
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTandem Peterson olefination and chemoselective asymmetric hydrogenation of β-hydroxy silanes†
Suppachai
Krajangsri‡
a,
Haibo
Wu‡
 a,
Jianguo
Liu‡
a,
Wangchuk
Rabten
a,
Thishana
Singh
b and
Pher G.
Andersson
*ab
a,
Jianguo
Liu‡
a,
Wangchuk
Rabten
a,
Thishana
Singh
b and
Pher G.
Andersson
*ab
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, 106 91, Stockholm, Sweden. E-mail: pher.andersson@su.se
bSchool of Chemistry and Physics, University of Kwazulu-Natal, Private Bag X54001, Durban, 4000, South Africa
First published on 4th February 2019
Abstract
Here, we report the first Ir–N,P complex catalyzed tandem Peterson olefination and asymmetric hydrogenation of β-hydroxy silanes. This reaction resulted in the formation of chiral alkanes in high isolated yields (up to 99%) and excellent enantioselectivity (up to 99% ee) under mild conditions. Modification of the reaction conditions provides a choice to transform either an olefin or the β-hydroxy silane in a chemoselective manner. Additionally, based on this method, an expedient enantioselective synthesis of (S)-(+)-α-curcumene, from a simple ketone, was accomplished in two steps with 75% overall yield and 95% ee.
Introduction
The chiral benzylic hydrocarbon motif is widespread in various functional molecules and bioactive natural products.1 Examples of such molecules include the anaesthetic propofol analog HSK3486,2 the bisabolanes sesquiterpenes (S)-(+)-curcumene and (S)-(+)-ar-turmerone with antibacterial activity,3 (+)-helianane with antifungal and cytotoxic activities4 and ileabethoxazole with strongly antimycobacterial activity5 (Fig. 1a). Notably, the induction of the benzylic chiral center is the crucial but challenging step in the synthesis of these compounds and has attracted considerable attention.6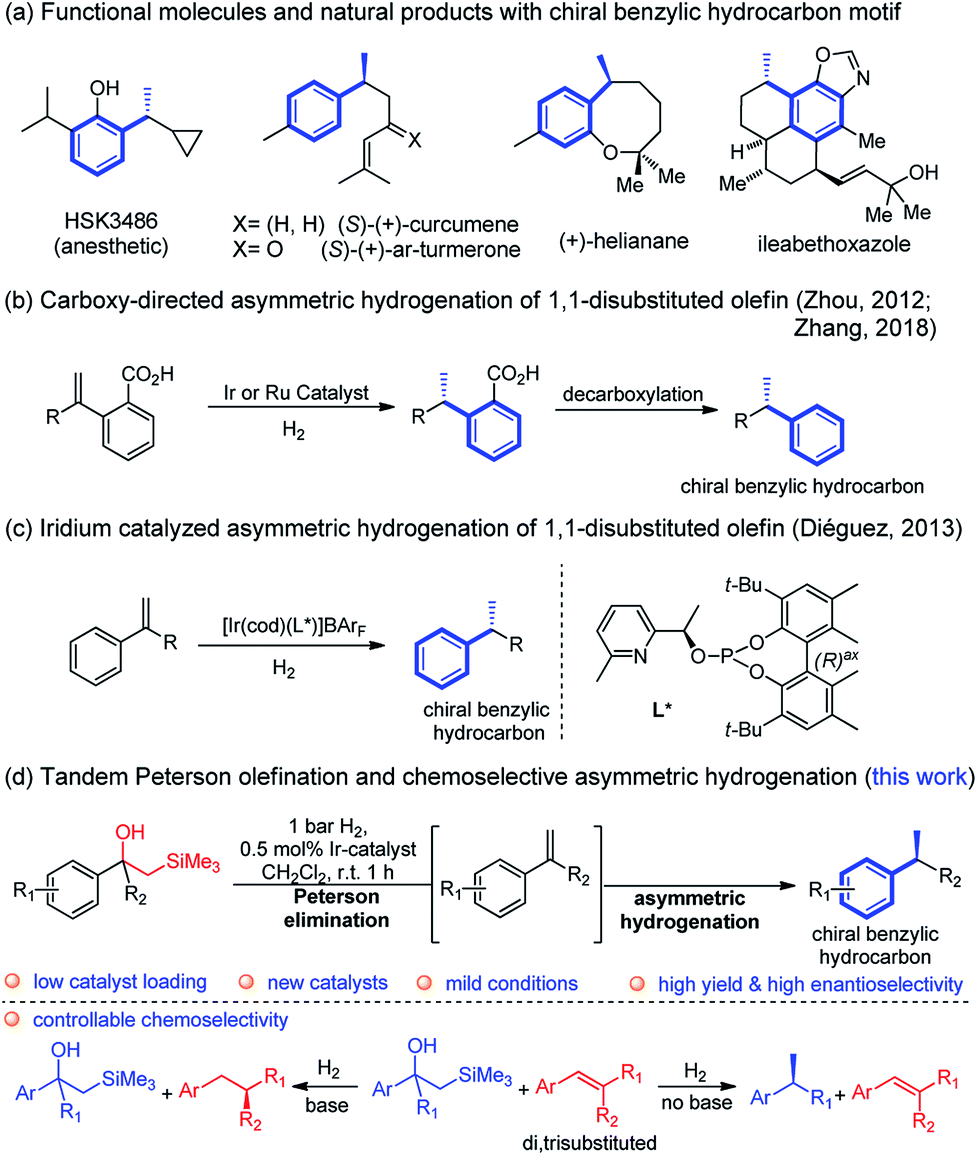 | ||
| Fig. 1 Asymmetric hydrogenation to construct chiral benzylic hydrocarbon motif. | ||
Asymmetric hydrogenation has, over the past decades, been successfully developed to achieve excellent enantioselectivity for a wide range of substrates.7 The reduction of olefins usually involves iridium, ruthenium, or rhodium complexes.8 Iridium catalysts have the outstanding advantage of not requiring a chelating functional group adjacent to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond to achieve high reaction activity and selectivity. In addition to well-known Ir, Rh and Ru catalyst systems, cobalt, iron and nickel catalysts recently have recently proved to be an effective alternative method for asymmetric hydrogenation.9 Furthermore, chemoselectivity and regioselectivity are still challenging to attain and there are only a few reported examples highlighting this feature.10
C bond to achieve high reaction activity and selectivity. In addition to well-known Ir, Rh and Ru catalyst systems, cobalt, iron and nickel catalysts recently have recently proved to be an effective alternative method for asymmetric hydrogenation.9 Furthermore, chemoselectivity and regioselectivity are still challenging to attain and there are only a few reported examples highlighting this feature.10
Asymmetric hydrogenation of 1,1-disubstituted olefins was expected to be a direct route to access the benzylic chiral center, however, it is quite challenging to achieve excellent enantioselectivity11 since it is much more difficult to distinguish the two prochiral faces of the terminal olefin. Utilizing directing groups, Zhou's group12a,b and Zhang's group12c successfully attained asymmetric hydrogenation of 1,1-disubstituted olefins with high enantioselectivity (Fig. 1b). The use of novel iridium phosphite–pyridine catalysts, by the Diéguez's group12d resulted in highly selective asymmetric hydrogenation of a wide range of terminal olefins (Fig. 1c). We recently achieved significant improvement in the development of Ir–N,P-complexes for the asymmetric hydrogenation of di-, tri- and tetrasubstituted olefins. Among these, substrates with the silane group have raised much interest.10e,13 In a previous study that we conducted on the asymmetric hydrogenation of secondary allylic alcohols, a substrate containing the β-hydroxy silane moiety was evaluated (Fig. 2a).14 Instead of attaining the mono-hydrogenated product of trisubstituted olefin, the fully hydrogenated alkane was observed as the only product. In fact, a tandem reaction of Peterson elimination and asymmetric hydrogenation occurred due to the acidic medium15 created by activation of the iridium catalyst. Recently, Mazet and co-workers reported a vinylogous Peterson elimination of allylic alcohols catalyzed by an Ir–N,P complex, where hydrogen gas was involved in the activation of the catalyst to generate the iridium hydride species (Fig. 2b).16 The β-hydroxy silane compounds are well-known as important intermediates for olefination,17 however their use as substrates for asymmetric hydrogenation have never been reported. Herein, we describe the first iridium catalyzed tandem Peterson elimination/olefination and asymmetric hydrogenation of β-hydroxy silanes to provide efficient access to the chiral benzylic hydrocarbon motif. Under different conditions, controllable chemoselectivity between β-hydroxy silanes and other olefins could be obtained (Fig. 1d).
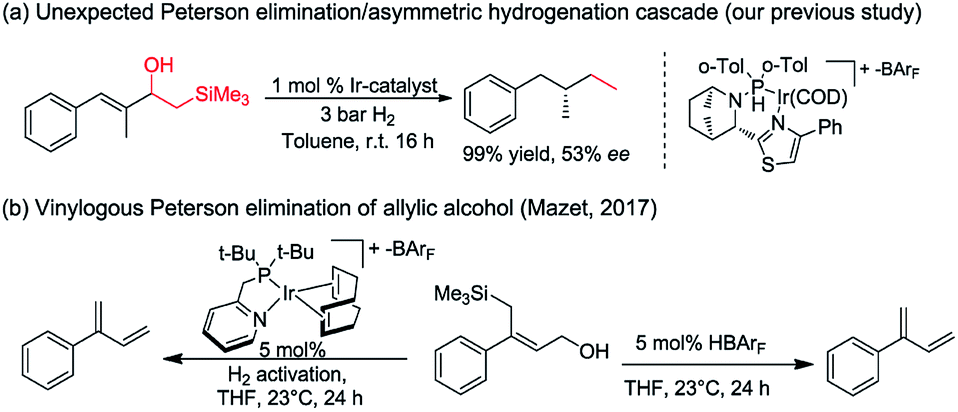 | ||
| Fig. 2 Peterson elimination and asymmetric hydrogenation. | ||
Results and discussion
Our initial investigation began with the cascade Peterson elimination and asymmetric hydrogenation of β-hydroxy silane 1 to optimize the reaction conditions (Table 1). It was found that CH2Cl2 was the best solvent for this reaction, giving high conversion within a short reaction time and excellent ee. Other solvents retarded the hydrogenation and resulted in an incomplete reaction that contained olefin 2a in the product mixture (entries 2–5). Regarding the effect of hydrogen pressure, it was found that increased H2 pressure lowered the enantioselectivity (entries 6–8). Hence, it was established that the ideal conditions for this reaction was 1 bar hydrogen with CH2Cl2 as the solvent.|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Pressure (bar) | 2a (yieldb/%) | 3a (yieldb/%) | 3a (eec/%) |
| a Reaction conditions: 0.05 mmol substrate, 0.5 mol% catalyst, 0.5 mL CH2Cl2. b Determined by 1H NMR spectroscopy of the crude product. c Determined by chiral GC analyses. | |||||
| 1 | CH2Cl2 | 1 | — | >99 | 95 |
| 2 | Benzene | 1 | 24 | 76 | 91 |
| 3 | PhCF3 | 1 | 25 | 75 | 93 |
| 4 | Toluene | 1 | 62 | 38 | 94 |
| 5 | THF | 1 | >99 | — | — |
| 6 | CH2Cl2 | 10 | — | >99 | 90 |
| 7 | CH2Cl2 | 20 | — | >99 | 83 |
| 8 | CH2Cl2 | 50 | — | >99 | 73 |
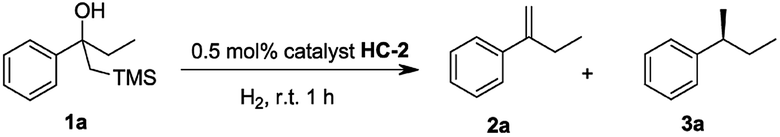
Initially, the new iridium phosphite–oxazoline complex HC-2 was found to be an efficient catalyst for the model reaction. To further evaluate the potential for steric and electronic tuning, the substituent on the oxazoline ring was changed from furan (HC-1,2,3,4,5) to thiophen (HC-6,7,8) and the heterocycle was attached to the ligand at different positions and substituents were also varied (Table 2). Variation of the heteroaromatic ring did not significantly affect the reactivity of β-hydroxy silane substrate, with all catalysts giving full conversion to the hydrogenated product in excellent yield. Formation of the isomerized, trisubstituted olefin was not observed in any of the reactions. An improvement in enantioselectivity was observed for catalyst HC-7 (97% ee) and catalyst HC-3 (98% ee).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Ir Cat. | Conv.b (%) | eec (%) | Entry | Ir Cat. | Conv.b (%) | eec (%) |
| a Reaction conditions: 0.05 mmol substrate, 0.5 mol% catalyst, 0.5 mL CH2Cl2. b Determined by 1H NMR spectroscopy of the crude product. c Determined by chiral GC analyses. | |||||||
| 1 | HC-1 | >99 | 95 | 5 | HC-5 | >99 | 96 |
| 2 | HC-2 | >99 | 97 | 6 | HC-6 | >99 | 96 |
| 3 | HC-3 | >99 | 98 | 7 | HC-7 | >99 | 97 |
| 4 | HC-4 | >99 | 95 | 8 | HC-8 | >99 | 96 |
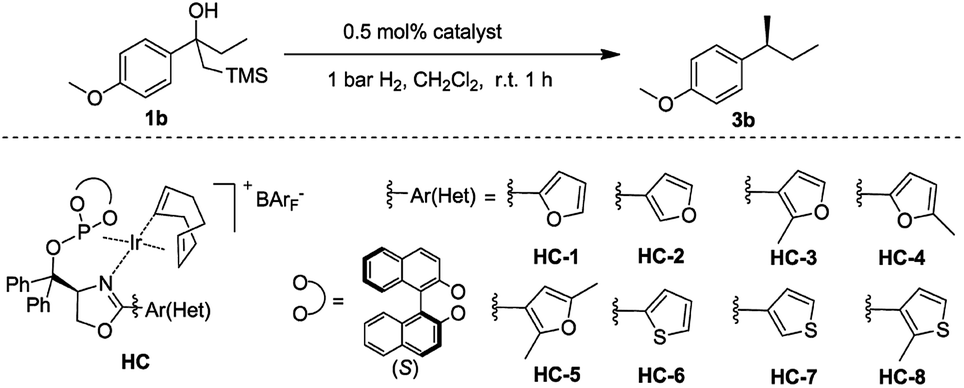
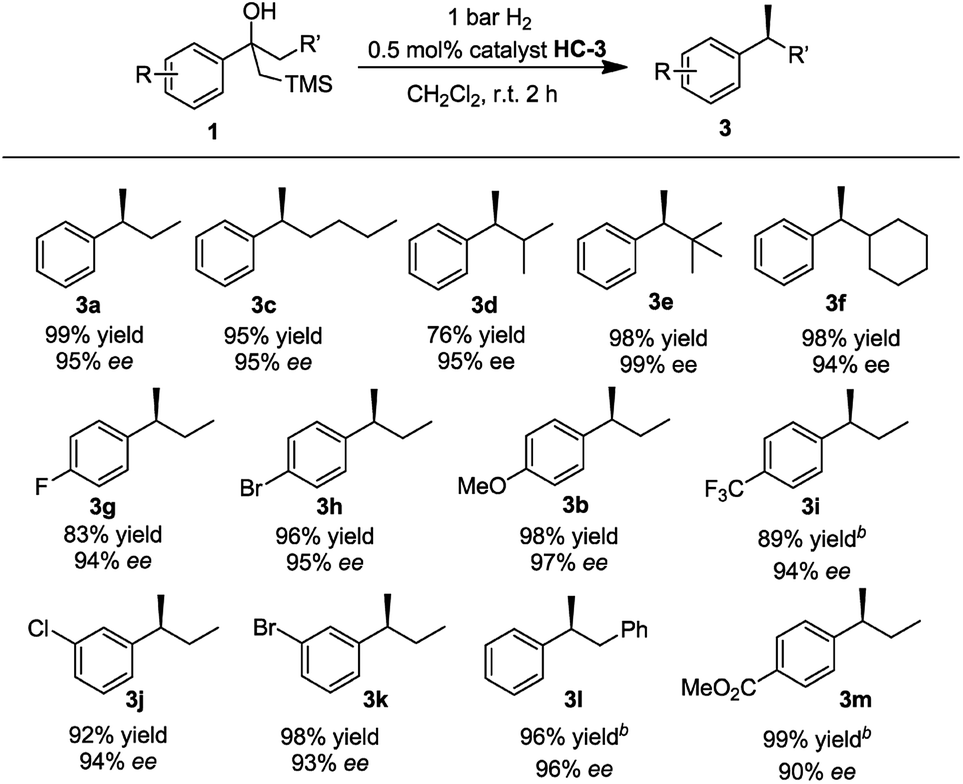
With the optimized reaction conditions established, we prepared and tested several different substrates to evaluate the scope of the reaction (Table 3). Substrates having aliphatic substituents such as ethyl, n-butyl, i-propyl, cyclohexyl, and t-butyl (3a–3f) provided the desired product in excellent yield and high enantioselectivity of up to 99% ee. The effect of varying the substituents on the phenyl ring was also investigated. Electron-withdrawing groups as well as electron-donating groups at the para-position (3g–3i) did not affect the efficiency of the reaction. Excellent isolated yields of up to 99% and ees up to 95% were achieved. Electron-withdrawing groups at the meta-position (3j, 3k) also gave very good results. Substrate 1l, containing the benzyl group, gave the desired product in excellent yield of 96% and high ee of 97%. No trace of the isomerized trisubstituted olefin was observed in the reaction. Substrate 1m, bearing the ester functional group was converted to the desired product in excellent yield of 99% with good ee of 90%.
Given the very mild reaction conditions and the high reaction rates for the Peterson elimination–hydrogenation, we were compelled to investigate the degree of chemoselectivity in the asymmetric hydrogenation of β-hydroxy silanes compared to some common olefins (Scheme 1). In this study, β-hydroxy silanes and different olefins were mixed and subjected to the standard reaction conditions. We observed that 1a yielded the completely hydrogenated product in a short reaction time with excellent ee while the olefins remained unaffected. The trans disubstituted olefins 4 and 5 were not hydrogenated whereas 1a was fully converted to the desired product (Scheme 1a). Trisubstituted olefins 6 and 7 were also tested in the hydrogenation, no hydrogenation of the olefins was detected, while full conversion of β-hydroxy silane 1e was observed (Scheme 1b).
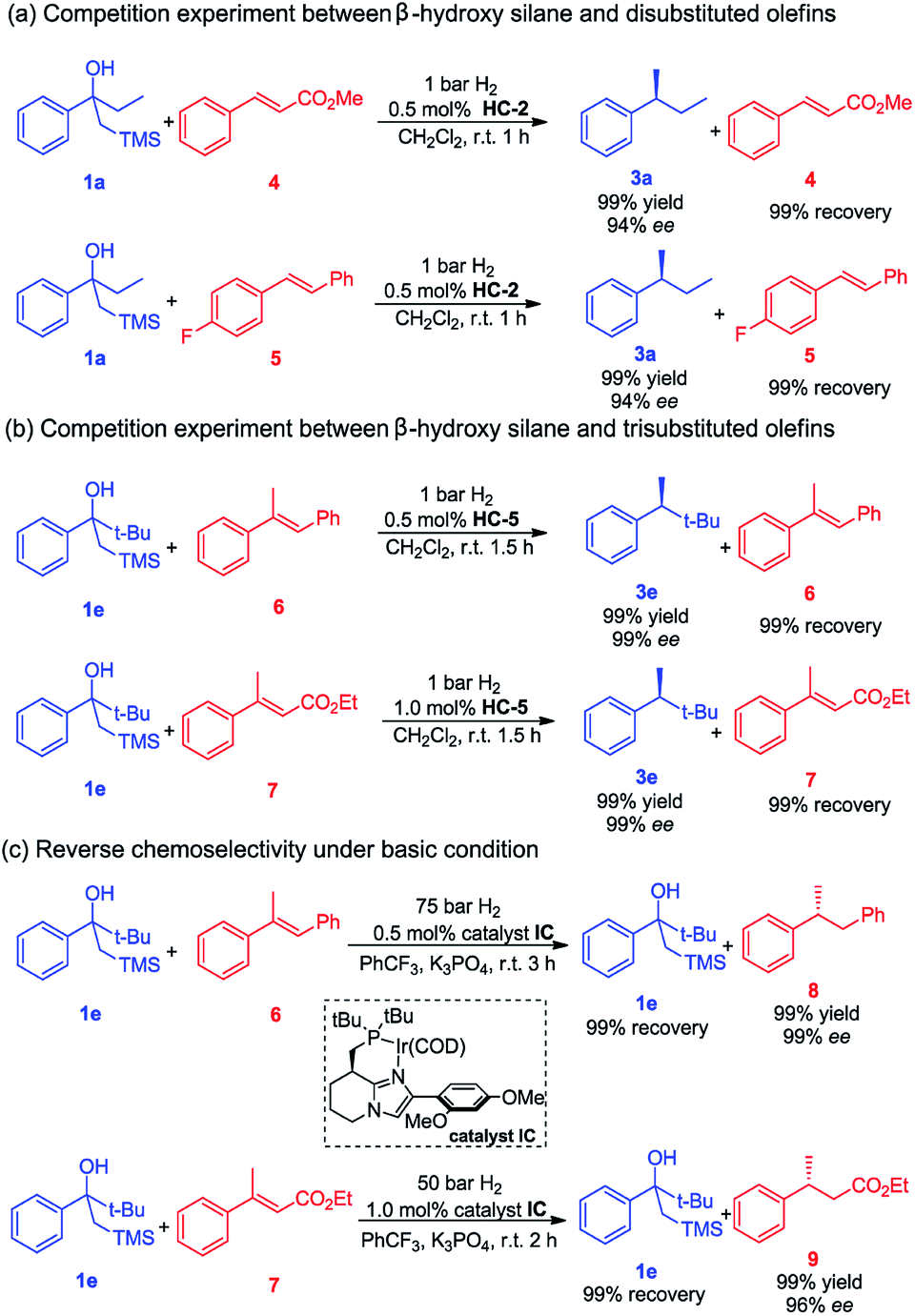 | ||
| Scheme 1 Chemoselective asymmetric hydrogenation. | ||
We also investigated the possibility of selective hydrogenation of an olefin in the presence of a β-hydroxy silane. Our rational was that, since the Peterson olefination requires acidic condition, and then a less acidic iridium catalyst would hydrogenate the olefin without catalyzing the Peterson reaction. When a mixture of trans-α-methylstilbene 6 or trans-β-methylcinnamate 7 and β-hydroxy silane 1e was subjected to a higher hydrogen pressure using catalyst IC in the presence of K3PO4, as expected, these reactions gave a complete change in chemoselectivity and resulted in a selective hydrogenation of the olefin, leaving the β-hydroxy silane intact (Scheme 1c). The basic additive (K3PO4) has proved to be efficient in preventing the decomposition of the acid-sensitive starting material or hydrogenated product under the acidic environment, which was generated in the iridium catalyzed asymmetric hydrogenation. These results demonstrate that a highly chemoselective transformation of either an olefin or a β-hydroxy silane is possible by selecting the appropriate reaction conditions.
Based on the completely opposite chemoselectivity that was obtained upon addition of base, we assumed that the in situ generated iridium dihydride catalyst acted as a Brønsted acid in the elimination step.15d Alternatively, the iridium dihydride intermediate might be directly involved in promoting the Peterson elimination.16 However, since Peterson elimination of β-hydroxy silane 1e did not occur during the hydrogenation of olefins 6 and 7 under basic conditions (Scheme 1c), it is suggested that the iridium dihydride is not directly responsible for the elimination. In a control experiment it was found that the addition of a bulky, non-coordinating base such as DTBMP completely inhibited the elimination (see the ESI† for details).
The potential of the reaction was investigated by testing a substrate that contained both an olefin as well as an ester group (Scheme 2). The unsaturated keto ester 10 reacted selectively with TMSCH2MgCl to give 11 in 77% isolated yield. Using catalyst HC-3, under standard reaction conditions, substrate 21 underwent Peterson olefination/hydrogenation to give the desired product in a highly chemoselective manner with good ee (95%).
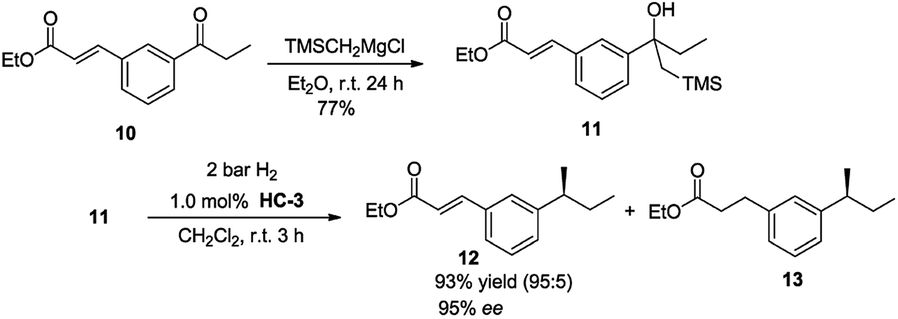 | ||
| Scheme 2 Intramolecular chemoselective functionalization. | ||
To further demonstrate the practical application of this methodology, a gram scale reaction was carried out. The reaction was conducted in CH2Cl2 with 1 bar H2 at room temperature in the presence of 1.0 mol% catalyst HC-3, affording the desired product 3f in high yield and excellent ee (Scheme 3a). When compared to the small scale reaction, it was noted that the yield was not affected and the enantioselectivity slightly increased.
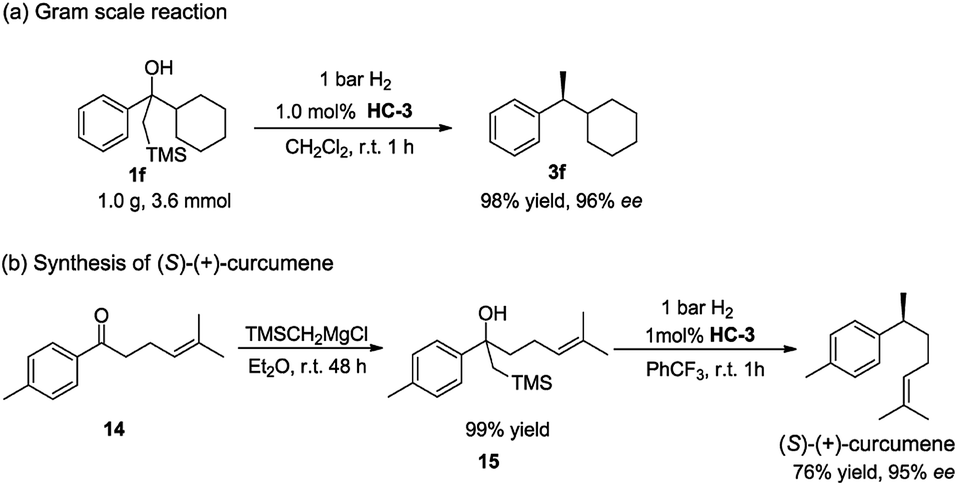 | ||
| Scheme 3 Scale-up preparation and synthesis application. | ||
The synthetic utility of this method was highlighted by the facile synthesis of bisabolane sesquiterpene (S)-(+)-curcumene (Scheme 3b). Despite its simple structure, the benzylmethyl chiral center poses a significant challenge in the asymmetric synthesis, and it has evolved as a popular test target for demonstrating the proficiency of new asymmetric processes.18 The synthesis began with treatment of simple ketone 14 with Peterson reagent smoothly affording the β-hydroxy silane compound 15 in quantitative yield. Subsequent asymmetric hydrogenation of 15 directly furnished the target natural product (S)-(+)-curcumene with 76% yield and 95% ee. This two-step, highly enantioselective synthesis represents one of the most efficient ways to access (S)-(+)-curcumene.
Conclusions
In summary, we have demonstrated an iridium catalyzed tandem Peterson olefination and asymmetric hydrogenation of β-hydroxy silanes under particularly mild conditions and low catalyst loading using novel Ir–N,P catalysts. The method we have developed provided efficient access to the chiral benzylic hydrocarbon motif. It was found that the reaction is highly chemoselective and that it could be tuned to selectively transform either an olefin or a β-hydroxy silane. By use of this methodology, we achieved the asymmetric syntheses of natural product (S)-(+)-curcumene in two steps with high yield. Further application of this strategy for the synthesis of more structurally related natural products is ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Swedish Research Council (VR) and Stiftelsen Olle Engkvist Byggmästare supported this work. J. L. thanks the Guangzhou Elite Scholarship Council for the PhD fellowship.Notes and references
- For selected examples, see: (a) A. E. Wright, S. A. Pomponi, O. J. McConnell, S. Kohmoto and P. J. McCarthy, J. Nat. Prod., 1987, 50, 976 CrossRef CAS; (b) H. Matsuda, T. Morikawa, K. Ninomiya and M. Yoshikawa, Bioorg. Med. Chem., 2001, 9, 909 CrossRef CAS PubMed; (c) A. D. Rodríguez and C. Ramírez, J. Nat. Prod., 2001, 64, 100 CrossRef; (d) F. A. Macías, R. M. Varela, A. Torres, J. M. G. Molinillo and F. R. Fronczek, Tetrahedron Lett., 1993, 34, 1999 CrossRef; (e) V. Roussis, Z. Wu, W. Fenical, S. A. Strobel, G. D. Van Duyne and J. Clardy, J. Org. Chem., 1990, 55, 4916 CrossRef CAS; (f) C. A. Harvis, M. T. Burch and W. Fenical, Tetrahedron Lett., 1988, 29, 4361 CrossRef CAS; (g) S. A. Look, W. Fenical, R. S. Jacobs and J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 6238 CrossRef CAS PubMed; (h) S.-e. Muto, M. Bando and K. Mori, Eur. J. Org. Chem., 2004, 2004, 1946 CrossRef; (i) S. P. Chavan and H. S. Khatod, Tetrahedron: Asymmetry, 2012, 23, 1410 CrossRef CAS.
- (a) L. Qin, L. Ren, S. Wan, G. Liu, X. Luo, Z. Liu, F. Li, Y. Yu, J. Liu and Y. Wei, J. Med. Chem., 2017, 60, 3606 CrossRef CAS PubMed; (b) Y. Wei, G. Qiu, B. Lei, L. Qin, H. Chu, Y. Lu, G. Zhu, Q. Gao, Q. Huang, G. Qian, P. Liao, X. Luo, X. Zhang, C. Zhang, Y. Li, S. Zheng, Y. Yu, P. Tang, J. Ni, P. Yan, Y. Zhou, P. Li, X. Huang, A. Gong and J. Liu, J. Med. Chem., 2017, 60, 8580 CrossRef CAS PubMed.
- (a) V. K. Honwad and A. S. Rao, Tetrahedron, 1964, 20, 2921 CrossRef CAS; (b) V. K. Honwad and A. S. Rao, Tetrahedron, 1965, 21, 2593 CrossRef CAS; (c) F. J. McEnroe and W. Fenical, Tetrahedron, 1978, 34, 1661 CrossRef CAS; (d) D. Del Prete, E. Millán, F. Pollastro, G. Chianese, P. Luciano, J. A. Collado, E. Munoz, G. Appendino and O. Taglialatela-Scafati, J. Nat. Prod., 2016, 79, 267 CrossRef CAS PubMed.
- (a) B. Harrison and P. Crews, J. Org. Chem., 1997, 62, 2646 CrossRef CAS PubMed; (b) M. J. Martín, F. Berrué, P. Amade, R. Fernández, A. Francesch, F. Reyes and C. Cuevas, J. Nat. Prod., 2005, 68, 1554 CrossRef PubMed; (c) J. C. Green, S. Jiménez-Alonso, E. R. Brown and T. R. R. Pettus, Org. Lett., 2011, 13, 5500 CrossRef CAS PubMed.
- (a) I. I. Rodríguez, A. D. Rodríguez, Y. Wang and S. G. Franzblau, Tetrahedron Lett., 2006, 47, 3229 CrossRef; (b) T. J. Heckrodt and J. Mulzer, Top. Curr. Chem., 2005, 244, 1 CrossRef CAS; (c) T. W. Johnson and E. J. Corey, J. Am. Chem. Soc., 2001, 123, 4475 CrossRef CAS PubMed.
- For selected examples, see: (a) R. R. Cesati III, J. de Armas and A. H. Hoveyda, J. Am. Chem. Soc., 2004, 126, 96 CrossRef PubMed; (b) A. Zhang and T. V. RajanBabu, Org. Lett., 2004, 6, 3159 CrossRef CAS PubMed; (c) S. Nave, R. P. Sonawane, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 17096 CrossRef CAS PubMed; (d) B. L. H. Taylor, E. C. Swift, J. D. Waetzig and E. R. Jarvo, J. Am. Chem. Soc., 2011, 133, 389 CrossRef CAS PubMed; (e) A. Wilsily, Y. Nguyen and E. Fillion, J. Am. Chem. Soc., 2009, 131, 15606 CrossRef CAS PubMed; (f) A. López-Pérez, J. Adrio and J. C. Carretero, Org. Lett., 2009, 11, 5514 CrossRef PubMed; (g) S. Drissi-Amraoui, M. S. T. Morin, C. Crévisy, O. Baslé, R. Marcia de Figueiredo, M. Mauduit and J.-M. Campagne, Angew. Chem., 2015, 127, 11996 CrossRef.
- (a) W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998 CrossRef CAS; (b) R. Noyori, Adv. Synth. Catal., 2003, 345, 15 CrossRef CAS; (c) A. Pfaltz and W. J. Drury, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5723 CrossRef CAS PubMed; (d) T. L. Church and P. G. Andersson, Coord. Chem. Rev., 2008, 252, 513 CrossRef CAS.
- For some recent reviews, see: (a) X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272 CrossRef CAS PubMed; (b) S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402 CrossRef CAS PubMed; (c) J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130 CrossRef CAS PubMed; (d) C. Margarita and P. G. Andersson, J. Am. Chem. Soc., 2017, 139, 1346 CrossRef CAS PubMed.
- (a) M. R. Friedfeld, M. Shevlin, J. M. Hoyt, S. W. Krska, M. T. Tudge and P. J. Chirik, Science, 2013, 342, 1076 CrossRef CAS PubMed; (b) S. Monfette, Z. R. Turner, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2012, 134, 4561 CrossRef CAS PubMed; (c) M. R. Friedfeld, M. Shevlin, G. W. Margulieux, L.-C. Campeau and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 3314 CrossRef CAS PubMed; (d) J. Chen, C. Chen, C. Ji and Z. Lu, Org. Lett., 2016, 18, 1594 CrossRef CAS PubMed; (e) M. Shevlin, M. R. Friedfeld, H. Sheng, N. A. Pierson, J. M. Hoyt, L.-C. Campeau and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 3562 CrossRef CAS PubMed; (f) T. Hibino, K. Makino, T. Sugiyama and Y. Hamada, ChemCatChem, 2009, 1, 237 CrossRef CAS.
- (a) D. Valentine, K. K. Johnson, W. Priester, R. C. Sun, K. Toth and G. Saucy, J. Org. Chem., 1980, 45, 3698 CrossRef CAS; (b) H. Takaya, T. Ohta, N. Sayo, H. Kumobayashi, S. Akutagawa, S. Inoue, I. Kasahara and R. Noyori, J. Am. Chem. Soc., 1987, 109, 15967 CrossRef; (c) L. Panella, B. L. Feringa, J. G. de Vries and A. J. Minnaard, Org. Lett., 2005, 7, 4177 CrossRef CAS PubMed; (d) B. K. Peters, J. Liu, C. Margarita, W. Rabten, S. Kerdphon, A. Orebom, T. Morsch and P. G. Andersson, J. Am. Chem. Soc., 2016, 138, 11930 CrossRef CAS PubMed; (e) J. Liu, S. Krajangsri, T. Singh, G. De Seriis, N. Chumnanvej, H. Wu and P. G. Andersson, J. Am. Chem. Soc., 2017, 139, 14470 CrossRef CAS PubMed.
- (a) O. Pàmies, P. G. Andersson and M. Diéguez, Chem.–Eur. J., 2010, 16, 14232 CrossRef PubMed; (b) S. McIntyre, E. Hörmann, F. Menges, S. P. Smidt and A. Pfaltz, Adv. Synth. Catal., 2005, 347, 282 CrossRef CAS; (c) D. H. Woodmansee and A. Pfaltz, Chem. Commun., 2011, 47, 7912 RSC; (d) J. Mazuela, P.-O. Norrby, P. G. Andersson, O. Pàmies and M. Diéguez, J. Am. Chem. Soc., 2011, 133, 13634 CrossRef CAS PubMed; (e) J. Mazuela, J. J. Verendel, M. Coll, B. Schäffner, A. Börner, P. G. Andersson, O. Pàmies and M. Diéguez, J. Am. Chem. Soc., 2009, 131, 12344 CrossRef CAS PubMed; (f) M. C. Perry, X. Cui, M. T. Powell, D.-R. Hou, J. H. Reibenspies and K. Burgess, J. Am. Chem. Soc., 2003, 125, 113 CrossRef CAS PubMed.
- (a) S. Song, S.-F. Zhu, Y.-B. Yu and Q.-L. Zhou, Angew. Chem., Int. Ed., 2013, 52, 1556 CrossRef CAS PubMed; (b) S. Yang, S.-F. Zhu, N. Guo, S. Song and Q.-L. Zhou, Org. Biomol. Chem., 2014, 12, 2049 RSC; (c) S. Wen, C. Chen, S. Du, Z. Zhang, Y. Huang, Z. Han, X.-Q. Dong and X. Zhang, Org. Lett., 2017, 19, 6474 CrossRef CAS PubMed; (d) J. Mazuela, O. Pàmies and M. Diéguez, Adv. Synth. Catal., 2013, 355, 2569 CrossRef CAS.
- W. Rabten, C. Margarita, L. Eriksson and P. G. Andersson, Chem.–Eur. J., 2018, 24, 1681 CrossRef CAS PubMed.
- J. Liu, S. Krajangsri, J. Yang, J.-Q. Li and P. G. Andersson, Nat. Catal., 2018, 1, 438 CrossRef.
- (a) R. H. Crabtree, Science, 1998, 282, 2000 CrossRef CAS; (b) P. Brandt, C. Hedberg and P. G. Andersson, Chem.–Eur. J., 2003, 9, 339 CrossRef CAS PubMed; (c) G. S. McGrady and G. Guilera, Chem. Soc. Rev., 2003, 32, 383 RSC; (d) Y. Zhu, Y. Fan and K. Burgess, J. Am. Chem. Soc., 2010, 132, 6249 CrossRef CAS PubMed.
- H. Li, D. Fiorito and C. Mazet, ACS Catal., 2017, 7, 1554 CrossRef CAS.
- (a) D. J. Peterson, J. Org. Chem., 1968, 33, 780 CrossRef CAS; (b) L. F. v. Staden, D. Gravestock and D. J. Ager, Chem. Soc. Rev., 2002, 31, 195 RSC; (c) T. A. Hamlin, C. B. Kelly, R. M. Cywar and N. E. Leadbeater, J. Org. Chem., 2014, 79, 1145 CrossRef CAS PubMed.
- For selected recent examples, see: (a) T. Nishimura, Y. Yasuhara, T. Sawano and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 7872 CrossRef CAS PubMed; (b) V. K. Aggarwal, L. T. Ball, S. Carobene, R. L. Connelly, M. J. Hesse, B. M. Partridge, P. Roth, S. P. Thomas and M. P. Webster, Chem. Commun., 2012, 48, 9230 RSC; (c) I. Ibrahem, G. Ma, S. Afewerki and A. Córdova, Angew. Chem., Int. Ed., 2013, 52, 878 CrossRef CAS PubMed; (d) K. Spielmann, R. M. de Figueiredo and J.-M. Campagne, J. Org. Chem., 2017, 82, 4737 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05261a |
| ‡ Suppachai Krajangsri, Haibo Wu and Jianguo Liu contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |