
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcyclic 1,2-dimagnesioethanes/-ethene derived from magnesium(I) compounds: multipurpose reagents for organometallic synthesis†
Deepak
Dange
a,
Andrew R.
Gair
ab,
Dafydd D. L.
Jones
 a,
Martin
Juckel
a,
Simon
Aldridge
b and
Cameron
Jones
*a
a,
Martin
Juckel
a,
Simon
Aldridge
b and
Cameron
Jones
*a
aSchool of Chemistry, Monash University, PO Box 23, VIC, 3800, Australia
bInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: cameron.jones@monash.edu; Web: http://www.monash.edu/science/research-groups/chemistry/jonesgroup
First published on 4th February 2019
Abstract
Reactions of three magnesium(I) dimers, [{(ArNacnac)Mg–}2] (ArNacnac = [(ArNCMe)2CH]−; Ar = xylyl (Xyl), mesityl (Mes) or 2,6-diethylphenyl (Dep)), with either 1,1-diphenylethylene (DPE), α-methylstyrene (MS), trans-stilbene (TS) or diphenylacetylene (DPA) led to the 1,2-addition of the Mg–Mg bond across the substrate, giving rise to the 1,2-dimagnesioethanes, [{(XylNacnac)Mg}2(μ-DPE)], [{(DepNacnac)Mg}2(μ-MS)], [{(ArNacnac)Mg}2(μ-TS)] (Ar = Mes or Dep); and a 1,2-dimagnesioethene, [{(MesNacnac)Mg}2(μ-DPA)]. The reactions involving the 1,1-substituted alkenes are shown to be readily redox reversible, in that the reaction products are in equilibrium with a significant proportion of the starting materials at room temperature. Variable temperature NMR spectroscopy and a van't Hoff analysis point to low kinetic barriers to these weakly exergonic reactions. [{(MesNacnac)Mg}2(μ-DPE)] and [{(MesNacnac)Mg}2(μ-DPA)] behave as 1,2-di-Grignard reagents in their reactions with very bulky amido-zinc bromides, yielding the first examples of a 1,2-dizincioethane, [(L*Zn)2(μ-DPE)] (L* = –N(Ar*)(SiPri3); Ar* = C6H2Me{C(H)Ph2}2-4,2,6), and a 1,2-dizincioethene, [(TBoLZn)2(μ-DPA)] (TBoL = –N(SiMe3){B(DipNCH)2}, Dip = 2,6-diisopropylphenyl), respectively. Divergent reactivity is shown for [{(MesNacnac)Mg}2(μ-DPE)], which behaves as a two-electron reducing agent when treated with amido-cadmium and amido-magnesium halide precursors, yielding the cadmium(I) and magnesium(I) dimers, [PhBoLCdCdPhBoL] (PhBoL = –N(SiPh3){B(DipNCH)2}) and [L†MgMgL†] (L† = –N(Ar†)(SiMe3); Ar† = C6H2Pri{C(H)Ph2}2-4,2,6), respectively. A further class of reactivity for [{(MesNacnac)Mg}2(μ-DPE)] derives from its reaction with the bulky amido-germanium chloride, L*GeCl, which gives a magnesio-germane, presumably via intramolecular C–H activation of a highly reactive magnesiogermylene intermediate, [:Ge(L*){Mg(MesNacnac)}]. [{(MesNacnac)Mg}2(μ-DPE)] can be considered as acting as a two-electron reducing, magnesium transfer reagent in this reaction.
Introduction
Over the last 15 years or so, interest in low oxidation state p-block systems has rapidly grown, largely as a result of the fact that many such compounds have been shown capable of activating a variety of small molecules (e.g. H2, CO, CO2, alkenes etc.) under mild conditions.1 These activations are often closely related to fundamental steps in important synthetic transformations that are typically catalysed by late transition metal complexes. Although the involvement of low oxidation state p-block complexes in analogous catalytic transformations is generally prevented by the irreversibility of their oxidative small molecule activations, reports of reversible redox processes, and associated catalysis, at p-block element centres are becoming increasingly common.2Given the more electropositive nature of s-block metals, it is perhaps not surprising that reversible redox processes involving those metals at room temperature were unknown until recently. Indeed, for such processes to occur, low oxidation state s-block metal compounds would need to be accessible. The only stable, well developed examples of compounds in this category are magnesium(I) dimers, LMg–MgL (L = anionic ligand), first described by us in 2007.3,4 Since that time, approximately 20 examples of magnesium(I) compounds have come forward, and their utility as widely applicable, selective, soluble reducing agents in many areas of inorganic and organic synthesis has been extensively exploited.5 This work has included the reductive activation of a wide array of small molecules and unsaturated substrates, none of which are reversible in the absence of an external reducing agent.
This situation changed in 2017 when we reported that reactions between two magnesium(I) dimers and the activated alkene, 1,1-diphenylethylene (DPE), led to 1,2-addition of the Mg–Mg bonds across the alkene, and the formation of the 1,2-dimagnesioethane compounds, 1 and 2 (Scheme 1).6 Remarkably, these reactions were shown to readily and rapidly reversible at room temperature, leading to equilibrium mixtures of products and reactants. As such, they represented the first examples of reversible redox processes for s-block metal complexes. Also of interest is the fact that 1 and 2 can be considered as analogues of acyclic 1,2-di-Grignard reagents,7 which are unknown despite attempts to prepare them since the time of Grignard himself.8 Preliminary reactivity studies of 1 and 2 revealed that to some extent they do behave as Grignard reagents, but the high charge density on their doubly reduced C–C cores leads to them behaving as more highly activated magnesium alkyls. This was demonstrated through their facile reactions with H2, CO and ethylene which lead to hydrogenolysis of an Mg–C bond, C–C coupling of two CO molecules with the DPE dianion, and insertion of ethylene into an Mg–C bond, respectively. The high reactivity of 1 and 2 is evidenced by the fact that magnesium alkyls have previously proved unreactive towards these three gaseous substrates in the absence of catalysts. The activated nature of 1 and 2 is comparable to that of a trans-stilbene dianion bridged bis(amidinato-calcium) complex 3 (ref. 9 and 10) and a benzene dianion bridged bis(β-diketiminato-magnesium) complex 4,11 which Harder and co-workers have recently shown to activate dihydrogen, C–F and C–H bonds, in processes in which the bridging dianion behaves as either a Brønsted base or as a two-electron reductant.
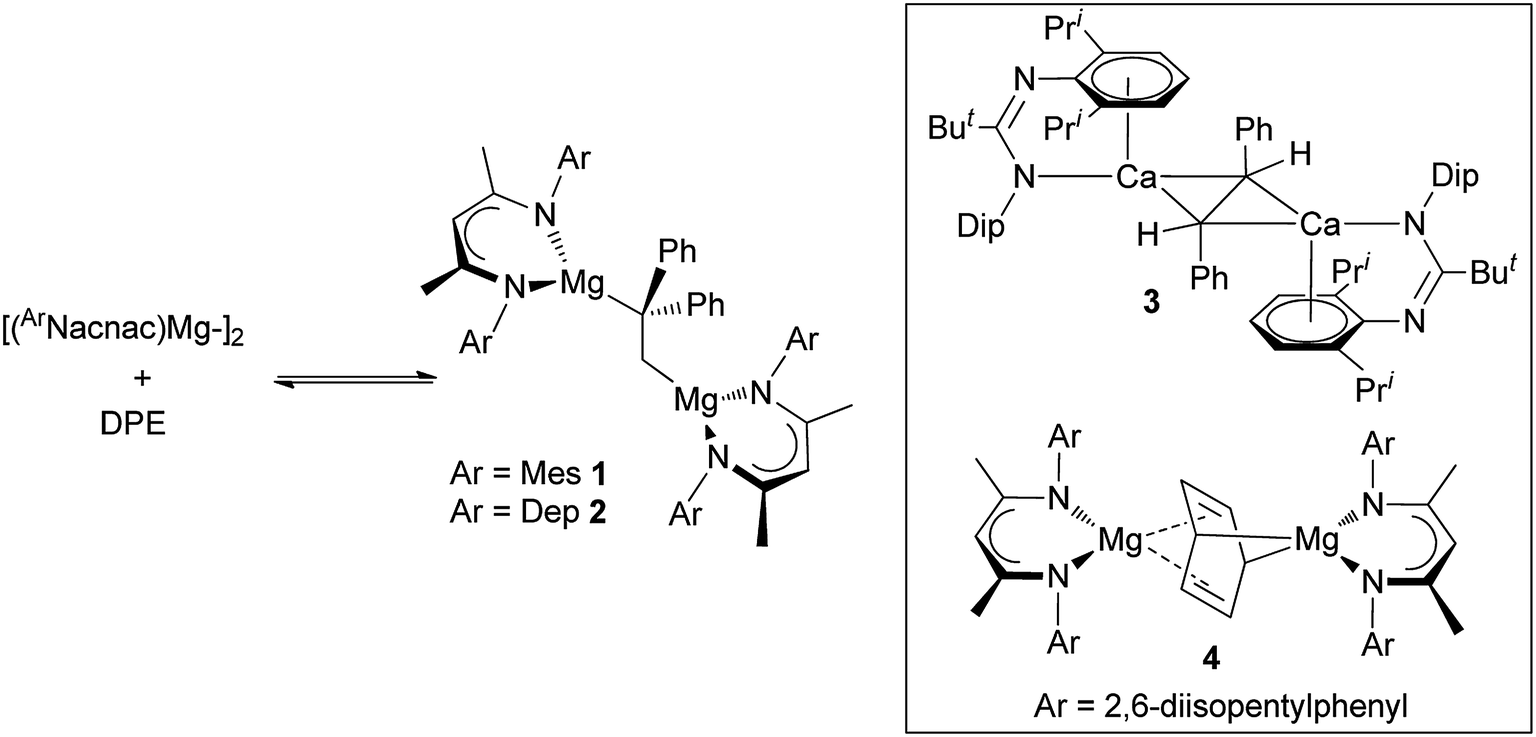 | ||
| Scheme 1 Equilibrium reaction mixtures involving compounds 1 and 2 (left, DPE = 1,1-diphenylethylene), and organyl dianion bridged compounds 3 and 4 (inset). | ||
Considering the unprecedented nature of the redox reversible reactions that gave 1 and 2, and the unusual reactivity of those compounds, we wished to test the generality of 1,2-addition reactions between magnesium(I) dimers and unsaturated organic molecules other than DPE. Here we describe a number of comparable additions of Mg–Mg bonds across alkenes and one alkyne, and show that these too can be readily reversible under mild conditions. In addition, we explore applications of the generated magnesium alkyls in organometallic synthesis. This work has revealed the compounds to be multi-functional reagents which can act as either 1,2-di-Grignard reagents, two-electron reductants, or reductive magnesium transfer reagents in their reactions with bulky amido-metal halide compounds.
Results and discussion
(i) 1,2-additions of magnesium(I) dimers across alkenes and an alkyne
As was the case with the synthesis of 1 and 2, the reactions of three β-diketiminate substituted magnesium(I) dimers, [{(ArNacnac)Mg–}2] (ArNacnac = [(ArNCMe)2CH]−; Ar = xylyl (Xyl), mesityl (Mes) or 2,6-diethylphenyl (Dep)), with either DPE, α-methylstyrene, trans-stilbene or diphenylacetylene afforded the 1,2-dimagnesioethane/-ethene complexes, 5–9, as orange to red crystalline solids in moderate to good isolated yields (Scheme 2). It is of note that we previously reported that attempts to prepare the trans-stilbene dianion bridged system, 7, resulted in an intractable product mixture.6 It is now believed that in that case the stilbene starting material was contaminated with impurities. Of further note is the fact that treatment of the bulkier 2,6-diisopropylphenyl (Dip) substituted magnesium(I) dimer, [{(DipNacnac)Mg–}2], with α-methylstyrene led to no reaction. This magnesium(I) compound showed a similar lack of reactivity towards DPE.6 Its inertness towards these olefins is most likely a function of its greater steric bulk relative to [{(ArNacnac)Mg–}2] (Ar = Xyl, Mes or Dep), which hinders approach of the olefin to its Mg centres. | ||
| Scheme 2 Synthesis of compounds 5–9. Secondary Mg⋯C interactions omitted. Depicted reaction equilibria only exist for mixtures containing 5 and 6. | ||
All compounds 5–9 are thermally stable in the solid state. In solution, their NMR spectral patterns are largely consistent with their solid state structures (see below). Of note are the signals for the alkenic derived protons in the 1H NMR spectra of 5–8 (viz. δ 0.37 ppm 5, δ 0.16 ppm 6, δ 2.50 ppm 7, δ 2.39 ppm 8) which are considerably upfield from the corresponding signals in the olefin starting material, suggesting reduction of the double bonds in those substrates. With that said, NMR spectroscopic assignments for 7 and 8 are only tentative as, when even analytically pure samples were dissolved in non-coordinating deuterated solvents, spectra consistent with compound mixtures (possibly co-crystallised diastereoisomers), not including the alkene and magnesium(I) starting materials, eventuated. Variable temperature NMR spectroscopic studies of these mixtures led to little change in the appearance of their spectra.
Similar to 7 and 8, the variable temperature NMR spectra for 9 did not indicate an equilibrium in solution between this compound and the starting materials that led to it. In contrast, and analogous to the situation with 1 and 2, dissolution of crystals of 5 and 6 in C6D6 led to equilibrium mixtures of the compounds, and the magnesium(I) and olefin reactants. At 293 K the ratio of products to reactants for the equilibrium involving 5 was 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13, which is close to the 80:20 ratio reported for sterically slightly larger 1.6 In contrast, the product:reactant ratio for the equilibrium mixture containing 6 was 43:57 at 298 K. This is consistent with the sterically more hindered DepNacnac ligand disfavouring the product 6, but inconsistent with the smaller size of α-methylstyrene relative to DPE. Of course, electronic differences between the two olefins could play a part in the positions of these equilibria.
:13, which is close to the 80:20 ratio reported for sterically slightly larger 1.6 In contrast, the product:reactant ratio for the equilibrium mixture containing 6 was 43:57 at 298 K. This is consistent with the sterically more hindered DepNacnac ligand disfavouring the product 6, but inconsistent with the smaller size of α-methylstyrene relative to DPE. Of course, electronic differences between the two olefins could play a part in the positions of these equilibria.
As previously observed for 1 and 2,6 heating solutions of 5 or 6, led to an increase in the proportion of starting materials in the equilibria, while cooling them increased the amount of product. In these experiments, equilibria were rapidly established, suggesting the kinetic barrier to the redox reversible insertion of the olefin into the Mg–Mg bond is low. Indeed in a DFT analysis of the reaction profile that gave 1, no barrier to this insertion could be located.6 A van't Hoff analysis of the variable temperature NMR data for equilibrium mixture containing 5 was carried out, and this gave values of ΔH = −13.2(1.3) kcal mol−1, ΔS = −0.0326(0.004) kcal mol−1 K−1 and ΔG293 = −3.7(0.4) kcal mol−1, which are close to the respective values for the equilibrium in which 1 is the product (ΔH = −13.7(1.4) kcal mol−1, ΔS = −0.0353(0.004) kcal mol−1 K−1 and ΔG298 = −3.2(0.3) kcal mol−1), and show that the reaction that gives 5 is only mildly exergonic. A meaningful van't Hoff analysis of the reaction that gave 6 could not be carried out due to overlapping of product and reactant resonances over the temperature range studied.
All of the compounds 5–9 were crystallographically characterised, and the molecular structures of 5, 6, 8 and 9 are depicted in Fig. 1 (see Table 1 for selected metrical parameters, and ESI† for the structure of 7). All of the complexes are bimetallic, and confirm insertion of the alkene or alkyne moiety into the Mg–Mg bond of the magnesium(I) starting material. Such insertions of alkenes into M–M bonds are rare for main group elements, but non-reversible examples have been reported for p-block elements.12 The C–C bond lengths of the inserted fragments in 5–9 suggest the full reduction of those fragments, giving rise to 1,2-dimagnesioethane/-ethene complexes. The structures of 5 and 6 are similar to that of 1 in that they exhibit primary Mg–C bond lengths in the normal range,13 but also close secondary Mg–C interactions (dotted bonds in Fig. 1) with one opposing carbon of the reduced CC fragment (5 and 6), and one phenyl carbon of the DPE dianion in 5. As with 1, this leads to heavily distorted tetrahedral geometries at the carbon centres of the reduced alkene unit. This is not unreasonable considering the likely polarised, and low covalent, character of the primary δ+Mg–Cδ− bonds.
 | ||
| Fig. 1 Molecular structures of (a) 5, (b) 6, (c) 8 and (d) 9 (25% thermal ellipsoids are shown; hydrogen atoms, except alkenic protons, omitted). See Table 1 for selected metrical parameters. | ||
| 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|
| a Involving primary Mg–C interaction. Narrowest angle listed first. | |||||
| C–Calkene/yne | 1.552(2) | 1.58(1) | 1.491(7) | 1.545(3) | 1.370(6) |
| Mg–Calkene/yne (primary) | 2.137(2) | 2.084(7) | 2.153(3) | 2.156(2) | 2.104(5) |
| 2.142(2) | 2.16(1) | — | — | 2.106(5) | |
| Mg–Calkene/yne (secondary) | 3.329(2) | 2.324(1) | 2.401(3) | 2.552(2) | — |
| Mg⋯Carene (ipso) | — | — | 2.518(3) | 2.572(2) | 2.666(5) |
| Mg⋯Carene (ortho) | 2.566(2) | — | 2.721(4) | 2.537(2) | 2.727(5) |
| Mg–C–Ca | 76.47(9) | 77.3(5) | 62.1(2) | 57.4(1) | 103.3(3) |
| 101.2(1) | 110.4(5) | 80.2(2) | 85.5(1) | 112.5(3) | |
The reduced trans-stilbene complexes, 7 and 8, share similar structural features to 5 and 6, but are also closely related to structurally characterised complexes in which the trans-stilbene dianion bridges two lithium, aluminium or calcium centres in [{(pmdeta)Li}2{μ-C(H)(Ph)C(H)(Ph)}] (pmdeta = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine),14 [{(DAB)(THF)Al}{μ-C(H)(Ph)C(H)(Ph)}{Al(DAB)}] (DAB = (DipNCH)2)12b and 3,9 respectively. It is noteworthy that in the complexes involving the more electropositive metals, lithium and calcium, the alkenic derived carbons of the bridging trans-stilbene dianion exhibit considerable sp2-character (shorter C–C bonds than in 7 and 8), and the negative charges of the dianion are delocalised over the stilbene fragment. In contrast, in the less electropositive aluminium complex, the alkenic derived carbon centres appear to be close to sp3-hybridised, due to the more covalent M–C bonds in that compound. It is reasonable to conclude from the structural parameters displayed by 7 and 8, and the intermediate electronegativity of Mg, that the charge delocalisation in their bridging dianions lie somewhere between the cases mentioned above.9 As far as we are aware, compound 9 represents the first structurally characterised example of bimetallic, doubly reduced diphenylacetylene complex, incorporating s-block metals, and it displays a regioselective trans-disposition between its two (MesNacnac)Mg fragments, presumably due to steric reasons. It is worthy of mention that a number of complexes closely related to 9, but incorporating p-block metals have been reported,13 and the reduction of diphenyl acetylene with magnesium vapour has given the tetramer, [{–Mg(THF)C(Ph)C(Ph)–}4], as a mixture of trans- and cis-isomers.15
(ii) Reactivity of 1,2-dimagnesioethanes/-ethene towards amido-metal halide complexes
Taking into account the diverse and unprecedented reactivity the 1,2-dimagnesioethanes, 1 and 2, have shown towards small molecule substrates, it was seen of interest to explore the application of those compounds, and their analogues reported here, as reagents in organometallic synthesis. This is especially interesting as the compounds can be viewed as acyclic 1,2-di-Grignard reagents, which are unknown to date, but would have significant synthetic potential if accessible.7 In this respect, we believed reactions between the pseudo 1,2-di-Grignard reagents and extremely bulky amido metal halide complexes, developed in our group, would be a worthwhile starting point. Our reasoning here was that the bulky amide ligands in these precursor molecules have demonstrated abilities to kinetically stabilise low oxidation state metal systems,5 which might eventuate from the proposed reaction studies, if 1, 2, and 5–9 act as reducing agents, cf.4.11Initially we explored the 1:2 reaction of 1 (as an exemplar for 1,2-dimagnesioethanes) with the bulky amido zinc halide, L*ZnBr (L* = –N(Ar*)(SiPri3); Ar* = C6H2Me{C(H)Ph2}2-4,2,6). This precursor was chosen as we have previously shown its reaction with the magnesium(I) dimer, [{(MesNacnac)Mg–}2], leads to double reduction of the substrate and formation of the Zn–Mg bonded bimetallic complex, [(MesNacnac)Mg–ZnL*].16 Considering that 1 is in equilibrium in solution with [{(MesNacnac)Mg–}2], a similar outcome was possible here. In contrast, however, the reaction led to a double salt metathesis reaction and the formation of the first structurally characterised example of a 1,2-dizincioethane complex, 10, in good isolated yield (Scheme 3). Clearly, 1 is acting as 1,2-di-Grignard reagent in this reaction, and not as a two-electron reducing agent, either through the presence of [{(MesNacnac)Mg–}2] in the reaction mixture, or directly through electron transfer from its bridging DPE dianion, cf.4.11 It is noteworthy, however, that the reaction was carried out at −80 °C, at which temperature negligible [{(MesNacnac)Mg–}2] should be present in the mixture. Moreover, when the reaction was repeated in a 1:1 stoichiometry, only an approximately 50:50 mixture of 10 and unreacted L*ZnBr were returned (as determined by 1H NMR spectroscopic monitoring), which indicates that the possible heterobimetallic reaction intermediate, [L*Zn(μ-DPE)Mg(MesNacnac)], is not accessible.
 | ||
| Scheme 3 Synthesis of 1,2-dizincioethane 10 and 1,2-dizincioethene 11 (see text for ligand definitions). | ||
Like 5–8, 1,2-dizincioethanes, such as 10, have potential for use as di-Grignard like reagents in both organic and inorganic syntheses. However, the lower electronegativity of zinc relative to magnesium should make them milder, and potentially more controllable, nucleophiles in synthesis, as is the case for monometallic zinc alkyls compared to mono-Grignard reagents.17 So as to demonstrate a degree of generality to the accessibility of 1,2-dizincioorganyls, compound 9 was treated with two equivalents of the bulky amido zinc halide, [TBoLZnBr(THF)] (TBoL = –N(SiMe3){B(DipNCH)2}),18 which led to colourless crystalline 11 in good isolated yield (Scheme 3). Compound 11 is the first structurally characterised example of a 1,2-dizincioethene, and differs from 10 in that it incorporates a boryl substituted amide ligand, which is less bulky than the L* amide in 10, but which we have previously exploited in the kinetic stabilisation of low oxidation state main group compounds.19 Compounds 10 and 11 are thermally stable in both solution and the solid state. In solution, their NMR spectra are reminiscent of their solid state structures (see below), in that two sets of ligand resonances are observed for unsymmetrical 10, while only one set of ligand signals are seen for centrosymmetric 11. The alkene derived protons on the bridging DPE dianion resonate at a similar field (δ 0.65 ppm) to those in the 1,2-dimagnesioethanes, 1, 2, 5, and 6. The molecular structures of 10 and 11 are depicted in Fig. 2 and show each compound to possess two near linear two-coordinate zinc centres, having Zn–N and Zn–C distances in the normal ranges.13 In 10, the central C–C distance, and the tetrahedral geometry for each of the carbons, confirms the bond involving those carbons to be single. The C(1)–C(1)′ bond length in 11 is that of a double bond, as expected for the proposed 1,2-dizincioethene formulation of the compound. The two bulky zinc amide fragments are trans- to each other in this compound. In contrast to the magnesium organyls, 1 and 9, from which they were derived, there are no significant metal⋯arene interactions, or interactions between the metal and the opposing carbon of the central C2 unit in 10 and 11.
 | ||
| Fig. 2 Molecular structures of (a) 10 and (b) 11 (25% thermal ellipsoids are shown; hydrogen atoms, except alkenic protons, omitted). Selected bond lengths (Å) and angles (°) for 10: Zn(1)–N(1) 1.860(4), Zn(1)–C(43) 1.998(5), Zn(2)–N(2) 1.863(4), Zn(2)–C(56) 1.955(5), C(43)–C(56) 1.533(8), Zn(1)–N(1) 1.860(4), Zn(1)–C(43) 1.998(5), Zn(2)–N(2) 1.863(4), Zn(2)–C(56) 1.955(5), C(43)–C(56) 1.533(8), N(1)–Zn(1)–C(43) 163.9(2), N(2)–Zn(2)–C(56) 179.0(2), C(56)–C(43)–Zn(1) 105.3(3), C(43)–C(56)–Zn(2) 118.6(4). Selected bond lengths (Å) and angles (°) for 11: Zn(1)–N(1) 1.856(2), Zn(1)–C(1) 1.945(2), C(1)–C(1)′ 1.348(4), N(1)–Zn(1)–C(1) 174.16(8), C(1)′–C(1)–Zn(1) 123.2(2). | ||
Attempts were made to extend the utility of 1 and 9 as pseudo 1,2-di-Grignard reagents to the preparation of cadmium and mercury analogues of 10 and 11. To this end, reactions of 1 and 9 with a number of bulky aryl/silyl and boryl/silyl amide substituted cadmium and mercury halide precursor complexes were carried out. However, in all cases, corresponding 1,2-dicadmio- or 1,2-dimercurioorganyls were not isolated, and the reactions typically gave product mixtures which included the elemental group 12 metal. One exception was the 2:1 reaction between the very bulky boryl/silyl amide substituted compound, PhBoLCdI (PhBoL = –N(SiPh3){B(DipNCH)2}) and 1 at −80 °C. This gave a moderate isolated yield (46%) of the yellow crystalline cadmium(I) dimer 12 after work-up (Scheme 4). Contrasting to the synthesis of 10, reagent 1 appears to act as a two-electron reductant in the formation of 12, rather than as a 1,2-di-Grignard reagent. Although it is known that at −80 °C, spectroscopically immeasurable amounts of the magnesium(I) dimer [{(MesNacnac)Mg–}2] are in equilibrium with 1 in solution, the participation of the magnesium(I) compound as the reducing agent in the formation of 12, rather than 1, cannot be ruled out. Indeed, we found that the direct reduction of PhBoLCdI with [{(MesNacnac)Mg–}2] also yielded the cadmium(I) species, 12, in a similar yield. That said, this observation does not disprove the ability of 1 to act as a two-electron reductant in its own right.
 | ||
| Scheme 4 Synthesis of cadmium(I) dimer 12 and magnesium(I) dimer, [L†MgMgL†] (see text for ligand definitions). | ||
To examine this possibility further, we investigated the reaction of the bulky amido-magnesium(II) iodide complex, L†MgI (L† = –N(Ar†)(SiMe3); Ar† = C6H2Pri{C(H)Ph2}2-4,2,6) with 1. We have previously reported that reduction of this precursor with KC8 yielded a rare example of a two-coordinate magnesium(I) dimer, [L†MgMgL†], in moderate yield (67%).20 Similarly, reduction of L†MgI with 1 afforded a considerably higher isolated yield (80%) of [L†MgMgL†], and generated free DPE (which is unreactive towards [L†MgMgL†]). Importantly, attempts to reduce L†MgI directly with [{(MesNacnac)Mg–}2] led to no reaction occurring, even when the reaction mixtures were heated to 60 °C.21 This provides compelling evidence that 1 is acting as a two-electron reductant in its own right, and not via the magnesium(I) dimer [{(MesNacnac)Mg–}2] that it is in equilibrium with (cf. the use of 4 as a two electron reductant11). Accordingly, the variable synthetic utility of 1 as either a pseudo 1,2-di-Grignard reagent, or a two-electron reductant has been demonstrated.
Cadmium(I) dimer 12 is thermally stable, both in solution and the solid state. The 1H and 13C{1H} NMR spectra for the complex are consistent with its centrosymmetric structure, in that they exhibit one set of amide ligand resonances each. The relatively low solubility of the compound in C6D6 precluded confident assignment of its 113Cd NMR spectrum. The molecular structure of 12 is depicted in Fig. 3, which shows it to possess two essentially linear, two-coordinate cadmium centres, with no meaningful Cd⋯arene interactions. The Cd–Cd distance at 2.633(1) Å is similar to those in the only other two-coordinate cadmium(I) dimers ([L*CdCdL*] 2.5786(6) Å (ref. 16) and [Ar′CdCdAr′] 2.6257(5) Å, Ar′ = C6H3(C6H3Pri2-2,6)2-2,6 (ref. 22)), and there is no crystallographic (or spectroscopic) evidence for the presence of hydride ligands bridging the two Cd centres. It is noteworthy that the magnesium(I) dimer [L†MgMgL†] crystallised in a different polymorph to that already reported (see ESI† for crystallographic details),20 though there are no significant geometric differences between the two polymorphs.
 | ||
| Fig. 3 Molecular structure of 12 (25% thermal ellipsoids are shown; hydrogen atoms omitted). Selected bond lengths (Å) and angles (°): Cd(1)–N(3) 2.129(4), Cd(1)–Cd(1)′ 2.6328(8), B(1)–N(3) 1.395(8), Si(1)–N(3) 1.730(5), N(3)–Cd(1)–Cd(1)′ 176.70(11), B(1)–N(3)–Si(1) 126.4(4). | ||
As a final comparison between the further chemistry of [{(MesNacnac)Mg–}2] and 1, we examined the reaction of the latter with the bulky amido-germanium chloride complex, [L*GeCl]. We have previously shown that the almost equivalent complex, [L′GeCl] (L′ = –N(Ar†)(SiPri3)) is readily reduced with [{(MesNacnac)Mg–}2] to give the amido-digermyne, [L′GeGeL′], in good yield.23 In contrast, reaction of [L*GeCl] with 1 in 1:1 or 2:1 stoichiometries did not yield a digermyne, or a 1,2-digermaethane (cf.10), but instead gave free DPE and the unusual magnesiogermane, 13, as colorless crystals in up to a 56% isolated yield (Scheme 5). It is highly likely that this reaction proceeds via a two-electron reduction of [L*GeCl] by 1, with concomitant transfer of the Mg(MesNacnac) fragment to the Ge centre, to give the transient magnesio-germylene intermediate, 14 (not observed spectroscopically). This then rapidly undergoes an intramolecular phenyl C–H activation, ultimately yielding 13. The high reactivity of 14 is not surprising when it is considered that we have recently reported that, although isolable, a closely related zinca-germylene, (TBoL)(L*Zn)Ge:, possesses a very narrow HOMO–LUMO gap, and rapidly activates dihydrogen at room temperature to give the germane, [(TBoL)(L*Zn)GeH2].19b,24 The preparation of 13, via14, reveals a third mode of reactivity for 1,2-dimagnesioethanes towards amido-metal halides, namely their use as two-electron reducing, magnesium transfer reagents.
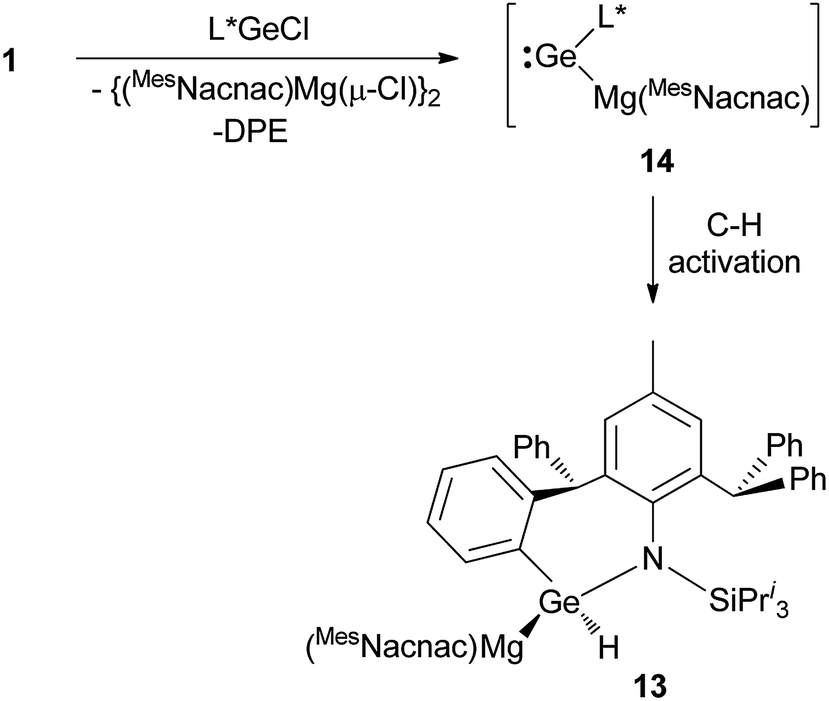 | ||
| Scheme 5 Synthesis of magnesiogermane 13via transient magnesiogermylene 14 (see text for ligand definitions). | ||
In solution, thermally stable 13 exhibits complicated 1H and 13C{1H} NMR spectra, as would be expected for this unsymmetrical compound. The highlight of its FTIR spectrum is a strong Ge–H stretching band centred at ν = 1942 cm−1. The molecular structure of 13 is illustrated in Fig. 4, which shows it to be monomeric with a tetrahedral Ge centre (hydride ligand located from difference maps and freely refined). Although only a handful of magnesiogermanes have been previously structurally characterised,25 examples bearing Ge–H moieties are unknown. The Mg–Ge distance (2.701(2) Å) is in the known range (2.626–2.766 Å),13 and the Ge(1)–C(42) separation (2.004(4) Å) clearly signifies a covalent bond.
 | ||
| Fig. 4 Molecular structure of 13 (25% thermal ellipsoids are shown; hydrogen atoms, except hydride H(1), omitted). Selected bond lengths (Å) and angles (°): Ge(1)–N(3) 1.907(3), Ge(1)–C(42) 2.004(4), Ge(1)–Mg(1) 2.7006(15), Ge(1)–H(1) 1.40(4), Mg(1)–C(52) 2.715(5), Mg(1)–C(53) 2.819(5), Mg(1)–C(51) 2.844(4), Mg(1)–Ge(1)–H(1) 117.2(19), N(3)–Ge(1)–C(42) 105.88(16). | ||
Conclusion
In summary, reactions of three magnesium(I) dimers with several alkenes and one alkyne have led to the 1,2-addition of Mg–Mg bonds across the double or triple bond of the substrate. In the cases of the 1,1-substituted alkenes, the reaction products were shown to be in equilibrium with a significant proportion of the starting materials at room temperature. Variable temperature NMR spectroscopy showed these equilibria to be rapidly reached, which indicates low kinetic barriers to these rare examples of redox reversible processes involving group 2 metals. Moreover, a van't Hoff analysis of the results of one variable temperature NMR spectroscopic study revealed the 1,2-addition reaction to be only weakly exergonic.The use of 1,2-dimagnesioethane and 1,2-dimagnesioethene complexes as reagents in organometallic chemistry was explored. This study showed the complexes to have multifunctional, and potentially very useful, capabilities in this realm. For example, both magnesium reagents behave as 1,2-di-Grignard transfer reagents in their reactions with amido-zinc bromide precursors, giving rise to the first examples of a 1,2-dizincioethane and a 1,2-dizincioethene. The importance of this result is compounded by the fact that acyclic 1,2-di-Grignard reagents are unknown, despite attempts to prepare them going back more than 100 years. Divergent reactivity has been shown by the 1,2-dimagnesioethane, which behaved as a two-electron reducing agent when treated with amido-cadmium and amido-magnesium halide precursors, yielding a new cadmium(I) dimer and a known magnesium(I) dimer. At least in the case of the formation of the amido-magnesium(I) dimer, it was confirmed that the reducing ability of the 1,2-dimagnesioethane was not derived from the β-diketiminate substituted magnesium(I) compound it is in equilibrium with. A further class of reactivity for the 1,2-dimagnesioethane was uncovered when it was treated with an amido-germanium chloride. Here it behaved as a two-electron reducing, magnesium transfer reagent in the synthesis of a magnesiogermane, which presumably proceeds via a highly reactive magnesio-germylene transient intermediate. We continue to explore the reactivity of 1,2-dimagnesioethanes and related activated magnesium(I) systems, and will report on this in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CJ and SA thank the Australian Research Council. CJ thanks the US Air Force Asian Office of Aerospace Research and Development (grant FA2386-18-1-0125) for funding. ARG acknowledges financial support from the Old Members' Trust of University College, University of Oxford. Part of this research was undertaken on the MX1 beamline at the Australian Synchrotron, Victoria, Australia.References
-
(a) C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS
; (b) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed
- Examples of formal redox and related Frustrated Lewis Pair based catalytic protocols for the p-block elements:
(a) N. L. Dunn, M. Ha and A. T. Radosevich, J. Am. Chem. Soc., 2012, 134, 11330–11333 CrossRef CAS PubMed
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed
- Several calcium(I) and beryllium(0) complexes are also known. See for example:
(a) S. Kriek, H. Görls, L. Yu, M. Reiher and M. Westerhausen, J. Am. Chem. Soc., 2009, 131, 2977–2985 CrossRef PubMed
-
(a) C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS
- A. J. Boutland, A. Carroll, C. A. Lamsfus, A. Stasch, L. Maron and C. Jones, J. Am. Chem. Soc., 2017, 139, 18190–18193 CrossRef CAS PubMed
- Reviews:
(a) F. Bickelhaupt, Angew. Chem., Int. Ed. Engl., 1987, 26, 990–1005 CrossRef
- L. Tissier and V. Grignard, C. R. Hebd. Seances Acad. Sci., 1901, 132, 835–837 Search PubMed
- A. Causero, H. Elsen, G. Ballmann, A. Escalona and S. Harder, Chem. Commun., 2017, 53, 10386–10389 RSC
- For a related DPE dianion bridged dicalcium systems, see: A. S. S. Wilson, M. S. Hill and M. F. Mahon, Organometallics, 2019, 38, 351–360 CrossRef CAS
- T. X. Gentner, B. Rösch, G. Ballmann, J. Langer, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 607–611 CrossRef CAS PubMed
- See for example:
(a) T. Sasamori, T. Sugahara, T. Agou, K. Sugamata, J.-D. Guo, S. Nagase and N. Tokitoh, Chem. Sci., 2015, 6, 5526–5530 RSC
- As determined from a survey of the Cambridge Crystallographic Database,† January, 2019.
- M. Walczak and G. Stucky, J. Am. Chem. Soc., 1976, 98, 5531–5539 CrossRef CAS
- M. A. G. M. Tinga, G. Schat, O. S. Akkerman, F. Bickelhaupt, E. Horn, H. Kooijman, W. J. J. Smeets and A. L. Spek, J. Am. Chem. Soc., 1993, 115, 2808–2817 CrossRef CAS
- J. Hicks, E. J. Underhill, C. E. Kefalidis, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2015, 54, 10000–10004 CrossRef CAS PubMed
-
The Chemistry of Organozinc Compounds, ed. Z. Rappoport and I. Marek, Wiley, Chichester, 2006 Search PubMed
- M. J. C. Dawkins, E. Middleton, C. E. Kefalidis, D. Dange, M. M. Juckel, L. Maron and C. Jones, Chem. Commun., 2016, 52, 10490–10492 RSC
-
(a) T. J. Hadlington, J. A. B. Abdalla, R. Tirfoin, S. Aldridge and C. Jones, Chem. Commun., 2016, 52, 1717–1720 RSC
- A. J. Boutland, D. Dange, A. Stasch, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2016, 55, 9239–9243 CrossRef CAS PubMed
- Reduction of magnesium(II) halide complexes with magnesium(I) dimers has previously afforded new magnesium(I) compounds, A. Stasch, Angew. Chem., Int. Ed., 2014, 53, 10200–10203 CrossRef CAS PubMed
- Z. Zhu, R. C. Fischer, J. C. Fettinger, E. Rivard, M. Brynda and P. P. Power, J. Am. Chem. Soc., 2006, 128, 15068–15069 CrossRef CAS PubMed
- T. J. Hadlington, M. Hermann, J. Li, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2013, 52, 10199–10203 CrossRef CAS PubMed
- For related activated germylenes, see: M. Usher, A. V. Protchenko, A. Rit, J. Campos, E. L. Kolychev, R. Tirfoin and S. Aldridge, Chem.–Eur. J., 2016, 22, 11685–11698 CrossRef CAS PubMed
- See for example: D. Matioszek, N. Katir, S. Ladeira and A. Castel, Organometallics, 2011, 30, 2230–2235 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for all new compounds. CCDC 1890169–1890179. Crystal data, details of data collections and refinements. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00200f |
| This journal is © The Royal Society of Chemistry 2019 |