
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic radical difluoromethoxylation of arenes and heteroarenes†
Johnny W.
Lee‡
 a,
Weijia
Zheng‡
a,
Cristian A.
Morales-Rivera
b,
Peng
Liu
*b and
Ming-Yu
Ngai
*a
a,
Weijia
Zheng‡
a,
Cristian A.
Morales-Rivera
b,
Peng
Liu
*b and
Ming-Yu
Ngai
*a
aDepartment of Chemistry, Institute of Chemical Biology and Drug Discovery, Stony Brook University, Stony Brook, NY 11794, USA. E-mail: ming-yu.ngai@stonybrook.edu
bDepartment of Chemistry, University of Pittsburgh, Pittsburgh, PA 15260, USA. E-mail: pengliu@pitt.edu
First published on 11th February 2019
Abstract
Intermolecular C–H difluoromethoxylation of (hetero)arenes remains a long-standing and unsolved problem in organic synthesis. Herein, we report the first catalytic protocol employing a redox-active difluoromethoxylating reagent 1a and photoredox catalysts for the direct C–H difluoromethoxylation of (hetero)arenes. Our approach is operationally simple, proceeds at room temperature, and uses bench-stable reagents. Its synthetic utility is highlighted by mild reaction conditions that tolerate a wide variety of functional groups and biorelevant molecules. Experimental and computational studies suggest single electron transfer (SET) from excited photoredox catalysts to 1a forming a neutral radical intermediate that liberates the OCF2H radical exclusively. Addition of this radical to (hetero)arenes gives difluoromethoxylated cyclohexadienyl radicals that are oxidized and deprotonated to afford the products of difluoromethoxylation.
Introduction
Modern drug discovery and development involves extensive fine-tuning of physicochemical properties of drug candidates. A common approach to control these properties involves incorporation of fluorine-containing functional groups such as the difluoromethoxy (OCF2H) group into drug candidates.1 The OCF2H moiety is a privileged functional group in medicinal chemistry because molecules bearing the OCF2H group have dynamic lipophilicity, where they can adjust their lipophilicity to adapt to the chemical environment via simple bond rotations.2 In addition, OCF2H-containing aromatic compounds can have an orthogonal structural geometry that enriches molecular spatial complexity and provides additional binding affinity to active sites in a target.3 Thus, incorporation of the OCF2H group into organic molecules often enhances their therapeutic efficacy by increasing metabolic stability, improving cellular membrane permeability, and altering pharmacokinetic properties.3 As a result, the OCF2H group is prevalent among pharmaceuticals and agrochemicals such as Pantoprazole® (a proton-pump inhibitor that is one of the top 100 selling drugs),4 Roflumilast®, Flucythrinate®, and Diflumetorim® (Scheme 1a).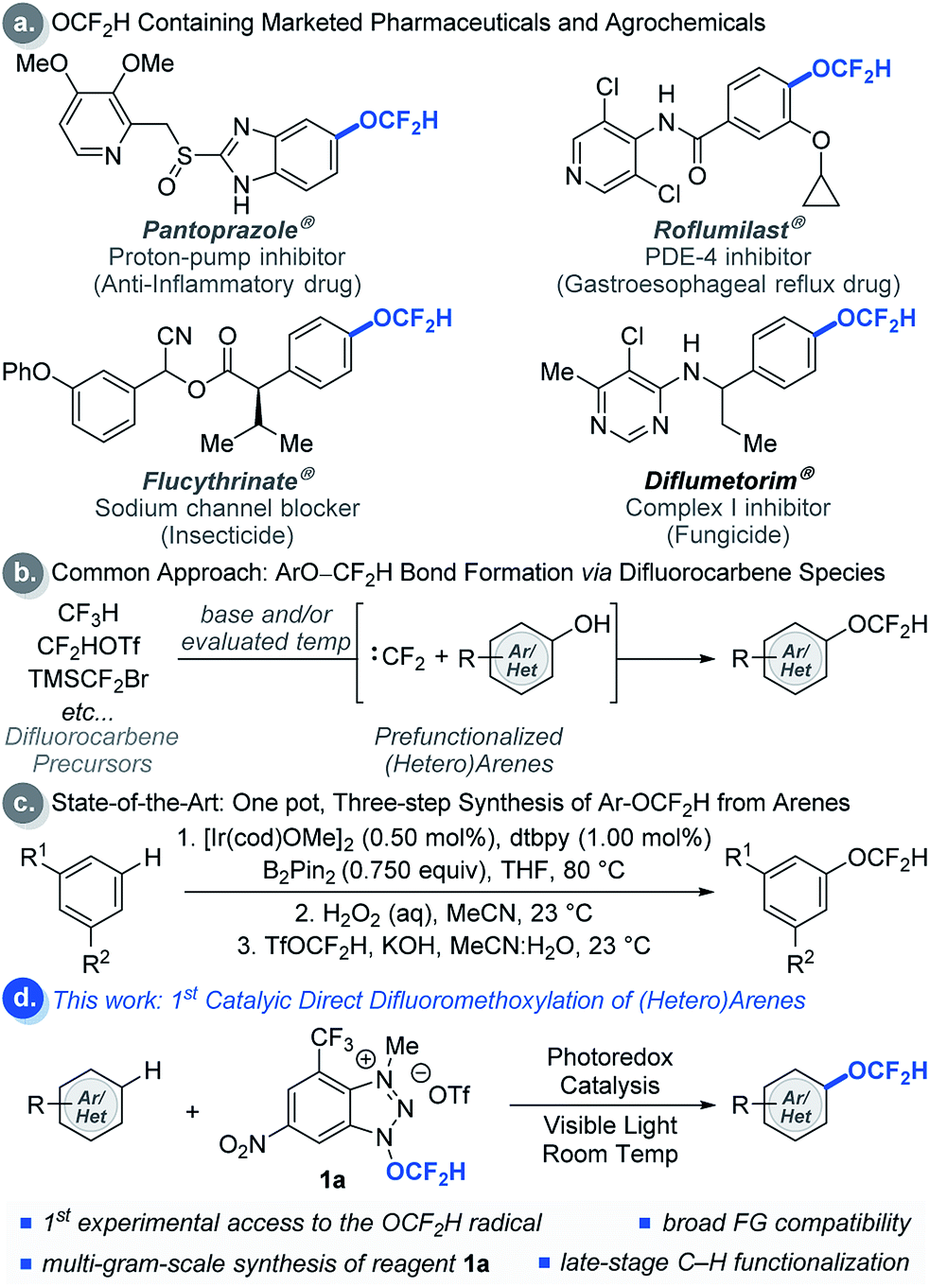 | ||
| Scheme 1 Applications and strategies for the synthesis of difluoromethoxylated (hetero)arenes. | ||
Even though numerous biologically active molecules have the OCF2H motif in an aromatic system, access to such analogues often requires the installation of the OCF2H group at an early stage of a multi-step synthetic sequence. The most common approach relies on O-difluoromethylation of phenols using different difluorocarbene precursors under basic and/or evaluated temperature conditions (Scheme 1b).5 This strategy has facilitated the site-selective synthesis of aryl difluoromethyl ethers, but identification of the ideal position of the OCF2H substitution in a drug candidate still requires parallel and laborious multi-step syntheses from aryl precursors bearing activating or directing groups at various positions in an aromatic ring. Hartwig et al. recently developed an elegant one-pot, three-step aryl C–H difluoromethoxylation protocol involving (i) catalytic C–H borylation of arenes, (ii) oxidation of the boronate esters, and (iii) difluoromethylation of phenols (Scheme 1c).5h Although this method has advanced the state-of-the-art, a catalytic, direct intermolecular C–H difluoromethoxylation of (hetero)arenes remains elusive.
As a part of our ongoing program to access and harness the reactivity of heteroatom radicals,6 we questioned whether a radical-mediated aromatic substitution using the OCF2H radical would allow direct introduction of the OCF2H group to a drug candidate generating multiple regioisomers in a single chemical operation. Such an approach is appealing because it obviates the need for laborious synthetic effort and the pre-functionalization of aromatic compounds. Moreover, the preparation and isolation of regioisomers would allow rapid assays of the biological activity of OCF2H analogues, a feature which would be particularly beneficial to modern drug discovery programs. Herein, we report the development of redox-active difluoromethoxylating reagents for late-stage, direct difluoromethoxylation of unactivated arenes and heteroarenes through a radical-mediated mechanism under visible light photocatalytic conditions at room temperature (Scheme 1d).7–9
Results and discussion
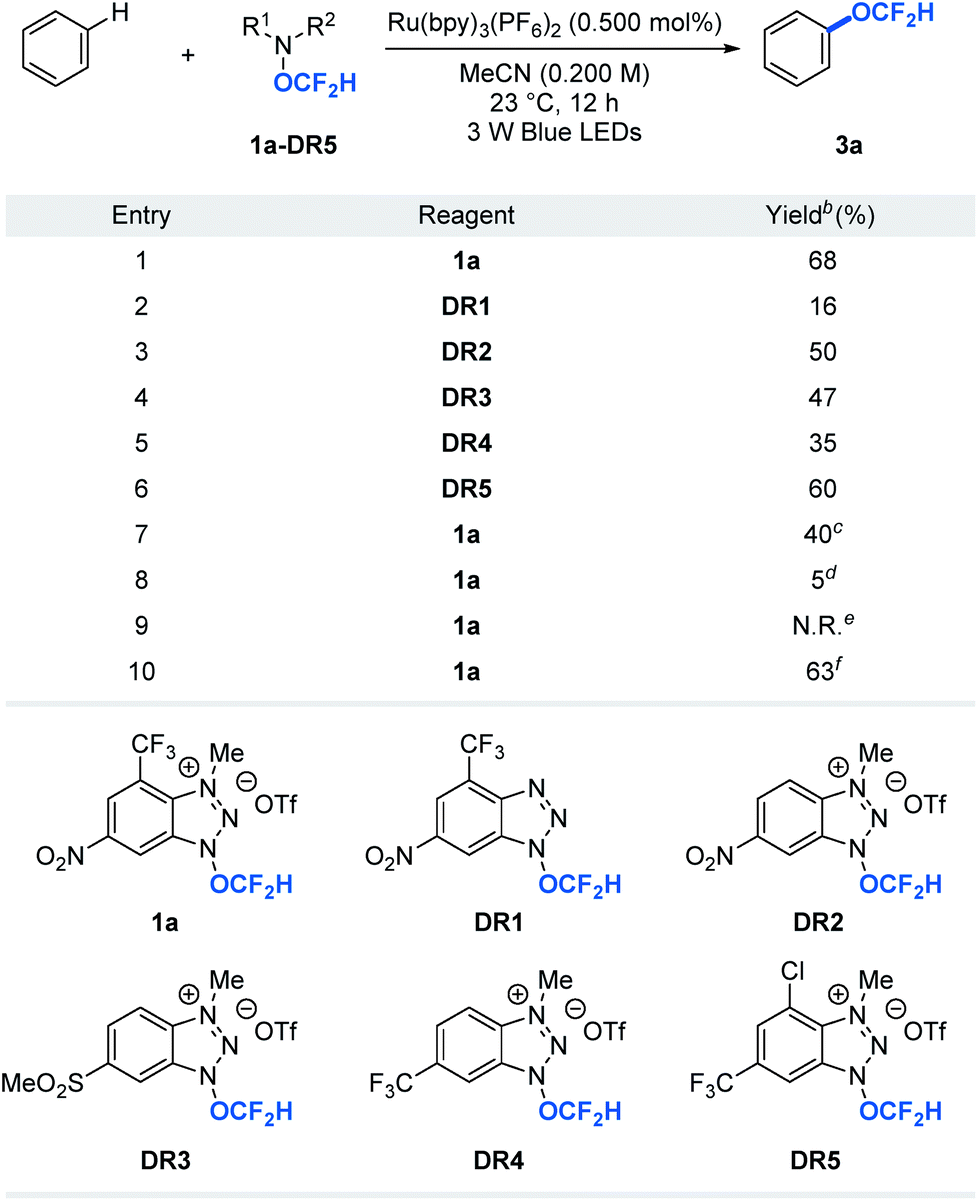
A key to the success of the proposed transformation is the ability to generate and trap the OCF2H radical under mild reaction conditions. Although computational studies of the OCF2H radical have been reported, experimental access to such a radical intermediate remains rare.10 We envision that the ability to generate the OCF2H radical in a controllable, catalytic, and selective manner under mild conditions will open a new reaction platform for the preparation of an important class of difluoromethoxylated molecules. Our recent success in the development of trifluoromethoxylating reagents by taking advantage of the weak N–O bond (ΔGN–O ≈ 57 kcal mol−1)6,11 prompted us to question whether we could develop difluoromethoxylating reagents for the first photocatalytic formation and utilization of the OCF2H radical in organic synthesis. Thus, we synthesized and examined a series of benzotriazole-based OCF2H reagents (1a, DR1–5, Table 1) for direct aryl C–H difluoromethoxylation of benzene. We found that cationic nature of the reagent is critical as it enhances the oxidizing power of the reagent and undergoes a photocatalytic single electron reduction to produce a neutral radical 1a′ that liberates the OCF2H radical selectively. Incorporation of electron deficient substituents on the benzotriazole ring prevents the addition of the OCF2H radical to the reagent byproducts and improves the reaction yields (entries 1, 3–6). Further reaction optimizations revealed that the reaction works with 1 equivalent of benzene albeit with diminished yield (40%) accompanied with additional 24% of bis(difluoromethoxylated) side products (entry 7). Control experiments showed that photoredox catalysts and light are essential, but the oxygen free environment is not required (entries 8–10). It is noteworthy that reagent 1a (mp = 153–154 °C) be prepared in a multi-gram scale and is thermally stable beyond 200 °C. Also, it can be manipulated and stored under ambient conditions without noticeable decomposition (see ESI†).| a Reactions were performed using 1 equivalent of reagent and 10 equivalents of benzene. b Yields were determined by 19F NMR spectroscopy using trifluorotoluene as an internal standard. c 1 equivalent of benzene. d Without Ru(bpy)3(PF6)2. e Without light. f The reaction was set-up under air atomsphere. |
|---|
|
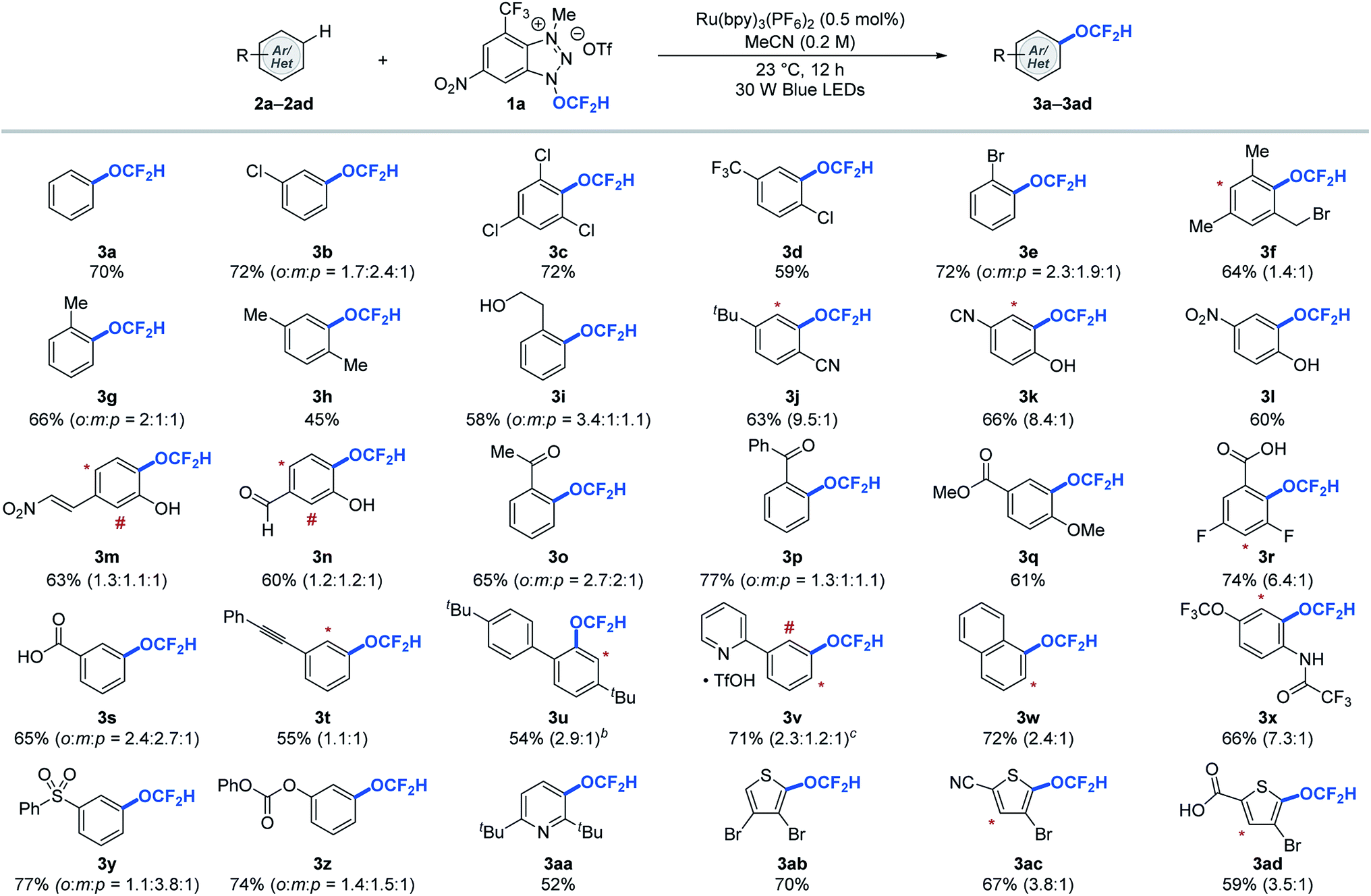
With the redox-active cationic difluoromethoxylating reagent 1a12 and optimized photoredox-catalysed aryl C–H difluoromethoxylation reaction conditions in hand, we then test the generality of the reaction against a wide array of arenes and heteroarenes. As shown in Table 2, a broad array of arenes and heteroarenes with diverse electronic properties and substitution patterns underwent photocatalytic (hetero)aryl C–H difluoromethoxylation under optimized reaction conditions using reagent 1a at room temperature. The reaction tolerated halide substituents such as fluoride (3r), chloride (3b–3d), and bromide (3e, 3f, 3ab–3ad), which is important from a synthetic perspective since these substituents provide useful handles for further structural elaboration through metal-catalysed coupling reactions. The weak benzylic C–H bond (BDE ≈ 88 kcal mol−1, 3f–3i),13 which is often a site for undesired reactivity in radical processes, proved compatible. More remarkably, unprotected alcohols (3i) and phenols (3k–3n) remained intact during the reaction. Carbonyl derivatives such as aldehydes (3n), ketones with or without enolizable protons (3o, 3p), carboxylic acids (3r, 3s, 3ad), esters (3q), amides (3x), and carbonates (3z) reacted smoothly to afford the desired products in good yields. Other functional groups such as trifluoromethyl (3d), methoxy (3q), trifluoromethoxy (3x), cyano (3j, 3k, 3ac), nitro (3l, 3m), sulfonyl (3y), and pyridinium (3v) were all well tolerated under the reaction conditions. Moreover, no competing radical addition to electron deficient olefin (3m) or alkyne (3t) was observed during the aryl difluoromethoxylation reaction. Heteroarenes such as pyridine (3aa) and thiophene (3ab–3ad) derivatives were also viable substrates. The reaction proceeded with one equivalent of arenes, but higher yields were obtained using ten equivalents of arenes.12 In such cases, we could recover 8.3–9.1 equivalents (see ESI†) of the aromatic substrates at the end of the reaction, which is critical for valuable aromatic compounds.
a Reactions were performed using 1.0 equivalent of reagent 1a and 10.0 equivalents of (hetero)arene. The asterisk (*) and number sign (#) denote functionalization of minor regioisomeric products. Overall yields and the ratio of the constitutional isomers were determined by 19F NMR spectroscopy using trifluorotoluene as an internal standard.
b Reaction performed with MeCN and CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.2 M).
c Reaction performed with 10.0 equiv. of TfOH. See ESI for experimental details. :1, 0.2 M).
c Reaction performed with 10.0 equiv. of TfOH. See ESI for experimental details.
|
|---|
|
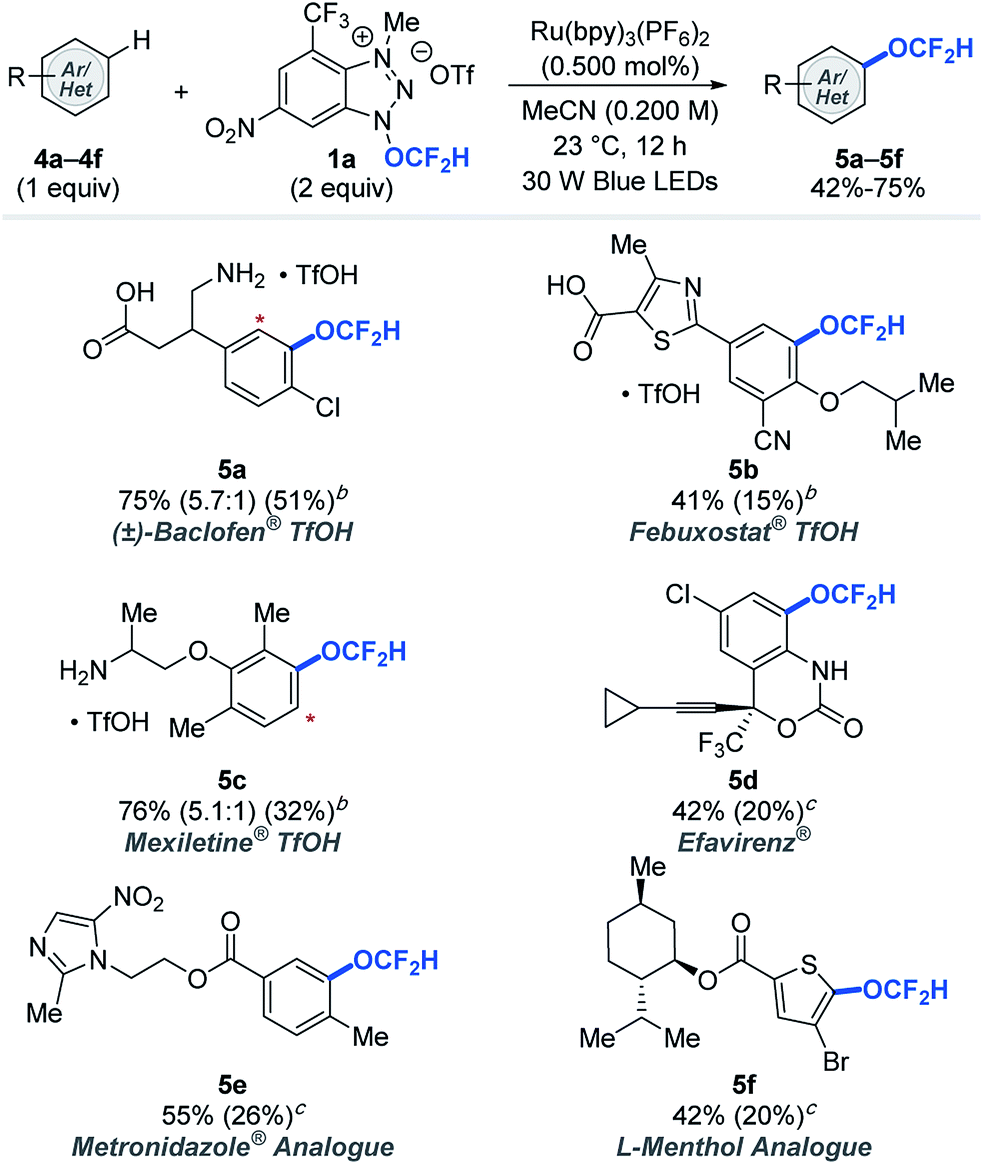
Late-stage modifications of biologically active molecules are often a key to identification of medicinal agents.14 To demonstrate the amenability of the photocatalytic difluoromethoxylation processes to late-stage synthetic applications, bio-relevant molecules were subjected to our standard reaction conditions using arenes as limiting reactants (Table 3). Approved drug molecules such as Baclofen® (muscle relaxant), Febuxostat® (anti-hyperuricemic), Mexiletine® (anti-arrhythmic), Efavirenz® (antiretroviral drug for treating HIV), as well as Metronidazole® (antiparasitic) and L-menthol (decongestants and analgesics) analogues were successfully difluoromethoxylated using reagent 1a to afford the desired products (5a–5f) in synthetically useful 42–76% yields, based on the recovery of the starting materials (BRSM). Our difluoromethoxylation strategy is applicable to a range of drug molecules and tolerates a number of sensitive functionalities, and this shows its potential utility in modern drug discovery programs.
| a Yields were determined based on the recovered starting material. The yield in parentheses is the isolated yield. The asterisk (*) denotes functionalization of a minor regioisomeric product. b Reaction performed with 1.00 equivalent of TfOH. c 1.00 equivalent of K2CO3. See ESI for experimental details. |
|---|
|
Our approach capable of forming multiple regioisomers in a single synthetic operation is complementary to the conventional site-selective protocols using phenols as substrates and could be useful in discovery chemistry. The regioselectivity of the reaction resembles that of radical-mediated aromatic substitution processes and is guided by the electronics of the substituent except in the case of a bulky substituent such as 3j, in which case the OCF2H radical adds preferably to the position distal from the tert-butyl group. If an aromatic substrate has multiple reaction sites, the OCF2H radical will add to these sites to form regioisomeric products, which could be separated to provide pure isomers (see ESI†). Such reactivity is particularly attractive from a drug discovery point of view because it allows rapid access to various OCF2H derivatives without labour-intensive, parallel multi-step analogue synthesis.14,15 More importantly, it will increase the efficiency of structure–activity relationship (SAR) studies of OCF2H analogues and can conveniently produce promising new candidates that might have never been evaluated otherwise.
We then performed a series of experiments and DFT calculations to better understand the reactivity of the OCF2H radical and the reaction mechanism (Scheme 2). The quantum yield of the reaction is 0.52, which supports that an extended radical chain mechanism is unlikely. This observation corroborates DFT calculations (see Fig. S24†). A series of Stern–Volmer quenching studies showed that only 1a quenched the excited *Ru(bpy)32+ efficiently (kq = 2.08 × 109 M−1 s−1) (Fig. S8†). To further probe the reaction mechanism, kinetic isotope effect (KIE) experiments were conducted using a 1:1 mixture of benzene and d6-benzene in the presence of reagent 1a, affording the desired products Ph-OCF2H and d5-Ph-OCF2H in a 1:1 ratio (Scheme 2b). This result excludes the possibility of H-atom abstraction/deprotonation as the rate-determining step. Moreover, intermolecular competition experiments using two electronically diverse arenes revealed that the OCF2H radical reacts more favourably with electron-rich arenes, and this confirms its electrophilic character (Scheme 2c). The formation of the OCF2H radical is the key for the success of the (hetero)aryl C–H difluoromethoxylation and is supported by (i) the regioselectivity of the reaction, and (ii) radical trap experiments using butylated hydroxytoluene (BHT) and 1,4-cyclohexadiene (Scheme 2d). Addition of 1 equivalent of BHT to the reaction mixture lowered the product yield from 70% to 29%. When 1,4-cyclohexadiene was used as a substrate, we observed the formation of the desired product 3a in 7% yield. Presumably, once the OCF2H radical is formed, it undergoes two consecutive H-atom abstraction from 1,4-cyclohexadiene, generating benzene as the product. Subsequently, this benzene can react with the OCF2H radical under photocatalytic conditions, furnishing the difluoromethoxylated product. A key feature of our cationic redox-active reagent 1a is its susceptibility to single electron reduction to form a neutral radical (1a′) that undergoes β-scission liberating the OCF2H radical exclusively (Scheme 3a). DFT calculations showed that both steps are energetically favourable in the presence of an excited photoredox catalyst, *Ru(bpy)32+. Once the OCF2H radical is formed, the subsequent steps (i.e., the addition of the OCF2H radical to an arene, oxidation of the resulting cyclohexadienyl radical by Ru(bpy)33+, and deprotonation) are all exergonic (Fig. S24†). We have determined the peak potential of reagent 1a [Ep(1a+/1a) = +0.109 V versus saturated calomel electrode (SCE) in MeCN, Fig. S6†], and so it can be reduced by the excited *Ru(bpy)32+ (Ered1/2 = −0.81 V versus SCE in MeCN).16
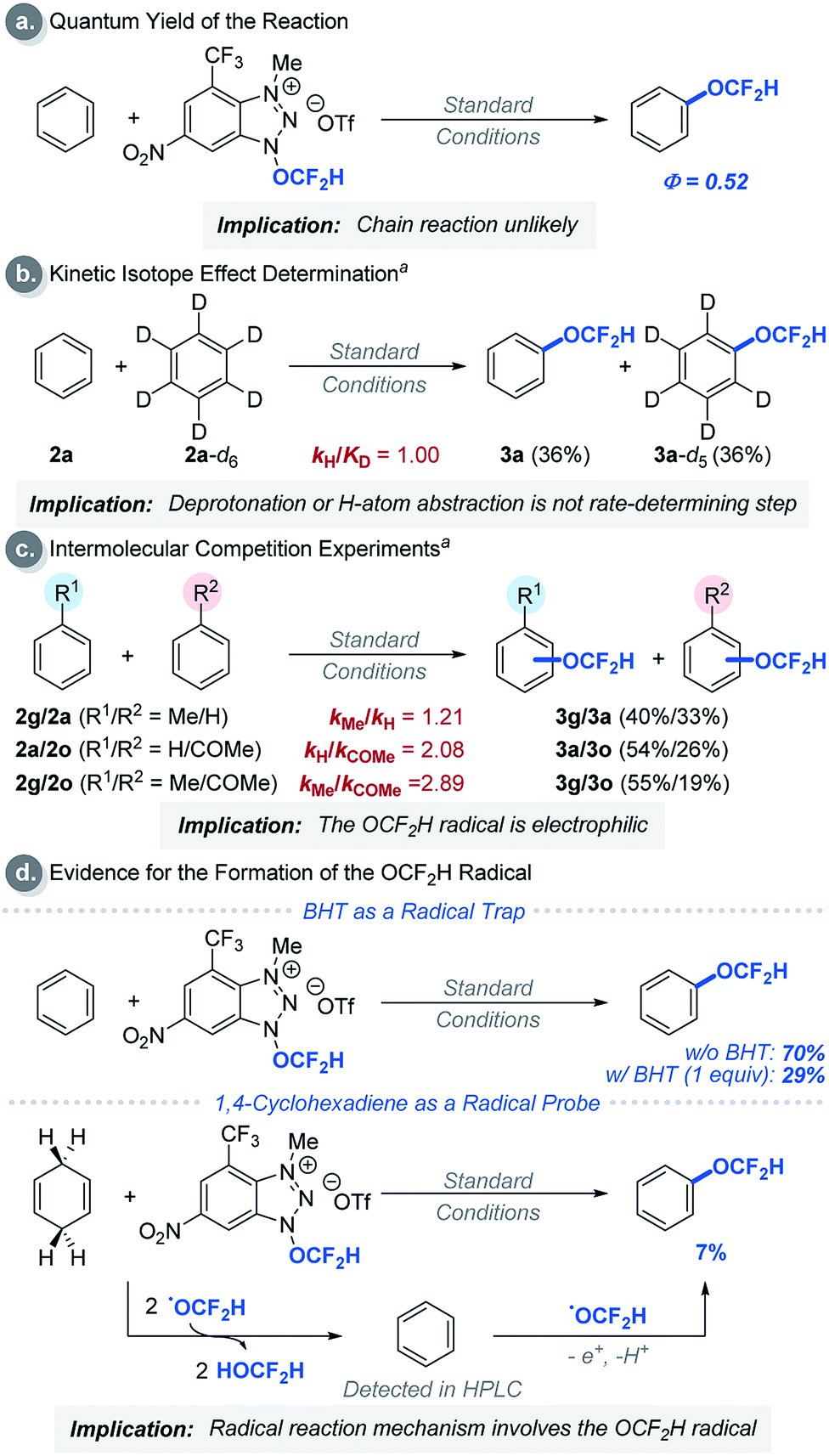 | ||
| Scheme 2 Experimental mechanism studies: areactions were performed using 5.00 equivalents of arenes each. See ESI† for experimental details. | ||
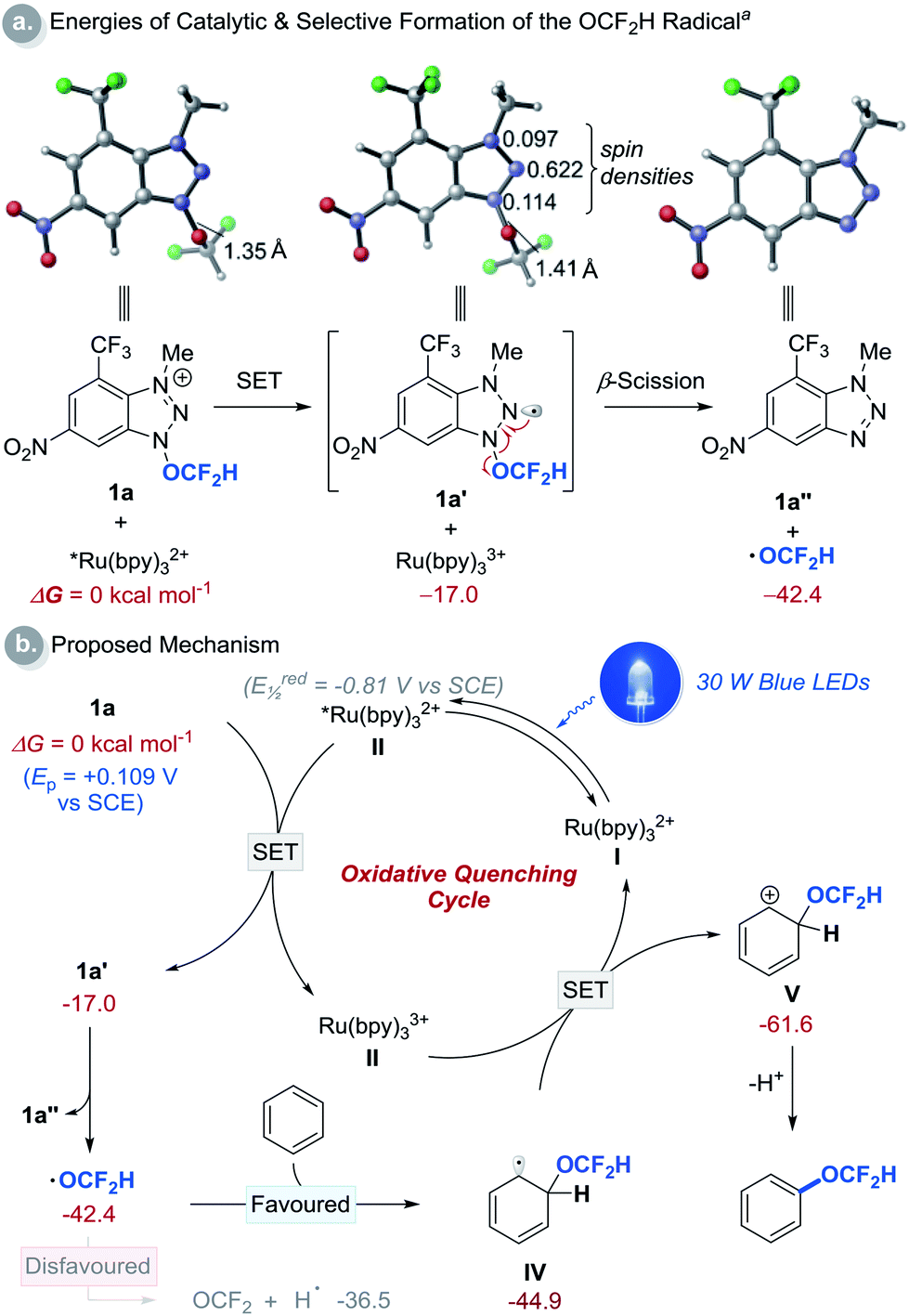 | ||
| Scheme 3 Computational studies and proposed reaction mechanism. aDFT calculations were performed at the M06-2X/6-311++G(d,p)/SMD(MeCN)//M06-2X/6-31+G(d) level of theory using reagent 1a and benzene as a substrate. All energies are in kcal mol−1 and are with respect to II and 1a. See (ESI†) for details. | ||
Based on these preliminary results, a catalytic cycle of this transformation was hypothesized and depicted in Scheme 3b. Initial excitation of the Ru(bpy)32+ photocatalyst (I, bpy = 2,2′-bipyridine) produces the long-lived triplet-excited state of *Ru(bpy)32+ (II, t1/2 = 1.1 μs).17 This catalyst (II) (Ered1/2 = −0.81 V versus SCE in MeCN)16 undergoes SET with the redox-active cationic reagent 1a (Ep of 1a = +0.109 V versus SCE in MeCN) generating Ru(bpy)33+ and neutral radical 1a′ that undergoes β-scission to liberate benzotriazole (1a′′) and the OCF2H radical. The addition of this radical to an arene to form cyclohexadienyl radical IV is thermodynamically more favourable than the decomposition of the OCF2H radical to fluorophosgene and hydrogen atom.10b Oxidation of IV by Ru(bpy)33+ (Ered1/2 = +1.28 V, versus SCE in MeCN) affords cyclohexadienyl cation V, which is deprotonated to give the desired C–H difluoromethoxylated arenes.
Conclusions
In summary, we have developed a redox-active cationic reagent 1a and identified photocatalytic conditions that allow facile difluoromethoxylation of arenes and heteroarenes without the need for aryl ring pre-functionalization or pre-activation. This radical-based aromatic substitution process provides rapid access to multiple regioisomers in a single synthetic operation, which will facilitate molecular screening and SAR studies of OCF2H analogues. The synthetic utility of our strategy has been highlighted by the late-stage difluoromethoxylation of bio-relevant molecules at ambient temperature and pressure. Notably, this report not only provides the first experimental access to and utilization of the OCF2H radical but also establishes the first photocatalytic and selective formation of the OCF2H radical. We expect that this reagent and protocol will create a new avenue for the design and development of difluoromethoxylation reactions of hydrocarbons to aid the discovery and synthesis of new pharmaceuticals.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support for this work was provided by NIH (R35GM119652 to M.-Y. N.), the NSF (CHE-1654122 to P. L.), and National Science Foundation Graduate Research Fellowship Program under Grant No. 1747452. We thanks Peng Zhao for help with measurement of the emission spectrum of the LED light and Zhixiu Liang for the CV measurements. Calculations were performed at the Center for Research Computing at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the NSF (ACI-1053575).Notes and references
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (b) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Blackwell Publishing Ltd, 2009 CrossRef; (c) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed; (d) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (e) W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Aceña, K. Izawa, H. Liu and V. A. Soloshonok, J. Fluorine Chem., 2014, 167, 37–54 CrossRef CAS; (f) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- (a) K. Müller, Chimia, 2014, 68, 356–362 CrossRef PubMed; (b) Q. A. Huchet, N. Trapp, B. Kuhn, B. Wagner, H. Fischer, N. A. Kratochwil, E. M. Carreira and K. Müller, J. Fluorine Chem., 2017, 198, 34–46 CrossRef CAS.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) L. Xing, D. C. Blakemore, A. Narayanan, R. Unwalla, F. Lovering, R. A. Denny, H. Zhou and M. E. Bunnage, ChemMedChem, 2015, 10, 715–726 CrossRef CAS PubMed.
- S. M. Cheer, A. Prakash, D. Faulds and H. M. Lamb, Drugs, 2003, 63, 101–133 CrossRef CAS PubMed.
- Synthesis of aryl difluoromethyl ethers from phenols. For a review on using carbene intermediate, see: (a) C. Ni and J. Hu, Synthesis, 2014, 46, 842–863 CrossRef; for selected examples of stoichiometric reactions, see: (b) L. Zhang, J. Zheng and J. Hu, J. Org. Chem., 2006, 71, 9845–9848 CrossRef CAS PubMed; (c) J. Zheng, Y. Li, L. Zhang, J. Hu, G. J. Meuzelaar and H.-J. Federsel, Chem. Commun., 2007, 5149–5151 RSC; (d) Y. Zafrani, G. Sod-Moriah and Y. Segall, Tetrahedron, 2009, 65, 5278–5283 CrossRef CAS; (e) F. Wang, W. Huang and J. Hu, Chin. J. Chem., 2011, 29, 2717–2721 CrossRef CAS; (f) F. Wang, L. Zhang, J. Zheng and J. Hu, J. Fluorine Chem., 2011, 132, 521–528 CrossRef CAS; (g) J. B. Sperry and K. Sutherland, Org. Process Res. Dev., 2011, 15, 721–725 CrossRef CAS; (h) P. S. Fier and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 2092–2095 CrossRef CAS PubMed; (i) C. S. Thomoson and W. R. Dolbier Jr, J. Org. Chem., 2013, 78, 8904–8908 CrossRef CAS PubMed; (j) L. Li, F. Wang, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2013, 52, 12390–12394 CrossRef CAS PubMed; for examples of catalytic O–CF2H bond forming reactions, see: (k) K. Levchenko, O. P. Datsenko, O. Serhiichuk, A. Tolmachev, V. O. Iaroshenko and P. K. Mykhailiuk, J. Org. Chem., 2016, 81, 5803–5813 CrossRef CAS PubMed; (l) J. Yang, M. Jiang, Y. Jin, H. Yang and H. Fu, Org. Lett., 2017, 19, 2758–2761 CrossRef CAS PubMed. For examples of other indirect, multi-step strategies, see: (m) Y. Hagooly, O. Cohen and S. Rozen, Tetrahedron Lett., 2009, 50, 392–394 CrossRef CAS; (n) W. R. Dolbier Jr, F. Wang, X. Tang, C. S. Thomoson and L. Wang, J. Fluorine Chem., 2014, 160, 72–76 CrossRef.
- (a) J. W. Lee, D. N. Spiegowski and M. Y. Ngai, Chem. Sci., 2017, 8, 6066–6070 RSC; (b) W. Zheng, C. A. Morales-Rivera, J. W. Lee, P. Liu and M. Y. Ngai, Angew. Chem., Int. Ed., 2018, 57, 9645–9649 CrossRef CAS PubMed; (c) W. Zheng, J. W. Lee, C. A. Morales-Rivera, P. Liu and M. Y. Ngai, Angew. Chem., Int. Ed., 2018, 57, 13795–13799 CrossRef CAS PubMed.
- Selected reviews on photoredox catalysis: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) M. D. Karkas, J. A. Porco Jr and C. R. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef CAS PubMed; (c) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (d) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (e) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed.
- Reviews on photocatalytic formation of alkoxy radicals: (a) J. Zhang and Y. Y. Chen, Acta Chim. Sin., 2017, 75, 41–48 CrossRef CAS; (b) J. J. Guo, A. H. Hu and Z. W. Zuo, Tetrahedron Lett., 2018, 59, 2103–2111 CrossRef CAS; (c) K. F. Jia and Y. Y. Chen, Chem. Commun., 2018, 54, 6105–6112 RSC.
- Selected examples of photocatalytic formation of alkoxy radicals via N–O bond cleavage: (a) V. Quint, F. Morlet-Savary, J. F. Lohier, J. Lalevee, A. C. Gaumont and S. Lakhdar, J. Am. Chem. Soc., 2016, 138, 7436–7441 CrossRef CAS PubMed; (b) C. Y. Wang, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 13495–13498 CrossRef CAS PubMed; (c) J. Zhang, Y. Li, F. Y. Zhang, C. C. Hu and Y. Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 1872–1875 CrossRef CAS PubMed; (d) J. Zhang, Y. Li, R. Y. Xu and Y. Y. Chen, Angew. Chem., Int. Ed., 2017, 56, 12619–12623 CrossRef CAS PubMed; (e) B. J. Jelier, P. F. Tripet, E. Pietrasiak, I. Franzoni, G. Jeschke and A. Togni, Angew. Chem., Int. Ed., 2018, 57, 13784–13789 CrossRef CAS PubMed; (f) A.-L. Barthelemy, B. Tuccio, E. Magnier and G. Dagousset, Angew. Chem., Int. Ed., 2018, 57, 13790–13794 CrossRef CAS PubMed.
- Computation studies of the OCF2H radical, see: (a) J. Rayez, M. Rayez, P. Halvick, B. Duguay and J. Dannenberg, Chem. Phys., 1987, 118, 265–272 CrossRef CAS; (b) J. Francisco and Y. Zhao, J. Chem. Phys., 1990, 93, 276–286 CrossRef CAS; (c) W. F. Schneider, B. I. Nance and T. J. Wallington, J. Am. Chem. Soc., 1995, 117, 478–485 CrossRef CAS.
- A. A. Tabolin and S. L. Ioffe, Chem. Rev., 2014, 114, 5426–5476 CrossRef CAS PubMed.
- See ESI† for detailed optimization tables.
- G. B. Ellison, G. E. Davico, V. M. Bierbaum and C. H. DePuy, Int. J. Mass Spectrom. Ion Processes, 1996, 156, 109–131 CrossRef CAS.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- D. A. Nagib and D. W. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed.
- C. Bock, T. Meyer and D. Whitten, J. Am. Chem. Soc., 1975, 97, 2909–2911 CrossRef CAS.
- A. Juris, V. Balzani, P. Belser and A. von Zelewsky, Helv. Chim. Acta, 1981, 64, 2175–2182 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05390a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |