in a PANalytical X'Pert PRO diffractometer using copper radiation (Cu Kα) with an X'Celerator detector, operating at 40 mA and 45 kV. Profiles were collected in the 2° < 2θ < 90° range with a step size of 0.013. Particle morphologies and dimensions were studied with a Hitachi S-4800 scanning electron microscope operating at 20 kV over metalized samples. Surface area, pore size and volume values were calculated from nitrogen adsorption–desorption isotherms recorded at 77 K on a Micromeritics 3Flex apparatus. Samples were degassed overnight at 150 °C and 10−6 Torr prior to analysis. Surface areas were estimated according to the BET model and pore size dimensions were calculated using Non-Linear Density Functional Theory (NLDFT) models for the adsorption branch assuming a cylindrical pore model. 13C-CP-MAS-NMR was carried out on a Bruker Avance III 400 WB Spectrometer. Samples were loaded in a 4 mm zirconia rotor and spun at 8 kHz. 1H-NMR spectra were run on a Bruker DRX300 spectrometer. See ESI† for additional experimental details on digestion of the solids for NMR study.
Synthesis of [Ti6O6(OiPr)6(4-tbbz)6] (Ti6)
The synthesis of the Ti6 cluster was carried out according to a previously reported procedure.47Synthesis of MIL-100(Ti)
A typical synthesis of MIL-100(Ti) was carried out by adding 7.2 mg of Ti6 (24 μmol of Ti) and 25.0 mg of H3btc (120 μmol) to 3 mL of a mixture of ACN:THF (3:1, v/v%) in a glass vial. Subsequently, 250 μL of acetic acid (180 equiv.) were added and the mixture was sonicated to get a homogeneous suspension. The vial was placed in the oven at 160 °C for 48 hours. After cooling down to room temperature, the white microcrystalline powder was recovered by centrifugation, rinsed with fresh DMF and MeOH and further washed by Soxhlet extraction with hot EtOH or MeOH for several hours. The solid was then allowed to dry under vacuum at room temperature.
Synthesis of MIL-100(Ti) from other Ti(IV) precursors (Cp2TiCl2, CpTiCl3, and Ti(OiPr)4)
The synthesis of MIL-100(Ti) from simple Ti(IV) precursors was carried out by adding 6.2 mg of Cp2TiCl2, 5.4 mg of CpTiCl3 or 7.2 μL of Ti(OiPr)4 (24 μmol of Ti(IV)), and 25.0 mg of H3btc (120 μmol) to 3 mL of a mixture of ACN:THF (3:1, v/v%) in a glass vial. Subsequently, 500 μL of acetic acid were added and the mixture was sonicated to get a homogeneous suspension. The vial was placed in the oven at 160 °C for 12 hours. After cooling down to room temperature, the white microcrystalline powder was recovered by centrifugation, rinsed with fresh DMF and MeOH and further washed by Soxhlet extraction with hot EtOH or MeOH for several hours. The solid was then allowed to dry under vacuum at room temperature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EU (ERC Stg Chem-fs-MOF 714122) and Spanish MINECO (MDM-2015-0538 & CTQ2017-83486-P). C. M.-G. and J. C.-G. thank the Spanish MINECO for a Ramón y Cajal Fellowship (RYC-2012-10894) and FPI Scholarship (CTQ2014-59209-P). N. M. P. thanks the European Union for a Marie Skłodowska-Curie Global Fellowship (H2020-MSCA-IF-2016-GF-749359-EnanSET). We thank the ISIS Facility for the access to synchrotron radiation, BSC-RES and Prof. Enrique Ortí for computational resources and Belén Lerma-Berlanga for technical help.Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M.-M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- A. Cadiau, Y. Belmabkhout, K. Adil, P. M. Bhatt, R. S. Pillai, A. Shkurenko, C. Martineau-Corcos, G. Maurin and M. Eddaoudi, Science, 2017, 356, 731–735 CrossRef CAS PubMed.
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 CrossRef CAS PubMed.
- D. Sheberla, J. C. Bachman, J. S. Elias, C.-J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed.
- J. E. Mondloch, M. J. Katz, W. C. Isley, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. Decoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Nat. Mater., 2015, 14, 512–516 CrossRef CAS PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- K. Park, Z. Ni, A. Côté, J. Choi, R. Huang, F. Uribe-Romo, H. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS PubMed.
- V. Colombo, S. Galli, H. J. Choi, G. D. Han, A. Maspero, G. Palmisano, N. Masciocchi and J. R. Long, Chem. Sci., 2011, 2, 1311 RSC.
- H. Assi, G. Mouchaham, N. Steunou, T. Devic and C. Serre, Chem. Soc. Rev., 2017, 46, 3431–3452 RSC.
- J. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC.
- M. Kim and S. M. Cohen, CrystEngComm, 2012, 14, 4096–4104 RSC.
- S. Wang, T. Kitao, N. Guillou, M. Wahiduzzaman, C. Martineau-Corcos, F. Nouar, A. Tissot, L. Binet, N. Ramsahye, S. Devautour-Vinot, S. Kitagawa, S. Seki, Y. Tsutsui, V. Briois, N. Steunou, G. Maurin, T. Uemura and C. Serre, Nat. Commun., 2018, 9, 1660 CrossRef PubMed.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Ferey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- S. Yuan, T.-F. Liu, D. Feng, J. Tian, K. Wang, J. qin, Q. Zhang, Y.-P. Chen, M. Bosch, L. Zou, S. J. Teat, S. J. Dalgarno and H.-C. Zhou, Chem. Sci., 2015, 6, 3926–3930 RSC.
- H. L. Nguyen, F. Gándara, H. Furukawa, T. L. H. Doan, K. E. Cordova and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 4330–4333 CrossRef CAS PubMed.
- C. Wang, C. Liu, X. He and Z.-M. Sun, Chem. Commun., 2017, 53, 11670–11673 RSC.
- B. Bueken, F. Vermoortele, D. E. P. Vanpoucke, H. Reinsch, C.-C. Tsou, P. Valvekens, T. De Baerdemaeker, R. Ameloot, C. E. A. Kirschhock, V. Van Speybroeck, J. M. Mayer and D. De Vos, Angew. Chem., Int. Ed., 2015, 54, 13912–13917 CrossRef CAS PubMed.
- S. Yuan, J.-S. Qin, H.-Q. Xu, J. Su, D. Rossi, Y. Chen, L. Zhang, C. Lollar, Q. Wang, H.-L. Jiang, D. H. Son, H. Xu, Z. Huang, X. Zou and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 105–111 CrossRef CAS PubMed.
- J. Castells-Gil, N. M. Padial, N. Almora Barrios, J. Albero, A. R. Ruiz-Salvador, J. Gonzalez-Platas, H. García and C. Martí-Gastaldo, Angew. Chem., Int. Ed., 2018, 57, 8453–8457 CrossRef CAS PubMed.
- J. Gao, J. Miao, P.-Z. Li, W. Y. Teng, L. Yang, Y. Zhao, B. Liu and Q. Zhang, Chem. Commun., 2014, 50, 3786–3794 RSC.
- N. T. T. Nguyen, H. Furukawa, F. Gándara, C. A. Trickett, H. M. Jeong, K. E. Cordova and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 15394–15397 CrossRef CAS PubMed.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- C. K. Brozek and M. Dincă, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed.
- L. Zou, D. Feng, T.-F. Liu, Y.-P. Chen, S. Yuan, K. Wang, X. Wang, S. Fordham and H.-C. Zhou, Chem. Sci., 2016, 7, 1063–1069 RSC.
- J. G. Santaclara, A. I. Olivos-Suarez, A. Gonzalez Nelson, D. Osadchii, M. A. Nasalevich, M. A. van der Veen, F. Kapteijn, A. M. Sheveleva, S. L. Veber, M. V. Fedin, A. T. Murray, C. H. Hendon, A. Walsh and J. Gascon, Chem. Mater., 2017, 29, 8963–8967 CrossRef CAS.
- M. S. Denny, L. R. Parent, J. P. Patterson, S. K. Meena, H. Pham, P. Abellan, Q. M. Ramasse, F. Paesani, N. C. Gianneschi and S. M. Cohen, J. Am. Chem. Soc., 2018, 140, 1348–1357 CrossRef CAS PubMed.
- V. Guillerm, S. Gross, C. Serre, T. Devic, M. Bauer and G. Ferey, Chem. Commun., 2010, 46, 767–769 RSC.
- S. Smolders, A. Struyf, H. Reinsch, B. Bueken, T. Rhauderwiek, L. Mintrop, P. Kurz, N. Stock and D. E. De Vos, Chem. Commun., 2018, 112, 869 Search PubMed.
- D. Feng, K. Wang, Z. Wei, Y.-P. Chen, C. M. Simon, R. K. Arvapally, R. L. Martin, M. Bosch, T.-F. Liu, S. Fordham, D. Yuan, M. A. Omary, M. Haranczyk, B. Smit and H.-C. Zhou, Nat. Commun., 2014, 5, 5723–5729 CrossRef CAS PubMed.
- G. Ferey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef CAS PubMed.
- P. Horcajada, S. Surblé, C. Serre, D.-Y. Hong, Y.-K. Seo, J.-S. Chang, J.-M. Grenèche, I. Margiolaki and G. Ferey, Chem. Commun., 2007, 2820–2822 RSC.
- Y. Keum, S. Park, Y.-P. Chen and J. Park, Angew. Chem., Int. Ed., 2018, 238, 37 Search PubMed.
- T. Loiseau, L. Lecroq, C. Volkringer, J. Marrot, G. Ferey, M. Haouas, F. Taulelle, S. Bourrelly, P. L. Llewellyn and M. Latroche, J. Am. Chem. Soc., 2006, 128, 10223–10230 CrossRef CAS PubMed.
- G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef CAS PubMed.
- A. Sonnauer, F. Hoffmann, M. Fröba, L. Kienle, V. Duppel, M. Thommes, C. Serre, G. Ferey and N. Stock, Angew. Chem., Int. Ed., 2009, 48, 3791–3794 CrossRef CAS PubMed.
- J. W. Yoon, Y.-K. Seo, Y. K. Hwang, J.-S. Chang, H. Leclerc, S. Wuttke, P. Bazin, A. Vimont, M. Daturi, E. Bloch, P. L. Llewellyn, C. Serre, P. Horcajada, J.-M. Grenèche, A. E. Rodrigues and G. Ferey, Angew. Chem., Int. Ed., 2010, 49, 5949–5952 CrossRef CAS PubMed.
- M. J. Kalmutzki, C. S. Diercks and O. M. Yaghi, Adv. Mater., 2018, 289, 1704304 CrossRef PubMed.
- M. Wickenheisser, A. Herbst, R. Tannert, B. Milow and C. Janiak, Microporous Mesoporous Mater., 2015, 215, 143–153 CrossRef CAS.
- F. Jeremias, A. Khutia, S. K. Henninger and C. Janiak, J. Mater. Chem., 2012, 22, 10148–10151 RSC.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, 85th edn, 2004, vol. 127 Search PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. M. Asiri and H. García, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed.
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D’arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed.
- K. Hong, W. Bak and H. Chun, Inorg. Chem., 2014, 53, 7288–7293 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1871195. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05218b |
| This journal is © The Royal Society of Chemistry 2019 |