
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReal-time mechanistic study of carbon nanotube anion functionalisation through open circuit voltammetry†
Adam J.
Clancy
 *ab,
Pichamon
Sirisinudomkit
bc,
David B.
Anthony
b,
Aaron Z.
Thong
c,
Jake L.
Greenfield
bd,
Maniesha K.
Salaken Singh
b and
Milo S. P.
Shaffer
*bc
*ab,
Pichamon
Sirisinudomkit
bc,
David B.
Anthony
b,
Aaron Z.
Thong
c,
Jake L.
Greenfield
bd,
Maniesha K.
Salaken Singh
b and
Milo S. P.
Shaffer
*bc
aDepartment of Chemistry, University College London, WC1E 7JE, UK. E-mail: a.clancy@ucl.ac.uk
bDepartment of Chemistry, Imperial College London, SW7 2AZ, UK. E-mail: m.shaffer@imperial.ac.uk
cDepartment of Materials, Imperial College London, SW7 2AZ, UK
dDepartment of Chemistry, University of Cambridge, CB2 1EW, UK
First published on 7th February 2019
Abstract
The mechanism of the functionalisation of reduced single walled carbon nanotubes with organobromides was monitored by open circuit voltammetry throughout the reaction and further elucidated through a series of comparative reactions. The degree of functionalisation was mapped against the reagent reduction potential, degree of electron donation of substituents (Hammett parameter), and energies calculated, ab initio, for dissociation and heterolytic cleavage of the C–Br bond. In contrast to the previously assumed reduction/homolytic cleavage mechanism, the reaction was shown to consist of a rapid association of carbon–halide bond to the reduced nanotube as a complex, displacing surface-condensed countercations, leading to an initial increase in the net nanotube surface negative charge. The complex subsequently slowly degrades through charge transfer from the reduced single-walled carbon nanotube to the organobromide, utilizing charge, and the carbon–halide bond breaks heterolytically. Electron density on the C–Br bond in the initial reagent is the best predictor for degree of functionalisation, with more electron donating substituents increasing the degree of functionalisation. Both the mechanism and the new application of OCV to study such reactions are potentially relevant to a wide range of related systems.
Introduction
Single-walled carbon nanotubes (SWCNTs) have superlative mechanical,1 conductive, and (opto)electronic2 properties, making them promising candidates for a host of applications including multi-functional composites, thin conductive films,3 transistors, gas sensors,4 and drug delivery vectors.5 As-synthesized SWCNTs are usually agglomerated into mechanically and electronically inferior bundles, and typically contain both carbonaceous and catalytic impurities. To maximize performance, they require individualisation and purification before application. Covalent functionalisation of SWCNTs can tune a wide variety of properties, including solubility,6 composite interfaces,1 biocompatibility,7 and protein nucleation.8 Functionalisation may be performed via condensation chemistry with carboxylic acids formed from oxidation of the SWCNT carbon framework, however, the necessary oxidation leads to framework vacancies which severely degrade intrinsic properties. Alternatively, high energy species (most commonly carboradicals, nitrenes, and azomethine ylides) may react directly with the SWCNT framework. Understanding the mechanism of such reactions is crucial to developing reagents, maximizing efficiency, and minimizing side products. Substantial progress has been made in understanding the mechanism of neutral SWCNT functionalisation, particularly with carbon-centric radicals (predominantly from diazonium salts,9–11 but also xanthates,12 and peroxides12), and the gas phase reactions with oxygen13 and fluorine.14 To perform solution phase reactions, the SWCNT must first be dispersed, through routes frequently involving harsh oxidation or sonication, damaging the intrinsic SWCNT properties. In addition, only a small fraction of SWCNTs are individualized, with the majority remaining as smaller bundles; separation of the individualized fraction involves the use of ultracentrifugation which is generally a small-scale process with low yield. Alternatively, SWCNTs can be processed through reduction to nanotubide anions,15 which can dissolve spontaneously16,17 in a variety of aprotic, polar solvents to give truly individualized SWCNTs in high yield. The use of sodium naphthalide (NaNp) in N,N-dimethylacetamide (DMAc) facilitates a single-step reduction and dissolution of SWCNTs which simplifies the creation of these nanotubide solutions.18Additionally, the heightened reactivity of nanotubide opens a new family of functionalisations, using several reactive moieties, most commonly organohalides, but also disulfides,19 epoxides,20 and molecular bromine21 amongst others. However, the mechanism of reductive SWCNT functionalisation remains relatively unstudied. Catalyst impurities are known to impede the reaction,22 whilst steric occlusion at the nanotube surface is known to limit the extent of functionalisation, particularly for reactions with polymers.23 Functionalisation is thought to be electronically preferable alpha to previous defects,24 although larger species are unlikely to be capable of functionalizing adjacent carbons. The only proposed mechanism (originally proposed by Chattopadhyay et al.19 and expanded upon by Voiry et al.25) is the single electron transfer (SET) reduction of the alkyl halide to an anionic radical which cleaves to form a halide anion and an alkyl radical which spontaneously reacts with the SWCNT sidewall (Fig. 1). The SET mechanism is based on that established for the reaction of reduced polyaromatics with alkyl halides.26 Since bis-termination of the proposed radical intermediate is expected to yield an organic dimer byproduct, the observation of trace biphenyl,25 4,4′-di-t-butylbiphenyl,27 and dicumene19 from nanotubide reactions with iodobenzene, 4-t-butyliodobenzene, and cumyl phenyl sulfide, respectively, is consistent with the SET mechanism. However, light-induced homolytic cleavage or decomposition of alkyl halide with excess reductant28 provide alternative explanations for the traces of these dimers. In addition, reductively-functionalized graphenide (negatively-charged graphene sheets) has been proposed cleave homolytically at low temperatures, leading to dimer formation post-functionalisation.29
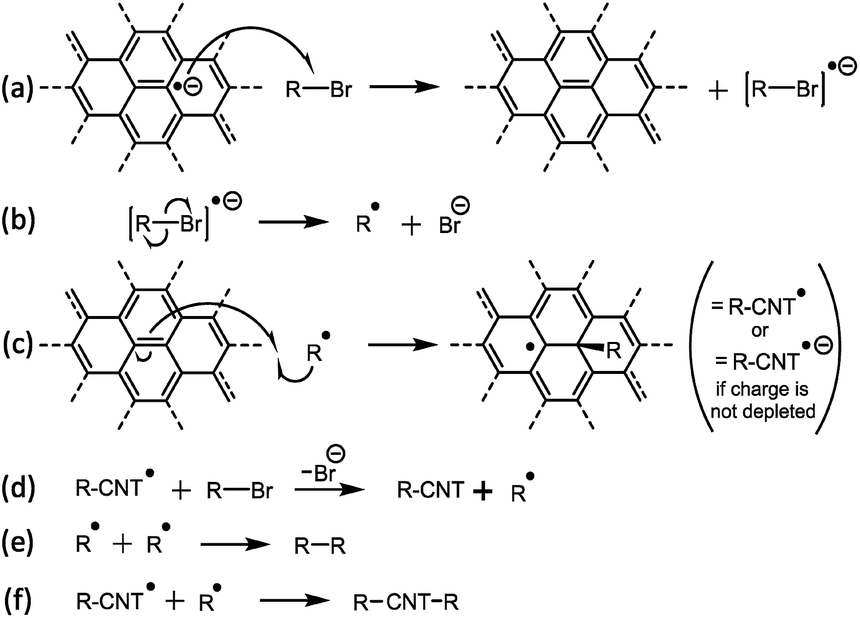 | ||
| Fig. 1 SET mechanism for reductive functionalization of SWCNTs, as proposed by Voiry et al.25 | ||
The SET mechanism implies that the extent of reaction will depend on the redox potential of the electrons at the Fermi level of the (reduced) SWCNTs, relative to the redox potential of the reagent (for example, the alkyl halide). During the reaction, the most reducing electrons on the nanotubide will be consumed, until the Fermi level balances the reduction potential of the alkyl halide; at this point, the SWCNT will still be charged relative to the neutral SWCNT species. Qualitatively, the degree of functionalisation has indeed been linked to the charge stoichiometry,30 SWCNT band structure,31 and reactivity of both organic18 and inorganic32 reagents. Furthermore, the unutilized residual charge is known to initiate further adventitious reactions30 if not removed through careful discharging,33,34 or can be used deliberately to initiate a second functionalisation through later addition of a more easily reduced reagent.30
However, there are some difficulties with the SET mechanism for nanotubide reactions. Most obviously the nanotubide Fermi level, is not thought sufficient to reduce many of the organohalides applied. Mildly charged SWCNTs are known to react with an array of alkyl halides18 despite modest reduction potentials e.g. E°[C20Na] ≈ −0.45 V (all values vs. SHE) as calculated from integrated density of states (more complete description and comparison to literature potentials provided in the ESI†). Even highly charged nanotubide (C5Na ≈ −1.43 V) have reduction potentials lower than some known reagents, notably organochlorides18,35,36 (ca. −2.8 to −3.1 V).37 Whilst absolute Fermi levels for specific (reduced) SWCNTs remain uncertain, even at the absolute limit, the maximum nanotubide Fermi level cannot exceed the reduction potential of the reductant (−2.71 V for sodium) which is still lower than the reactive alkyl chlorides. An additional difficulty is that the degree of functionalisation never exceeds the charging stoichiometry, as might be expected if the mechanism involves radical propagation steps (Fig. 1c and d); further, the proposed CNT-bound radical (Fig. 1c) does not lead to nanotube crosslinking termination, as is seen in other systems involving CNT bound radicals.38 For these reasons, we have studied the mechanism of reductive functionalisation in more detail to provide greater insight into these versatile reactions.
Results and discussion
Open circuit voltammetry (OCV) measures the potential of a working electrode versus a reference in the absence of a flowing current, providing a direct measurement of the potential on surface. OVC is used as a characterisation tool for redox rates, commonly for characterising the performance of energy storage devices39 and quantifying corrosion resistance,40 in addition to being a common measurement parameter in chemical sensors.41 Here, OCV is instead applied to monitoring the SWCNT organo-functionalisation in real time. The measured OCV potential must relate to the degree of charging, which shifts the Fermi level of the SWCNTs, modulated by the counterions condensed in the Stern layer (as modelled by Manning-Oosawa theory,42 and applied to SWCNTs15,17,43). By using an assembly of SWCNTs as the working electrode, the effective net charge on the nanotube surface may be monitored, during both the reduction step (addition of sodium naphthalide) and the subsequent functionalisation (addition of alkyl halide).A preweighed ‘buckypaper’ (a dense film of entangled SWCNTs) of reductively purified44 SWCNTs was submerged in 0.1 mM NaClO4/DMAc and used as the working electrode in a 3 electrode setup, alongside a Pt counter electrode and Ag/AgNO3 reference electrode (full details in the ESI†). Buckypapers were soaked in electrolyte for ≥24 h prior to use to minimize temperature and ion gradients between electrode interior and bulk electrolyte. Nevertheless the absolute initial OCVs of SWCNTs varied and showed some drift (ESI, Fig. S3†). Once the OCV was consistent and stable (typically 15 min), NaNp/DMAc solution (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 C/Na) was added to the cell (Fig. 2). The potential dropped rapidly, indicating nanotubide formation from naphthalide charge transfer. The majority of the OCV decrease was completed within 1 h (ESI Fig. S3†), slower than analogous systems using SWCNT powders,22 a timescale most likely dominated by ion diffusion into the compact assembly of nanotubes. After charging with NaNp, the OCV was allowed to stabilize before adding alkyl halide (1-bromobutane, 3 eq. vs. NaNp) to the cell (Fig. 2). Although the reaction with alkyl bromides is expected to remove charge from the SWCNTs, surprisingly, the OCV became rapidly more negative (peak dV/dt = −91 mV h−1). The rate of voltage change then inverts to positive values indicating consumption of nanotubide charge, before levelling off as the reaction ceases. These data show that the nanotubide electrode becomes more negative upon alkylbromide addition. Electron transfer from (neutral) alkyl halide to (anionic) nanotubide can be reasonably excluded. Instead, the change in potential may be attributed to the displacement of counter cations from the Stern layer, by adsorption of the reagent. Two controls were tested: (i) a non-brominated alkane analogue was added to the charged SWCNT system (ESI Fig. S5†), and (ii) an alkyl halide was added to a neutral buckpaper (ESI Fig. S6†). Neither control showed the characteristic long-lived drop in reduction potential, thus, it can be seen that the polar C–Br bond is necessary to displace cations from the surface of the charged SWCNTs, consistent with the initial formation of a C–Br complex at the nanotubide surface. After this rapid complex formation, the positive OCV rate (and eventual increase in absolute OCV), on the hour timescale, implies a charge transfer away from the buckypaper, presumably in the form of the halide anions generated by the full grafting reaction. Interestingly, an analogous two-step mechanism involving fast reversible absorption followed by slow covalent reaction has been previously proposed for functionalisation of neutral SWCNTs with diazonium salts.9 After the reaction completed and the OCV restabilized, the SWCNTs remained more charged than the initial neutral electrode, consistent with the previously observed post-functionalisation residual charge,30 supporting a SWCNT charge threshold for functionalisation. The use of 1-bromohexane as the functionalizing moiety (ESI, Fig. S7†) demonstrated the same OCV and derivative trends as 1-bromohexane, but with lesser utilisation of charge, presumably due to a steric contribution.18
:1 C/Na) was added to the cell (Fig. 2). The potential dropped rapidly, indicating nanotubide formation from naphthalide charge transfer. The majority of the OCV decrease was completed within 1 h (ESI Fig. S3†), slower than analogous systems using SWCNT powders,22 a timescale most likely dominated by ion diffusion into the compact assembly of nanotubes. After charging with NaNp, the OCV was allowed to stabilize before adding alkyl halide (1-bromobutane, 3 eq. vs. NaNp) to the cell (Fig. 2). Although the reaction with alkyl bromides is expected to remove charge from the SWCNTs, surprisingly, the OCV became rapidly more negative (peak dV/dt = −91 mV h−1). The rate of voltage change then inverts to positive values indicating consumption of nanotubide charge, before levelling off as the reaction ceases. These data show that the nanotubide electrode becomes more negative upon alkylbromide addition. Electron transfer from (neutral) alkyl halide to (anionic) nanotubide can be reasonably excluded. Instead, the change in potential may be attributed to the displacement of counter cations from the Stern layer, by adsorption of the reagent. Two controls were tested: (i) a non-brominated alkane analogue was added to the charged SWCNT system (ESI Fig. S5†), and (ii) an alkyl halide was added to a neutral buckpaper (ESI Fig. S6†). Neither control showed the characteristic long-lived drop in reduction potential, thus, it can be seen that the polar C–Br bond is necessary to displace cations from the surface of the charged SWCNTs, consistent with the initial formation of a C–Br complex at the nanotubide surface. After this rapid complex formation, the positive OCV rate (and eventual increase in absolute OCV), on the hour timescale, implies a charge transfer away from the buckypaper, presumably in the form of the halide anions generated by the full grafting reaction. Interestingly, an analogous two-step mechanism involving fast reversible absorption followed by slow covalent reaction has been previously proposed for functionalisation of neutral SWCNTs with diazonium salts.9 After the reaction completed and the OCV restabilized, the SWCNTs remained more charged than the initial neutral electrode, consistent with the previously observed post-functionalisation residual charge,30 supporting a SWCNT charge threshold for functionalisation. The use of 1-bromohexane as the functionalizing moiety (ESI, Fig. S7†) demonstrated the same OCV and derivative trends as 1-bromohexane, but with lesser utilisation of charge, presumably due to a steric contribution.18
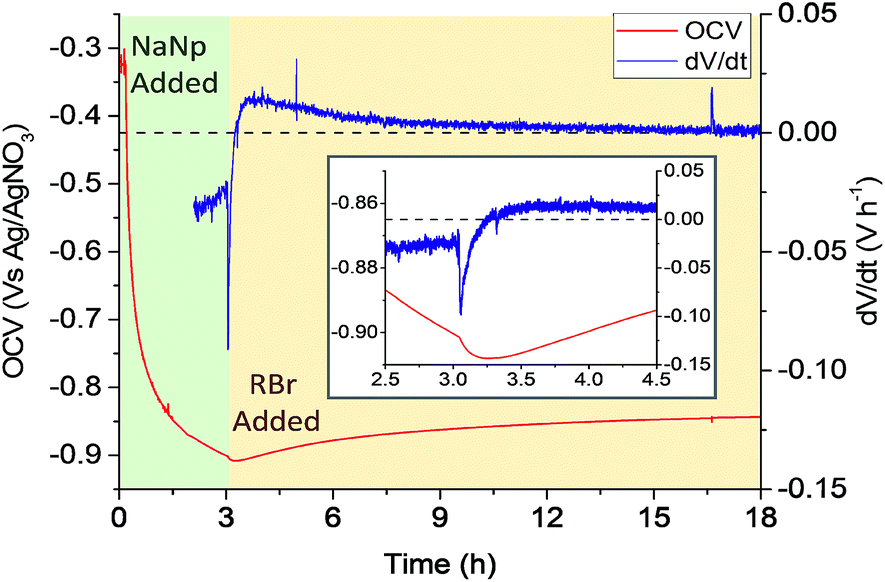 | ||
| Fig. 2 OCV (and derivative) versus time of a buckypaper working electrode during addition of sodium naphthalide solution (green), and addition of 1-bromobutane (3 h after NaNp addition, yellow). Dashed line represents dV/dt = 0. Inset shows zoomed region over 2 h around point of organic addition. Derivative before 2 h omitted for clarity with full range supplied in the ESI (Fig. S4†). | ||
To probe the nature of the proposed nanotubide/organohalide complex and the reaction mechanism, several stabilizing factors were considered. Firstly, the mechanism may proceed via either a concerted or stepwise mechanism. A concerted (SN2-like) mechanism would necessitate a carbon-centric trigonal bipyramidal transition state and would be dominated by steric effects due to the large size of the nucleophile (SWCNTn−). Conversely, a stepwise mechanism would involve an electron-poor intermediate, regardless of whether cleavage of the carbon–halogen was a heterolytic SN1-like step, or a homolytic radical step (as is proposed in the SET mechanism, Fig. 1b). The electron-poor species could be stabilized with hyperconjugation from bonds beta to the halide to stabilize the planar intermediate. To distinguish between concerted and stepwise mechanisms of the SWCNT-organohalide complex, different organohalides were used to probe the possible stabilizing factors. The primary, secondary, and tertiary isomers of bromopentane provide a convenient set (Fig. 3): 1-bromopentane (1), 2-bromopentane (2), and 2-bromo-2-methylbutane (3). While alkyl iodides lead to higher degrees of grafting in SWCNT reductive functionalisation than bromides,18 they introduce potentially complicating UV sensitivity.
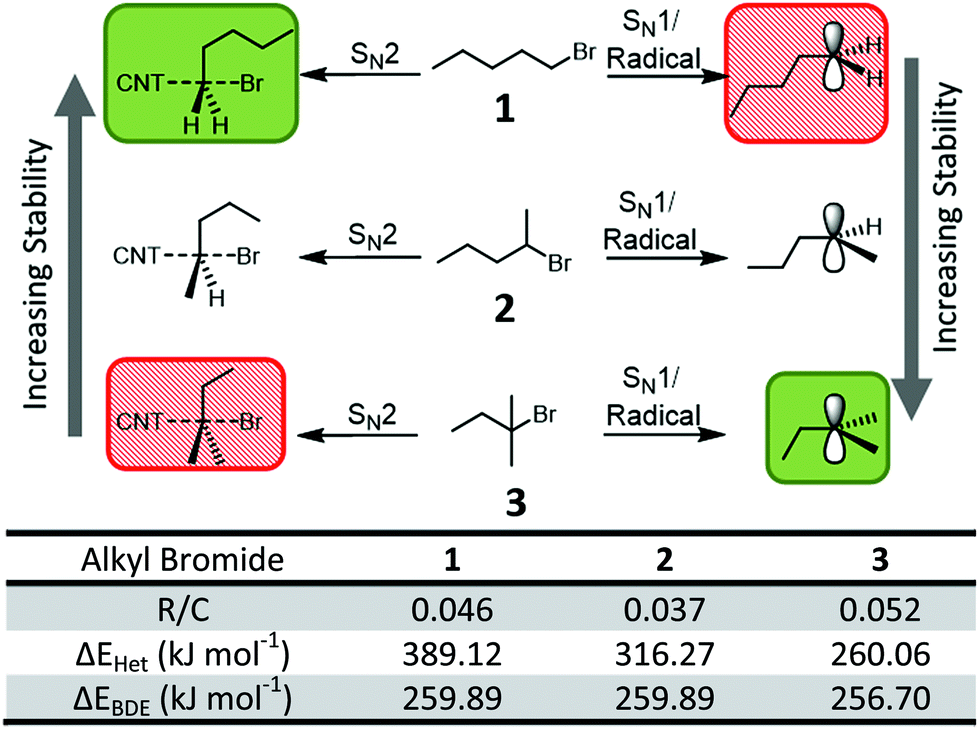 | ||
| Fig. 3 (Top) Schematic possible 5-valent and planar intermediates of reductive functionalization of BrC5H11 isomers (1–3) with nanotubide, illustrating stability trends. Most stable state shown with green background, least stable shown with hatched red background. (Bottom) Table of properties of SWCNTs functionalized with various alkyl bromides. | ||
The extent of steric interference is highly dependent on the substituents on the electrophilic carbon (here the brominated carbon), with occlusion small for primary (1, with two small hydrogens and one alkyl substituent) and maximized for tertiary (3, with three alkyls) reagents; thus the grafting trend 1 > 2 > 3 would be expected for a concerted (SN2-like) mechanism. Conversely, the opposite grafting trend 3 > 2 > 1 would be expected for a stepwise, purely electronically-driven mechanism, due to stabilisation from hyperconjugation of both radical and cationic intermediates, as confirmed by ab initio calculations45 (see ESI† for details) of the energies for heterolytic cleavage (ΔEHet) and bond dissociation (ΔEBDE).
The extent of functionalisation was quantified using thermogravimetric analysis (TGA, ESI Fig. S9†) with the grafting density (R/C) calculated as the number of grafted chains per SWCNT carbon atoms. The TGA data show the trend 3 (R/C 0.052) > 1 (0.046) > 2 (0.037). As the sterically hindered (but electronically stable) 3 leads to the highest grafting density, it can be assumed that electronic effects have a significant contribution, and importantly, that the mechanism involves multiple steps and is not concerted. The deviation of 1/2 from the predicted, purely electronic trend implies that steric interactions do play a role in at least one step (in support of previous work18,24) and dominates over the weak electronic contribution of a single methyl group.
The nature of the electronic component of the reaction's rate determining step was investigated in more depth, by comparing the degree of functionalisation with a set of model compounds to four factors: reduction potential (E°), ΔEBDE, ΔEHet, and electron density at the carbon–halide bond. A series of monosubstituted benzyl bromides were selected to facilitate the use of Hammett parameters (vide infra). para and meta substituted methyl, nitro, and trifluoromethyl benzyl bromides were reacted, alongside m-methoxy substituted, p-methylthio substituted, and unsubstituted benzyl bromide (N.B. p-methoxybenzyl bromide was too unstable for use, and m-methylthiobenzyl bromide was not commercially available). These reagents were selected to provide a broad range of electron-withdrawing/donating strengths and stabilities of resonance states of intermediates, with minimal variation in steric bulk of the functionalizing species. ortho-Functionalised benzyl bromides were not tested, as the varying steric influence of the substituents adjacent to the reactive C–Br bond would confound attempts to isolate the electronic effects. In fact, due to this very issue, Hammett parameters are not available for relevant ortho-functionalised species,46 adding a further hurdle to their use in this study.
E° values were measured through cyclic voltammetry; due to the organobromide's reduction peak overlapping with electrolyte reductive breakdown in some cases (masking the peak anodic current), the potential at the maximum of the dI/dV derivative was used to provide a trend. The first reduction peak is used in all cases, and all values are given versus the Ag/AgNO3(DMF) reference electrode (further information and voltammograms are provided in the ESI†). Homolytic and heterolytic bond cleavage energies were determined computationally through ab initio calculations as discussed earlier. Here, ΔEBDE most usefully provides an indirect measurement for the enthalpy change in forming R˙/Br− from the SET mechanism (Fig. 1b), offset by the electron affinity of atomic bromine, since homolytic cleavage is not anticipated (ESI, Fig. S8†). While the electron density and dipole of the pre-cleaved C–Br bond can be calculated, the use of benzyl bromides provides a simpler, experimentally verified route through comparison of Hammett parameters (σ). These values are a tool of organic chemistry that quantifies the electron withdrawing/donating nature of meta/para aryl substituents on benzyl species where more positive values imply more electron withdrawing species, and have recently been used in studying the reaction of aryl diazonium salts with molybdenum disulfide nanosheets.47 Hammett parameters used here are taken from the comprehensive library assembled by Hansch.48
All benzyl bromides successfully functionalized the SWCNTs, as monitored by TGA (ESI Fig. S10†), and the degree of functionalisation (R/C) was quantified. The R/C was plotted against each of the electronic factors (σ, E°, ΔEBDE, ΔEHet), to allow trends to be elucidated. A linear fit was applied and the coefficient of determination (R2) was used as a crude measure of correlation (Fig. 4). The strongest trends are seen for the Hammett parameter (R2 = 0.871) with more electron rich C–Br bonds being more reactive, and reduction potential (R2 = 0.812) with more easily reduced species functionalizing to a greater extent. These trends imply that the electron density at the C–Br bond is critical, likely in complex formation as seen in the OCV measurements. Further, the organobromide is directly reduced by the nanotubide, akin to the SET mechanism, explaining the residual change posited previously30 and confirmed by OCV in this work. The discrepancy between reduction potential of the alkyl halide and SWCNT Fermi level can be explained by the presence of the intermediate SWCNT–C–Br complex which may lower the energy barrier to reduction, and the more reducing potential of the SWCNT once the counter ions are displaced. However, while heterolytic bond cleavage energies showed a notable correlation (R2 = 0.765, with more easily cleaved C–Br showing greater reactivity), importantly, the homolytic cleavage showed virtually no correlation (R2 = 0.168) with the degree of functionalisation. Even the (weak) trend is in the ‘wrong’ direction with high ΔEBDE (a high enthalpy cost in forming the carboradical) linked to higher functionalisation. At its most simple, the trend implies there is no homolytic cleavage of C–Br, but as ΔEBDE is an indirect probe for the presence of a free R˙ intermediate (ESI Fig. S8†), it implies no such species is present, in contrast to the SET mechanism (Fig. 1b). Given the previous observation of bis-dimers in some reactions, it is possible that a homolytic C–Br cleavage is present as a non-rate-determining step, however, as alternative reactions may explain their presence, and ΔEHet shows a good correlation to functionalisation, the C–Br cleavage during reductive functionalisation is tentatively assigned as heterolytic decomposition of the complexed RBr on the SWCNT surface.
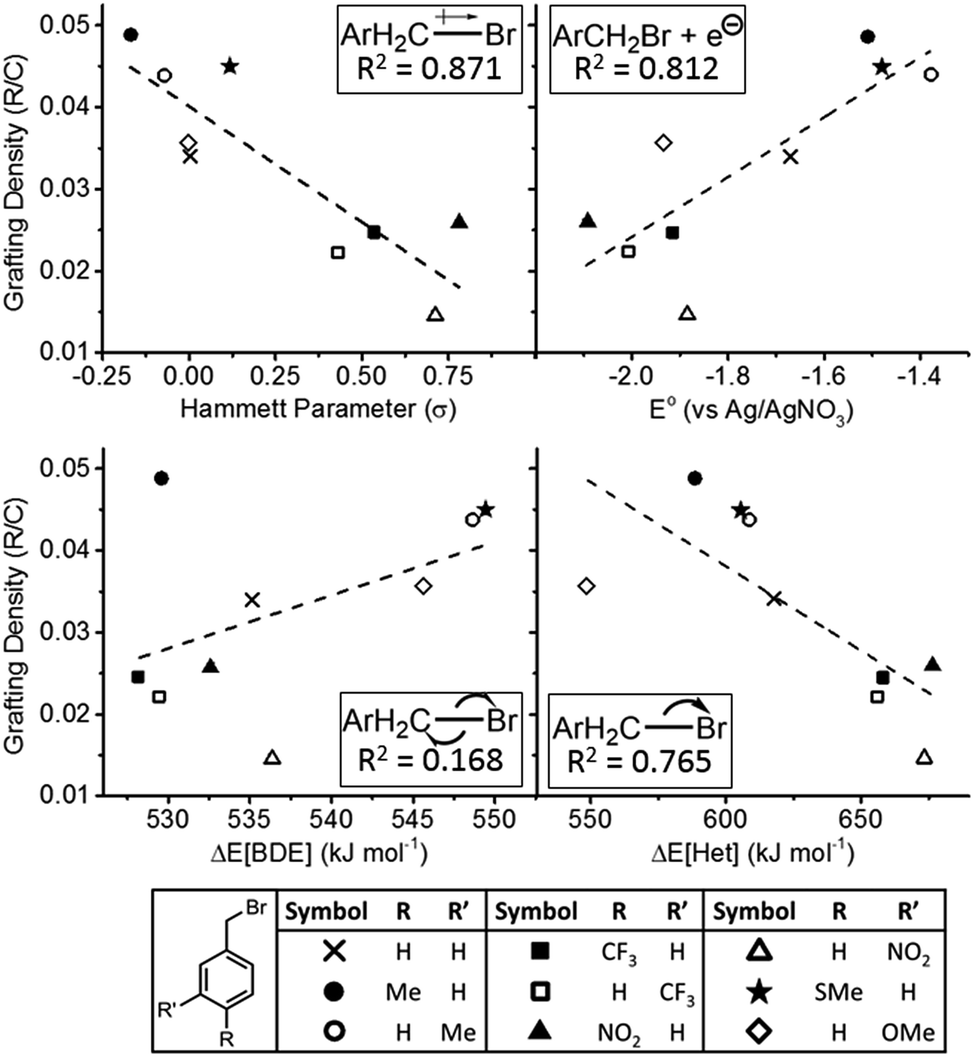 | ||
| Fig. 4 Grafting density (R/C) of monosubstituted benzyl bromides versus (clockwise from top left) Hammett parameter, reduction potential, heterolytic bond dissociation energy, and homolytic bond cleavage energy. Dashed line is a linear fit. Schematic of modelled parameter and coefficient of determination magnitude given inset in each graph. Full tabulated values are provided in the ESI.† | ||
Conclusions
In conclusion, the experiments indicate a development of the previously accepted mechanism reaction between nanotubide and organohalides. The reaction is initiated by rapid complexation of the organohalide to the nanotubide, displacing condensed counterions, before a second slower step involving degradation through halide reduction and cleavage of the carbon–halide bond to liberate the halide anion (Fig. 5). The absence of any nanotube or small molecule radical intermediates in the proposed mechanism is consistent with the lack of observation of either nanotube crosslinking or greater than stoichiometric grafting ratios as would be expected from mechanisms involving radical propagation steps (Fig. 1c and d). In agreement with previous work, steric occlusion of the organohalide is shown to play a significant role in determining the degree of functionalisation, assigned here to the packing of complexes on the SWCNT surface; however, for small molecules at least, steric effects are of lesser importance than electronic stabilisation of the intermediate. Furthermore, the degree of electron-donation from groups adjacent to the carbon–halide bond (i.e. electron density on the C–Br bond) is the strongest predictor for increasing the degree of functionalisation, with more electron donating substituents increasing the degree of functionalisation. Further investigation is required to elucidate a more detailed picture of the functionalisation mechanism, particularly the electronic stability of organohalide/nanotubide complex, the full reaction pathway, and detailed reaction kinetics. The concept of a surface adsorption activated grafting reaction is relevant more generally to other systems that span the molecular-continuum divide, including analogous reactions on graphenides and related materials. The development of OCV, as a method to study the organo-functionalisation, provides a new, simple, and readily accessible tool to investigate a wide range of such systems in real time. To use OCV to monitor surface chemical reactions, a material must be electronically conducting and physically stable in the relevant potential range, either as a cohesive electrode as seen here, or coated on an inert electrode surface. In principle, any reactions involving charge transfer between solvated species and the electrode may be monitored, although high specific surface areas may be needed to amplify the signal; with further refinement, more quantitative interpretation of the charge transfer and surface potentials may be possible. OCV may be useful not only for studying reactions on nanocarbons, but wide range of other systems, including functionalisation of conducting nanomaterials (e.g. transition metal dichalcogonides, nanodots, metallic nanoparticles, etc.), charge transfer to surface-bound ligands, as well as real-time monitoring pathways of heterogeneous catalytic systems.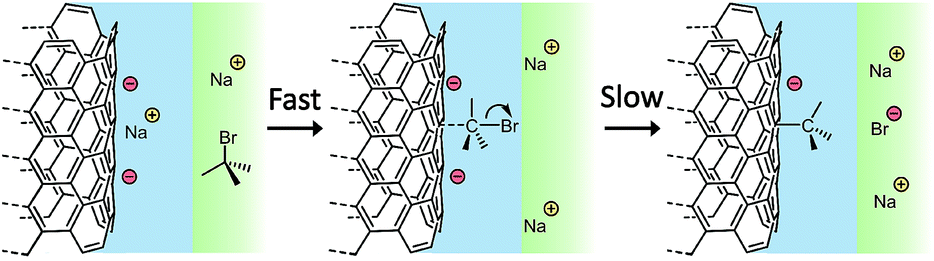 | ||
| Fig. 5 Proposed schematic of nanotubide reductive functionalization mechanism. Stern layer (blue), bulk solvent (green), positive charges (yellow), negative charges (red). Alkyl halide rapidly adsorbs onto charged nanotubide surface displacing cations condensed in the Stern layer and forming a more readily reduced complex between SWCNT, and C–Br bond. The complex slowly degrades, using nanotubide charge to break the C–Br bond. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the EPSRC (Doctoral Prize Fellowship, EP/M507878/1). Supporting data can be requested from the corresponding author, but may be subject to confidentiality obligations. P. S. appreciates the financial support from the Ministry of Science and Technology, Royal Thai Government.References
- J. N. Coleman, U. Khan, W. J. Blau and Y. K. Gun'ko, Small but Strong: A Review of the Mechanical Properties of Carbon Nanotube–Polymer Composites, Carbon, 2006, 44(9), 1624 CrossRef CAS.
- M. V. Kharlamova, Advances in Tailoring the Electronic Properties of Single-Walled Carbon Nanotubes, Prog. Mater. Sci., 2016, 77, 125 CrossRef CAS.
- D. D. Tune, A. J. Blanch, C. J. Shearer, K. E. Moore, M. Pfohl, J. G. Shapter and B. S. Flavel, Aligned Carbon Nanotube Thin Films from Liquid Crystal Polyelectrolyte Inks, ACS Appl. Mater. Interfaces, 2015, 7(46), 25857 CrossRef CAS PubMed.
- D. R. Kauffman and A. Star, Carbon Nanotube Gas and Vapor Sensors, Angew. Chem., Int. Ed., 2008, 47(35), 6550 CrossRef CAS PubMed.
- W. Zhang, Z. Zhang and Y. Zhang, The Application of Carbon Nanotubes in Target Drug Delivery Systems for Cancer Therapies, Nanoscale Res. Lett., 2011, 6(1), 1 Search PubMed.
- K. Shiral Fernando, Y. Lin and Y.-P. Sun, High Aqueous Solubility of Functionalized Single-Walled Carbon Nanotubes, Langmuir, 2004, 20(11), 4777 CrossRef.
- Z. Liu, C. Davis, W. Cai, L. He, X. Chen and H. Dai, Circulation and Long-Term Fate of Functionalized, Biocompatible Single-Walled Carbon Nanotubes in Mice Probed by Raman Spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(5), 1410 CrossRef CAS PubMed.
- H. S. Leese, L. Govada, E. Saridakis, S. Khurshid, R. Menzel, T. Morishita, A. J. Clancy, E. R. White, N. E. Chayen and M. S. Shaffer, Reductively PEGylated Carbon Nanomaterials and Their Use to Nucleate 3D Protein Crystals: A Comparison of Dimensionality, Chem. Sci., 2016, 7(4), 2916 RSC.
- M. L. Usrey, E. S. Lippmann and M. S. Strano, Evidence for a Two-Step Mechanism in Electronically Selective Single-Walled Carbon Nanotube Reactions, J. Am. Chem. Soc., 2005, 127(46), 16129 CrossRef CAS PubMed.
- T. Menanteau, E. Levillain and T. Breton, Spontaneous Grafting of Nitrophenyl Groups on Carbon: Effect of Radical Scavenger on Organic Layer Formation, Langmuir, 2014, 30(26), 7913 CrossRef CAS PubMed.
- A. J. Blanch, C. E. Lenehan and J. S. Quinton, Dispersant Effects in the Selective Reaction of Aryl Diazonium Salts with Single-Walled Carbon Nanotubes in Aqueous Solution, J. Phys. Chem. C, 2012, 116(2), 1709 CrossRef CAS.
- F. Pennetreau, C. Vriamont, B. Vanhorenbeke, O. Riant and S. Hermans, Covalent Functionalization of Carbon Nanotubes with Xanthates and Peroxides, Eur. J. Org. Chem., 2015,(8), 1804 CrossRef CAS.
- C.-Y. Moon, Y.-S. Kim, E.-C. Lee, Y.-G. Jin and K.-J. Chang, Mechanism for Oxidative Etching in Carbon Nanotubes, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65(15), 155401 CrossRef.
- S. l. Osuna, M. Torrent-Sucarrat, M. Sola, P. Geerlings, C. P. Ewels and G. V. Lier, Reaction Mechanisms for Graphene and Carbon Nanotube Fluorination, J. Phys. Chem. C, 2010, 114(8), 3340 CrossRef CAS.
- A. J. Clancy, M. K. Bayazit, S. A. Hodge, N. T. Skipper, C. A. Howard and M. S. P. Shaffer, Charged Carbon Nanomaterials: Redox Chemistries of Fullerenes, Carbon Nanotubes, and Graphenes, Chem. Rev., 2018, 118(16), 7363 CrossRef CAS PubMed.
- A. Pénicaud, P. Poulin, A. Derré, E. Anglaret and P. Petit, Spontaneous Dissolution of a Single-Wall Carbon Nanotube Salt, J. Am. Chem. Soc., 2005, 127, 8 CrossRef PubMed.
- D. Voiry, C. Drummond and A. Pénicaud, Portrait of Carbon Nanotube Salts As Soluble Polyelectrolytes, Soft Matter, 2011, 7(18), 7998 RSC.
- A. J. Clancy, J. Melbourne and M. S. P. Shaffer, A One-Step Route to Solubilised, Purified or Functionalised Single-Walled Carbon Nanotubes, J. Mater. Chem. A, 2015, 3(32), 16708 RSC.
- J. Chattopadhyay, S. Chakraborty, A. Mukherjee, R. Wang, P. S. Engel and W. E. Billups, SET Mechanism in the Functionalization of Single-Walled Carbon Nanotubes, J. Phys. Chem. C, 2007, 111(48), 17928 CrossRef CAS.
- Y. Martinez-Rubi, B. Ashrafi, J. Guan, C. Kingston, A. Johnston, B. Simard, V. Mirjalili, P. Hubert, L. Deng and R. Young, Toughening of Epoxy Matrices With Reduced Single-Walled Carbon Nanotubes, ACS Appl. Mater. Interfaces, 2011, 3(7), 2309 CrossRef CAS PubMed.
- F. Hof, F. Hauke and A. Hirsch, Brominated Single Walled Carbon Nanotubes As Versatile Precursors for Covalent Sidewall Functionalization, Chem. Commun., 2014, 50, 6582 RSC.
- K. S. Zhang, D. Pham, O. Lawal, S. Ghosh, V. S. Gangoli, P. Smalley, K. Kennedy, B. E. Brinson, W. E. Billups and R. H. Hauge, Overcoming Catalyst Residue Inhibition of the Functionalization of Single-Walled Carbon Nanotubes via the Billups–Birch Reduction, ACS Appl. Mater. Interfaces, 2017, 9(43), 37972 CrossRef CAS PubMed.
- A. J. Clancy, J. M. Serginson, J. L. Greenfield and M. S. Shaffer, Systematic Comparison of Single-Walled Carbon Nanotube/Poly(vinyl Acetate) Graft-To Reactions, Polymer, 2017, 133, 263 CrossRef CAS.
- S. Deng, Y. Zhang, A. Brozena, M. Mayes, P. Banerjee, W.-A. Chiou, G. Rubloff, G. Schatz and Y. Wang, Confined Propagation of Covalent Chemical Reactions on Single-Walled Carbon Nanotubes, Chem. Commun., 2011, 2, 382 Search PubMed.
- D. Voiry, O. Roubeau and A. Pénicaud, Stoichiometric Control of Single Walled Carbon Nanotubes Functionalization, J. Mater. Chem., 2010, 20, 4385 RSC.
- G. Sargent and G. Lux, Reactions of Aromatic Radical Anions. III. Evidence for an Alkyl Radical-Radical Anion Combination Mechanism for Alkylation of Sodium Naphthalenide With Alkyl Halides, J. Am. Chem. Soc., 1968, 90(25), 7160 CrossRef CAS.
- J. Chattopadhyay, A. K. Sadana, F. Liang, J. M. Beach, Y. Xiao, R. H. Hauge and W. E. Billups, Carbon Nanotube Salts. Arylation of Single-Wall Carbon Nanotubes, Org. Lett., 2005, 7(19), 4067 CrossRef CAS PubMed.
- V. Kulkarni and T. Cohen, Cumyl Phenyl Sulfide Forms Bicumyl Instead of Cumyllithium Upon Reductive Lithiation. Thiophenoxide as a Leaving Group in Nucleophilic Substitution by SET, Tetrahedron, 1997, 53(36), 12089 CrossRef CAS.
- G. Abellán, M. Schirowski, K. F. Edelthalhammer, M. Fickert, K. Werbach, H. Peterlik, F. Hauke and A. Hirsch, Unifying Principles of the Reductive Covalent Graphene Functionalization, J. Am. Chem. Soc., 2017, 139(14), 5175 CrossRef PubMed.
- F. Hof, S. Bosch, S. Eigler, F. Hauke and A. Hirsch, New Basic Insight Into Reductive Functionalization Sequences of Single Walled Carbon Nanotubes (SWCNTs), J. Am. Chem. Soc., 2013, 135(49), 18385 CrossRef CAS PubMed.
- D. Wunderlich, F. Hauke and A. Hirsch, Preferred Functionalization of Metallic and Small-Diameter Single-Walled Carbon Nanotubes by Nucleophilic Addition of Organolithium and-Magnesium Compounds Followed by Reoxidation, Chem.–Eur. J., 2008, 14(5), 1607 CrossRef CAS PubMed.
- S. A. Hodge, H. H. Tay, D. B. Anthony, R. Menzel, D. J. Buckley, P. L. Cullen, N. T. Skipper, C. A. Howard and M. S. Shaffer, Probing the Charging Mechanisms of Carbon Nanomaterial Polyelectrolytes, Farad. Discus., 2014, 172, 311 RSC.
- S. A. Hodge, D. J. Buckley, H. C. Yau, N. T. Skipper, C. A. Howard and M. S. Shaffer, Chemical Routes to Discharging Graphenides, Nanoscale, 2017, 9(9), 3150 RSC.
- P. Vecera, J. Holzwarth, K. F. Edelthalhammer, U. Mundloch, H. Peterlik, F. Hauke and A. Hirsch, Solvent-Driven Electron Trapping and Mass Transport in Reduced Graphites to Access Perfect Graphene, Nat. Commun., 2016, 7, 12411 CrossRef CAS PubMed.
- J. N. Coleman, M. Cadek, R. Blake, V. Nicolosi, K. P. Ryan, C. Belton, A. Fonseca, J. B. Nagy, Y. K. Gun'ko and W. J. Blau, High Performance Nanotube-Reinforced Plastics: Understanding the Mechanism of Strength Increase, Adv. Funct. Mater., 2004, 14(8), 791 CrossRef CAS.
- A. J. Clancy, D. B. Anthony, S. J. Fisher, H. Leese, C. S. Roberts and M. S. P. Shaffer, Reductive Dissolution of Supergrowth Carbon Nanotubes for Tougher Nanocomposites by Reactive Coagulation Spinning, Nanoscale, 2017, 9(25), 8764 RSC.
- F. L. Lambert and G. B. Ingall, Voltammetry of Organic Halogen Compounds. IV. The Reduction of Organic Chlorides at the Vitreous (Glassy) Carbon Electrode, Tetrahedron Lett., 1974, 15(36), 3231 CrossRef.
- Y. Gao, H. Chen, J. Ge, J. Zhao, Q. Li, J. Tang, Y. Cui and L. Chen, Direct Intertube Cross-Linking of Carbon Nanotubes at Room Temperature, Nano Lett., 2016, 16(10), 6541 CrossRef CAS PubMed.
- J.-P. Diard, B. Le Gorrec and C. Montella, EIS Study of Electrochemical Battery Discharge on Constant Load, J. Power Sources, 1998, 70(1), 78 CrossRef CAS.
- R. G. Kelly, J. R. Scully, D. Shoesmith and R. G. Buchheit, in Electrochemical Techniques in Corrosion Science and Engineering, CRC Press, 2002 Search PubMed.
- R. Sathiyamoorthi, R. Chandrasekaran, T. Mathanmohan, B. Muralidharan and T. Vasudevan, Study of Electrochemical Based Gas Sensors for Fluorine and Chlorine, Sens. Actuators, B, 2004, 99(2–3), 336 CrossRef CAS.
- B. O'Shaughnessy and Q. Yang, Manning-Oosawa Counterion Condensation, Phys. Rev. Lett., 2005, 94(4), 048302 CrossRef PubMed.
- A. Clancy, H. S. Leese, N. Rubio, D. J. Buckley, J. L. Greenfield and M. S. P. Shaffer, Depleting Depletion: Maintaining Single-Walled Carbon Nanotube Dispersions after Graft-to Polymer Functionalization, Langmuir, 2018, 34(50), 15396 CrossRef CAS PubMed.
- A. J. Clancy, E. R. White, H. Tay, H. Yau and M. Shaffer, Systematic Comparison of Conventional and Reductive Single-Walled Carbon Nanotube Purifications, Carbon, 2016, 108, 423 CrossRef CAS.
- E. I. Izgorodina, D. R. Brittain, J. L. Hodgson, E. H. Krenske, C. Y. Lin, M. Namazian and M. L. Coote, Should Contemporary Density Functional Theory Methods Be Used to Study the Thermodynamics of Radical Reactions?, J. Phys. Chem. A, 2007, 111(42), 10754 CrossRef CAS PubMed.
- M. Charton, The Application of the Hammett Equation to Ortho-Substituted Benzene Reaction Series, Can. J. Chem., 1960, 38(12), 2493 CrossRef CAS.
- E. E. Benson, H. Zhang, S. A. Schuman, S. U. Nanayakkara, N. D. Bronstein, S. Ferrere, J. L. Blackburn and E. M. Miller, Balancing the Hydrogen Evolution Reaction, Surface Energetics, and Stability of Metallic MoS2 Nanosheets via Covalent Functionalization, J. Am. Chem. Soc., 2017, 140(1), 441 CrossRef PubMed.
- C. Hansch, A. Leo and R. Taft, A Survey of Hammett Substituent Constants and Resonance and Field Parameters, Chem. Rev., 1991, 91(2), 165 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, a fuller modelling of nanotubide reduction potentials with comparison to literature values, raw thermograms, raw cyclic voltammograms, ab initio details, and additional OCV data are provided in the supplementary information. See DOI: 10.1039/c8sc04970j |
| This journal is © The Royal Society of Chemistry 2019 |