
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled living anionic polymerization of cyanoacrylates by frustrated Lewis pair based initiators†
Rubén
Sáez
a,
Ciaran
McArdle‡
b,
Fouad
Salhi
c,
Jordi
Marquet
 *a and
Rosa M.
Sebastián§
*a
*a and
Rosa M.
Sebastián§
*a
aChemistry Department, Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Barcelona, Spain. E-mail: rosamaria.sebastian@uab.es
bHenkel Ireland Operations and Research Ltd., Tallaght Business Park, Whitestown Road, Tallaght, Dublin 24, Ireland
cHenkel Corporation, Edifici Eureka, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Barcelona, Spain
First published on 1st February 2019
Abstract
A hydrogenated frustrated Lewis pair ([TMPH+][HB(C6F5)3−]) promotes controlled living ionic polymerization of cyanoacrylates. Controlled growth of various homopolymeric CAs through sequential monomer addition has been achieved, in addition to CA block copolymers with controlled block sequences for the first time in the long history of these materials.
The emergence of synthetic polymeric materials probably constitutes one of the major breakthroughs in materials science in the 20th century. Any new technology, ranging from composites used in supersonic aircraft and high-speed railway to components for surgical implants, requires the development of diverse materials with very specific properties, and synthetic polymers play a critical role.1 The specific structural and molecular parameters (molecular weight and molecular weight distribution), the presence of functional groups and their spatial location, or the presence of unreacted monomers, etc. define the suitability of a polymer for a particular application.2 So-called ‘living’ polymerization techniques have allowed the realization of well-defined structures such as block, star, macrocyclic, and telechelic copolymers, and represent some of the most important examples of molecular design and control in polymer science.3–5 The initiation step in an ideal living anionic polymerization is fast as compared with the propagation reaction, and no transfer or termination reactions occur. Active chain ends remain “living” and are only quenched upon addition of a terminating agent. The molecular weight of the resulting polymer is given by the monomer/initiator ratio, and narrow molecular weight distributions are obtained (Mw/Mn ≤ 1.1).4 Different mechanistic types of living polymerizations have been described since Szwarc reported the anionic polymerization of styrene, but nowadays living polymerization has also been achieved with cationic, radical, metal catalyzed, and group-transfer initiators.6
Cyanoacrylates, produced in high volume for adhesive applications, are highly reactive monomers that mainly polymerize by anionic initiation through a conjugate addition of anionic nucleophiles (hydroxyl or bromide groups for example) or bases such as amines7 and phosphines.8 In these last cases zwitterionic polymerization ensues with a carbanionic active propagating species.9 The use of tetrabutylammonium salts and fluorenyllithium as initiators has also been investigated. In some cases nearly ideal living polymerizations have been claimed; however, higher than expected molecular weight polymers (as calculated by the initial monomer/initiator ratio), together with broad molecular weight distributions are observed in most of the studies.10,11
The excellent adhesive properties of polycyanoacrylates (PCA) have enabled their widespread use for many household applications (Super Glue®), and, most recently for applications in medicine,12 surgery,13 ophthalmology,14 dentistry,15 tissue reconstruction,16 and for drug delivery,17 due to their biocompatibility and biodegradability. Various other applications for CAs include fingerprint detection for forensic applications, coating for lithographic printing plates, waveguides for optical sensors, photoresists and holographic recording media, which exploit their rapid polymerizability or the optical properties of their polymers.18 However their use as designer materials is scarce partly due to the difficulty of controlling their polymerization and copolymerization processes. Nevertheless some anionic copolymerizations of 2-cyanoacrylate monomers have been described,19–21 and a few of these, containing also poly(ethylene glycol) blocks, show certain control of the final polymer architecture achieved by a nearly living oxyanion-initiated polymerization (Mw/Mn > 1.2).22,23 Random cyanoacrylate copolymerization with vinyl monomers has been reported by radical,24,25 and metal catalysed initiators.26 Recent studies showed that the use of RAFT agents did not promote ideal living block polymerization of CA.27
To control the polymerization of CA a reduction of the exceptionally fast propagation rate is required. To the best of our knowledge, the concept of “group transfer” in anionic polymerization has never been applied before for CA. Frustrated Lewis pairs (FLPs), showing unusual reactivity,28 have been recently used as promoters for the polymerization of acrylic monomers.29 In these cases, sterically encumbered Lewis acid and Lewis base moieties work cooperatively to activate the monomer substrate and participate in chain initiation (1,4-conjugate additions are observed), as well as chain propagation and termination/transfer events. Hydrogenated FLPs obtained from hydrogen splitting reactions can reduce selectively the double bond in the α-position of a carbonyl group.30 With these interesting characteristics in mind we were curious to learn if FLPs or their hydrogenated derivatives could initiate, in a controlled manner, and participate somehow in the propagation steps of CA polymerization. In this work several Lewis pairs and FLPs have been studied and applied as promoters for the polymerization of various CA monomers. A hydrogenated derivative of a FLP has allowed us to achieve for the first time a controlled living ionic polymerization of CA that also enables the construction of block copolymers with designed structures.
To gain familiarity with CA chemistry, the polymerization of n-butyl cyanoacrylate (BCA) initiated by BuLi (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, BCA:BuLi) was firstly studied in THF at 25 °C to reproduce the experiments of Pepper.31 The choice of CA type and solvent is an important factor for subsequent unambiguous GPC characterization. A high molecular weight polymer was formed (105–106 Da) within very short reaction times but the appearance of daughter low polymers (10000–15000 Da) was observed through time as previously reported, due to degradative retro-Michael reactions (Table 1, entries 1 and 2).32–34 Other simple initiators such as sec-BuLi, Bu2CuLi and LiI were also tested in order to see if any effect on the rate of the initiation step or the limitation of daughter polymer formation could be observed, however similar results were obtained according to GPC analysis (Table 1, entries 3–5). The higher the amount of initiator used (80:10), the faster was the generation of the lower molecular weight daughter polymers. This rate was reduced using freshly distilled monomers. Only through the use of a non-ionic initiator, PPh3 (zwitterionic polymerization), the formation of daughter polymers was avoided, but without control of the process (Table 1, entry 6).
:1, BCA:BuLi) was firstly studied in THF at 25 °C to reproduce the experiments of Pepper.31 The choice of CA type and solvent is an important factor for subsequent unambiguous GPC characterization. A high molecular weight polymer was formed (105–106 Da) within very short reaction times but the appearance of daughter low polymers (10000–15000 Da) was observed through time as previously reported, due to degradative retro-Michael reactions (Table 1, entries 1 and 2).32–34 Other simple initiators such as sec-BuLi, Bu2CuLi and LiI were also tested in order to see if any effect on the rate of the initiation step or the limitation of daughter polymer formation could be observed, however similar results were obtained according to GPC analysis (Table 1, entries 3–5). The higher the amount of initiator used (80:10), the faster was the generation of the lower molecular weight daughter polymers. This rate was reduced using freshly distilled monomers. Only through the use of a non-ionic initiator, PPh3 (zwitterionic polymerization), the formation of daughter polymers was avoided, but without control of the process (Table 1, entry 6).
| Entry | Initiator | Time (min) | Calc. Mna | Exp. Mnb | Đ |
|---|---|---|---|---|---|
|
a Calculated through the experimental ratio [M]/[I] (80:1) assuming that every initiator molecule initiates one polymer chain. Expressed in g mol−1.
b Experimental data obtained by GPC, when more than one peak is observed, the data related to each polymeric species are included from low to high molecular weight. The same pattern is extended to the corresponding dispersities and the relative peak areas.
c Area of each peak: 53/47.
d Area of each peak: 89/11.
|
|||||
| 1 | BuLi | 20 | 12250 |
8990/3.2 × 105 | 1.5/1.4c |
| 2 | BuLi | 240 | 12250 |
15144/5.0 × 105 |
1.7/1.3d |
| 3 | sec-BuLi | 120 | 12250 |
18580/3.4 × 105 |
1.7/1.3 |
| 4 | Bu2CuLi | 120 | 12250 |
7699/3.1 × 105 | 1.7/1.4 |
| 5 | LiI | 120 | 12250 |
5050/1.4 × 105 | 1.2/1.4 |
| 6 | PPh3 | 120 | 12250 |
3.5 × 105 | 1.7 |
In order to obtain a controlled living polymerization of CA we sought an initiator with good nucleophilic properties (Lewis base) but, also with the ability to stabilize the propagating anion (Lewis acid) thereby reducing the propagation rate. The ideal initiator must further reduce the formation of daughter polymers, chain transfer, and termination steps while maintaining the living character of the polymeric chain end using a group transfer polymerization type approach. In this sense, we opted to study three species (NBu4Br, NBu4OH and NaSCN) that alone polymerize CA in an uncontrolled manner (Table 2, entries 1, 3 and 5), now in the presence of B(C6F5)3. However, these combinations formed classical Lewis pairs (CLPs) and the reaction did not proceed (Table 2, entries 2, 4 and 6). Clearly passivation of the nucleophile by the Lewis acid should be avoided for control polymerization to succeed. This led us to hypothesize that FLPs could be the ideal initiators for controlled CA polymerization. The combination of boranes or alanes with sterically hindered amines, phosphines or carbenes has been recently applied to the polymerization of regular acrylic monomers.29 We decided to use B(C6F5)3 (alane derivative is shock and air/moisture sensitive) in combination with PPh3, pyridine and 2,2,6,6-tetramethylpiperidine (TMP) as FLP initiators for the highly electron deficient cyanoacrylic monomers. In two experimental combinations with these initiators, polymerization occurs, but without control (Table 2, entries 7 and 8). Nevertheless, the mixture of B(C6F5)3/TMP, yielding polymers of 18900 Da (Đ = 1.6), that although of higher molecular weight than expected (12250 Da based on the ratio [monomer:initiator]), gave an indication that some control was taking place (Table 2, entry 10). The sterically hindered amine TMP, used alone, was able to initiate CA polymerization giving rise to high molecular weight polydisperse polymers after 15 min (54280 Da, Đ 2.2, Table 2, entry 9). In this case the formation of the degradation product daughter polymers was observed with time. However, when a stoichiometric amount of B(C6F5)3 was present in the mixture with TMP (Table 2, entry 10), lower molecular weight and stable polymers were obtained, showing an important effect of free borane in the process as a potential “group transfer agent”.
| Entrya | Initiator | Time (min) | Calc. Mna | Exp. Mnb | Đ |
|---|---|---|---|---|---|
|
a Calculated through the experimental ratio [M]/[I] (80:1) assuming that every initiator molecule initiates one polymer chain. Expressed in g mol−1.
b Experimental data obtained by GPC, when more than one peak is observed, the data related to each polymeric species are included from low to high molecular weight. The same pattern is extended to the corresponding dispersities and the relative peak areas.
|
|||||
| 1 | NBu4Br | 30 | 12250 |
2.4 × 105 | 1.6 |
| 2 | NBu4Br/B(C6F5)3 | 30 | 12250 |
271 | 1.01 |
| 3 | NBu4OH | 30 | 12250 |
4000/9.8 × 104 | 2.4/1.5 |
| 4 | NBu4OH/B(C6F5)3 | 30 | 12250 |
252 | 1.01 |
| 5 | NaSCN | 30 | 12250 |
3.3 × 105 | 1.6 |
| 6 | NaSCN/B(C6F5)3 | 30 | 12250 |
252 | 1.01 |
| 7 | PPh3/B(C6F5)3 | 30 | 12250 |
2.6 × 105 | 3.5 |
| 8 | Pyr/B(C6F5)3 | 30 | 12250 |
91150 |
3.5 |
| 9 | TMP | 15 | 12250 |
54280 |
2.2 |
| 10 | TMP/B(C6F5)3 | 30 | 12250 |
18900 |
1.6 |
| 11 | [TMPH+][HB(C6F5)3−] | 15 | 12250 |
11050 |
1.17 |
In order to achieve better control we contemplated the possibility of increasing the initiation rate using a hydride atom from a hydrogenated FLP as the initiator. As described in the literature, these compounds have the ability to reduce double bonds in the α-position of a carbonyl group.35 With this aim [TMPH+][HB(C6F5)3−] was prepared following the procedure described in the literature,36 and it was used to polymerize BCA, showing much better control of the reaction (Table 2, entry 11 and Scheme 1). The propagation rate was reduced, the experimental molecular weights were quite similar to the calculated ones together with narrower molecular weight distributions (1.17), and the formation of daughter polymers did not occur. To the best of our knowledge this is the first example of controlled living ionic polymerization of CA at practical temperatures even if the polydispersities must be further reduced. The hybridoborate anion, [HB(C6F5)3−], as the counterion of metallacyclic cations, has been previously proposed to catalyze the polymerization of conjugated polar alkenes, not including cyanoacrylates, by an H-shuttling mechanism in a non-controlled manner, a role completely different to that proposed in this work.37
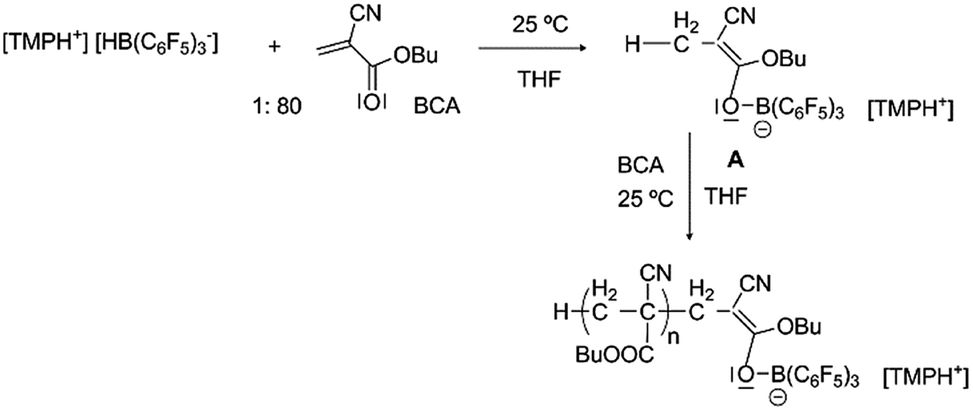 | ||
| Scheme 1 Proposed intermediates in the synthesis of poly(butyl cyanoacrylate) initiated by [TMPH+][HB(C6F5)3−]. | ||
A real living polymerization requires specific features such as full consumption of the monomer and initiator molecules, absence of irreversible termination and chain transfer reactions, initiation faster than propagation (almost inconceivable for CAs), targeted molecular weight, dispersities below 1.1, capability of growing in a controlled manner upon consecutive addition of monomers and the possibility to obtain block copolymers. An optimization of the different variables affecting the polymerization procedure of BCA was performed. A freshly distilled monomer and pure initiator were required to avoid the presence of oligomers in the final polymer. The addition order was also crucial; a concentrated solution of the initiator in THF was added in one shot directly to the solution of the monomer in THF. Under these conditions a polymer was obtained in less than 5 minutes showing polydispersities lower than 1.1, and daughter polymers were not observed after 60 min (Fig. 1, including comparison with BuLi initiation at similar reaction times). The reaction was carried out with different ratios of BCA: [TMPH+][HB(C6F5)3−], being possible to obtain polymers of different degrees of polymerization (DP 5, 35, 100, 215, 320 and 655 – MW ∼ 100 kg mol−1) with narrow polydispersities (1.02, 1.08, 1.09, 1.09, 1.07 and 1.11 respectively) (see ESI†). A negative dependence of the polymerization rate on temperature was observed in accordance with Pepper's studies performed on zwitterionic polymerization mechanisms some years ago and consistent with a complex composite initiation process that precedes propagation. This happened exclusively when tertiary amines were used (termed by Pepper “slowly initiated, non-terminated polymerizations”).38 Working at 60 °C, it was gratifying to see a unimodal GPC trace obtained after 5 minutes of reaction. At this temperature the monomer was not fully consumed, but it completely disappeared when experiments were conducted at 25 °C (faster reaction).
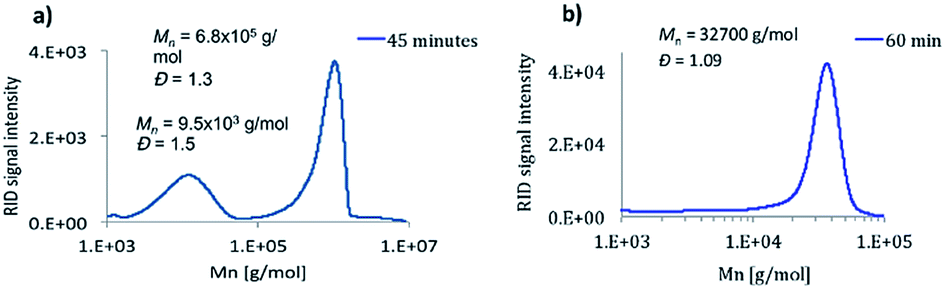 | ||
| Fig. 1 GPC traces of poly(n-butyl cyanoacrylate) (4.48 mmol distilled BCA in THF) initiated by [a] BuLi (0.056 mmol in THF, Exp. Mn ∼ 12250 Da) and [b] [TMPH+][HB(C6F5)3−] (0.021 mmol in THF, Exp. Mn ∼ 32678 Da) at 25 °C. | ||
An estimation of the relative polymerization rate was made (see ESI†). Calorimetric studies were performed for reactions at 25 °C but 1H NMR experiments were used to study the kinetics at 60 °C. A standard polymerization reaction using PPh3 as the initiator at 25 °C was used as the rate reference. A reduction of rate by at least two orders of magnitude was observed when the [TMPH+][HB(C6F5)3−] initiator was used at the same temperature. This rate was reduced by almost a further two orders of magnitude (i.e. four orders of magnitude with respect to the reference polymerization) when the reaction was carried out at 60 °C. These results would support the proposed mechanism of controlling the polymerization by lowering the polymerization rate through stabilization of the anionic chain end. Moreover, under these conditions, a linear evolution was observed in the Mnversus conversion-plot (see ESI†), supporting a controlled anionic living polymerization.
Two possible explanations can be envisaged to justify the inverse dependence of polymerization rate on temperature observed in our case. One would refer to the existence of a complex equilibrium composite process that precedes the actual initiation, related to the behavior described by Pepper and Ryan for zwitterionic polymerizations of CA with tertiary amines,38 but with the important difference that in our case we achieve total control of the polymerization. The second would be related to the variation of dielectric constant of the solvent (THF) with temperature.39 Hence, higher temperatures (lower dielectric constant of THF) would favor the formation of contact ion pairs, thus contributing to the reduction of the polymerization rate. Discrimination between those mechanistic explanations merits further studies that presently do not constitute the aim of this work.
No evidence was obtained for the presence of boron atoms linked to the polymer chains using various analytical techniques, most likely due to the low concentration of this atom in the sample. Other analyses were also performed in order to identify the formation of compounds of type A (Scheme 1) as promoters of the polymerization. A stoichiometric reaction between BCA and [TMPH+][HB(C6F5)3−] in THF at room temperature furnished a white solid that was analyzed by NMR. The disappearance of the double bond signals of BCA and the appearance of a new CH3 signal at 1.70 ppm in 1H NMR and 15.2 ppm in 13C NMR were observed. In the 11B NMR spectrum the disappearance of the B–H doublet signal observed at −23.9 ppm and the appearance of a new signal corresponding to B–O species at 4.02 ppm supported the existence of compounds of type A. Also in 19F NMR a clear shift of the signals was observed showing the new environment of fluorine atoms in agreement with the expected structures. Moreover direct evidence of the formation of A was obtained by MALDI-TOF MS analysis in the negative mode, where its signal at 666.1 g mol−1 was detected. Another interesting signal was identified at 819.2 g mol−1 corresponding to the dimer linked to the boron atom (Fig. 2). In our opinion, these data constitute direct evidence of the mechanism proposed for the living anionic polymerization of cyanoacrylates promoted by [TMPH+][HB(C6F5)3−], namely, hydride initiated polymers growing through a boron end-capping mediated controlled propagation, and shown in a simplified version in Scheme 1.
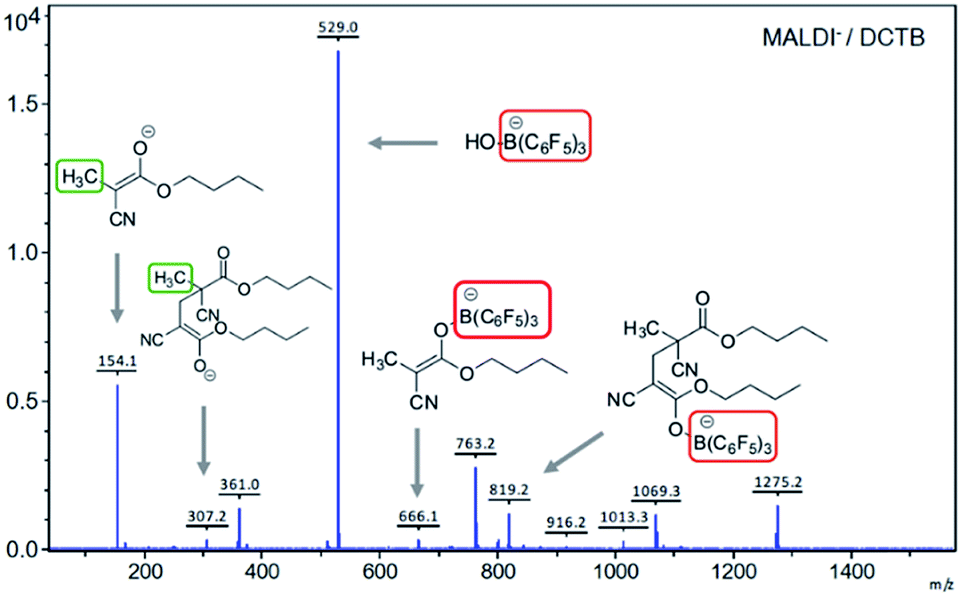 | ||
| Fig. 2 MALDI-TOF MS of the reaction crude of the stoichiometric reaction between BCA and [TMPH+][HB(C6F5)3−] in THF (negative mode), using DCTB as the matrix. | ||
In order to study the scope of the reaction two other CA monomers were polymerized under the best conditions found, ethyl cyanoacrylate (ECA) and β-methoxyethyl cyanoacrylate (MECA). The molecular weight of the obtained polymers was in agreement with the expected ones and narrow polydispersities were obtained (1.06 and 1.10 respectively, Fig. 3d and e, first addition) indicating a generality of use of such initiators with this class of monomers.
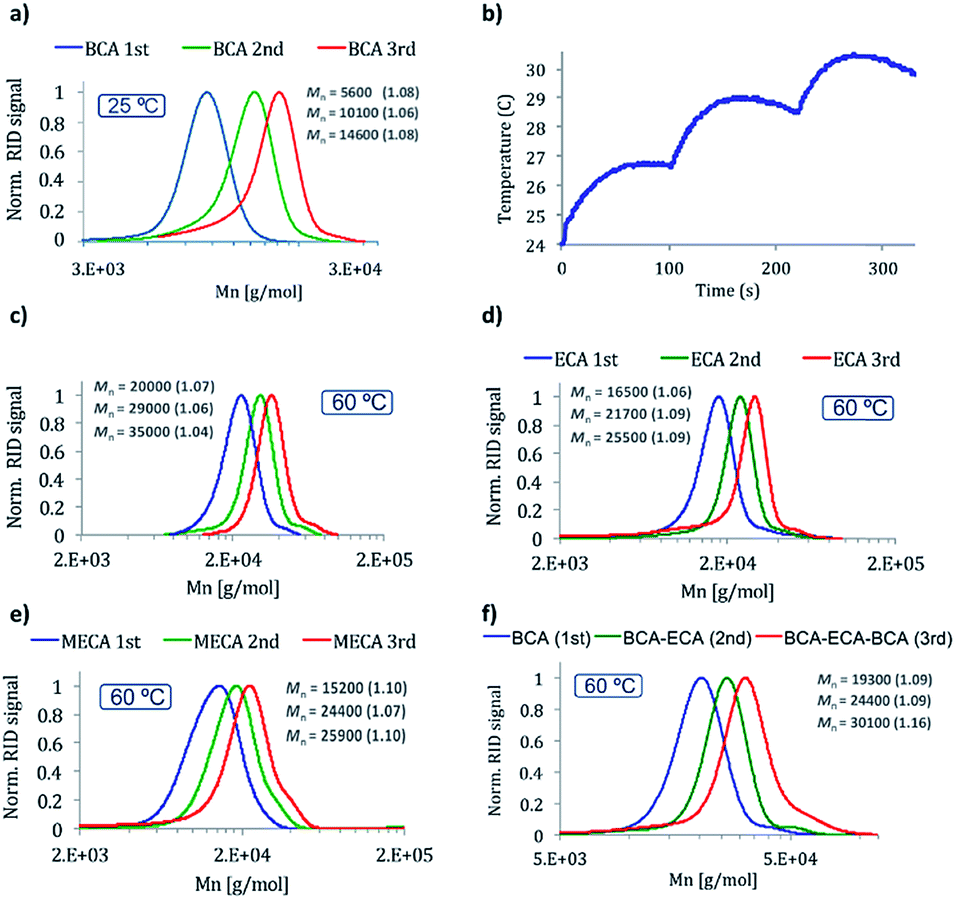 | ||
| Fig. 3 GPC traces of the polymers obtained in THF by three consecutive additions of BCA ([a] at 25 °C, [c] at 60 °C), ECA [d], MECA [e], and BCA + ECA + BCA [f] at 60 °C. Chart [b] shows the calorimetric traces corresponding to polymerizations in chart [a]. All reactions were initiated by [TMPH+][HB(C6F5)3−]. | ||
Although the synthesis of [TMPH+][HB(C6F5)3−] was performed in a glove box to avoid oxygen and moisture, the living polymerization process seemed to be resistant to significant amounts of water present in the media (10 × [initiator]), but always below the threshold range in which water could initiate itself CA polymerization. Acid pre-treated glassware or polypropylene flasks were required.
The living character of the polymer chain ends was confirmed by the preparation of polymers by up to three sequential monomer additions of either the same or different monomers (BCA, ECA or MECA) (Fig. 3a and c–f). To the best of our knowledge trace (f) in Fig. 3 represents the first ever example of controlled living ionic polymerization of CAs to produce bespoke block sequences even though these monomers first emerged for study in the late 1940's.40 Closer agreement between designed molecular weights and experimental ones was achieved once more working at 60 °C, where narrow polydispersities (≤1.1) were achieved in all cases. It was also possible to maintain the living polymer species dormant in the solid state.
Conclusions
To summarize, a hydrogenated frustrated Lewis pair ([TMPH+][HB(C6F5)3−]) initiator has allowed the controlled living ionic polymerization of highly reactive CA monomers (BCA, ECA and MECA) in THF. Homopolymers could be prepared by successive additions of monomers as could be block copolymers, and in all cases with narrow polydispersities. Formation of daughter polymers was avoided. An inverse dependence of the polymerization rate on temperature was observed with the hydrogenated FLP initiator, being four orders of magnitude lower than the rate given by a standard reference PPh3 initiator at 60 °C. Polymerization reaction intermediates containing living anions stabilized by B(C6F5)3 were detected by MALDI-TOF MS which supports the group transfer mechanism proposed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by Henkel KGaA & Co (contract for RS) and Ministerio de Economia, Industria y Competitividad (CTQ2015-65439-R, CTQ2016-81797-REDC). We also thank the DURSI-Generalitat de Catalunya for their recognition as consolidated research groups (2014 SGR 1105, 2014 SGR 560, and 2017 SGR 465). We thank Drs B. Ryan, C. Duffy, M. Phelan and B. Burns from Henkel KGaA & Co for their advice in the topic.Notes and references
- H. Morawetz, Polymers: The Origins and Growth of a Science, Courier Corporation, New York, USA, 2002 Search PubMed.
- R. O. Ebewele, Polymer Science and Technology,CRC Press LCC, Florida, USA, 2000 Search PubMed.
- O. W. Webster, Science, 1991, 251, 887 CrossRef CAS PubMed.
- W. F. Reed and A. M. Alb, Monitoring Polymerization Reactions: From Fundamentals to Applications, John Wiley and Sons. Inc., New Jersey, USA, 2014, ch. 2 Search PubMed.
- J. Smid, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2101 CrossRef CAS.
- (a) M. Szwarc, Nature, 1956, 178, 1168 CrossRef CAS; (b) H. F. Mark, Encyclopedia of Polymer Science and Engineering, John Wiley & Sons, New York, USA, 4th edn, 2014 Search PubMed.
- D. S. Johnston and D. C. Pepper, Macromol. Chem. Phys., 1981, 182, 421 CrossRef CAS.
- S. S. Johnston and D. C. Pepper, Makromol. Chem., 1981, 182, 393 CrossRef.
- P. Klemarczyk, Polymer, 2001, 42, 2837 CrossRef CAS.
- I. C. Eromosele and D. C. Pepper, Makromol. Chem., 1989, 190, 3085 CrossRef CAS.
- K. Ficht and C. D. Eisenbach, Makromol. Chem., Rapid Commun., 1993, 14, 669 CrossRef CAS.
- A. J. Domb and N. Kumar, Biodegradable Polymers in Clinical Use and Clinical Development, John Wiley and Sons. Inc., New Jersey, USA, 2011 Search PubMed.
- F. Scognamiglio, A. Travan, I. Rustighi, P. Tarchi, S. Palmisano, E. Marsich, M. Borgogna, I. Donati, N. de Manzini and S. Paoletti, J. Biomed. Mater. Res., Part B, 2016, 104, 626 CrossRef CAS PubMed.
- T. Kim and B. V. Kharod, Curr. Opin. Ophthalmol., 2007, 18, 39 CrossRef PubMed.
- P. A. Leggat, U. Kedjarune and D. R. Smith, Ind. Health, 2004, 42, 207 CrossRef CAS PubMed.
- D. Severian and V. Popa, Polymeric Biomaterials, CRC Press, Boca Ratón, USA, 2013, vol. II Search PubMed.
- S. Saranya and K. V. Radha, Polym.-Plast. Technol. Eng., 2014, 53, 1636 CrossRef CAS.
- J. Woods, Polycyanoacrylates, Encyclopedia of Polymer Science and Technology, John Wiley & Sons Inc., 2001, vol. 3 Search PubMed.
- M. Stein, J. Appl. Polym. Sci., 1992, 46, 2217 CrossRef CAS.
- C. Y. Huang and Y. D. Lee, Int. J. Pharm., 2006, 325, 132 CrossRef CAS PubMed.
- I. Szanka, A. Szanka, S. Sen, N. Nugay and J. P Kennedy, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1640 CrossRef CAS.
- X. Lin, R. Zhou, Y. Qiao, F. Jin, Y. Zhai, J. Xing, L. Deng and A. Dong, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7809 CrossRef CAS.
- X. Lin, L. Deng and A. Dong, J. Appl. Polym. Sci., 2012, 123, 3575 CrossRef CAS.
- H. K. Hall Jr, A. B. Padias, G. Chu, H. Y. Lee, I. Kalnin, M. Sansone and G. Breckenridge, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2341 CrossRef.
- H. Tang and N. V. Tsarevsky, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3683 CrossRef CAS.
- S. Gaikwad, S. S. Deshmukh, R. G. Gonnade, P. R. Rajamohanan and S. H. Chikkali, ACS Macro Lett., 2015, 4, 933 CrossRef CAS.
- C. Duffy, M. Phelan, P. B. Zetterlund and F. Aldabbagh, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1397 CrossRef CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 2 CrossRef PubMed.
- (a) M. Hong, J. Chen and E. Y. X. Chen, Chem. Rev., 2018, 118, 10551 CrossRef CAS PubMed; (b) J. Chen and E. X. Y. Chen, Molecules, 2015, 20, 9575 CrossRef CAS PubMed.
- J. S. Reddy, B. H. Xu, T. Mahdi, R. Fröhlich, G. Kehr, D. W. Stephan and G. Erker, Organometallics, 2012, 31, 5638 CrossRef CAS.
- D. C. Pepper, J. Polym. Sci., Polym. Symp., 1978, 62, 65 CAS.
- E. F. Donnelly and D. C. Pepper, Makromol. Chem., Rapid Commun., 1981, 442, 439 CrossRef.
- B. Ryan and G. McCann, Macromol. Rapid Commun., 1996, 17, 217 CrossRef CAS.
- D. R. Robello, T. D. Eldridge and M. T. Swanson, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4570 CrossRef CAS.
- J. S. Reddy, B. H. Xu, T. Mahdi, R. Fröhlich, G. Kehr, D. W. Stephan and G. Erker, Organometallics, 2012, 31, 5638 CrossRef CAS.
- V. Sumerin, F. Schulz, M. Nieger, M. Leskelä, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001 CrossRef CAS PubMed.
- E. Y. X. Chen, Top. Curr. Chem., 2013, 334, 239 CrossRef PubMed.
- D. C. Pepper and B. Ryan, Makromol. Chem., 1983, 184, 395 CrossRef CAS.
- C. Carvajal, K. J. Tölle, J. Smid and M. Szwarc, J. Am. Chem. Soc., 1965, 87, 5548 CrossRef CAS.
- A. E. Ardis, Preparation of monomeric alkyl alpha-cyano-acrylates, US Pat., 2,467,926, 1949.
Footnotes |
| † Electronic supplementary information (ESI) available: Devices, methods and procedures, analytical supporting material: NMR spectra, GPC traces and MALDI-TOF chromatograms. See DOI: 10.1039/c8sc04729d |
| ‡ Actual address: CTO-Afinitica Technologies s.l., Edifici Eureka, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Barcelona, Spain. |
| § All authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |