
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpace-confined indicator displacement assay inside a metal–organic framework for fluorescence turn-on sensing†
Ning-Ning
Yang
,
Li-Jiao
Zhou
,
Peng
Li
,
Qi
Sui
and
En-Qing
Gao
 *
*
Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, P. R. China. E-mail: eqgao@chem.ecnu.edu.cn
First published on 29th January 2019
Abstract
The indicator displacement assay (IDA) is for the first time performed within a metal–organic framework (MOF) to achieve ultrasensitive fluorescence turn-on sensing. A Zr(IV) ion MOF (UiO-67-DQ–PsO) furnished with electron-deficient diquat units (DQ2+, as the receptor) on the wall and electron-rich 1-pyrenesulfonate anions (PsO−, as the fluorescent indicator) in the pores was prepared by postsynthetic anion exchange. The MOF is capable of sensing alkylamines owing to the competing PsO−–DQ2+ and alkylamine–DQ2+ charge-transfer interactions, the former interaction causing a fluorescence OFF state and the latter displacing PsO− to trigger its emission. Significant advantages have been demonstrated for the IDA inside the MOF. The turn-on assay exhibits much higher sensitivity and anti-interference than the turn-off sensing using the MOF without indicators (the sensitivity is enhanced by as much as six orders of magnitude to the subnanomolar level). The integration of both the receptor and indicator in the porous solid enables facile regeneration and recyclability of the IDA ensemble. Furthermore, we show that the confined space provided by the MOF significantly enhances the supramolecular interactions to make possible the IDA impossible in solution. This work not only demonstrates a novel conceptual approach to fabricate superior fluorescence turn-on sensors using porous materials but also has important implications for supramolecular chemistry in porous materials.
Introduction
Luminescence is a simple, rapid, and highly sensitive signal transduction method, so there has been an ever-escalating interest in the development of chemosensors using luminescent materials. Metal–organic frameworks (MOFs), a huge class of crystalline porous materials built of metal ions/clusters and organic linkers, provide superior platforms for fabricating luminescence sensing systems.1 The frameworks can be imparted with tunable luminescence through use of variable linkers, metal nodes, guests or inter-component cooperation.2 The porous matrices can preconcentrate analytes, show size/shape effects, and be modified with diverse functionalities through pre- and post-synthetic methods to provide or improve sensing performance.1c,3 In particular, analyte-responsive receptors/reporters can be installed into the pores or on the surface through chemical bonds or supramolecular encapsulation.4 Furthermore, the confined space can allow for more efficient implementation of various sensing mechanisms through enhancing receptor–analyte interactions,5 promoting electron or energy transfer,6 and facilitating exciplex formation.7 A great number of MOFs have been explored as luminescent sensors for a variety of species pertinent to environmental pollution, public safety or medical health.8 Most of them involve luminescence quenching (turn-off). More attractive but much more challenging is luminescence turn-on sensing, where an analyte shows emission in a dark or weakly emissive state or causes an obvious shift in emission wavelength.9 The turn-on mode can have improved signal-to-noise ratios and anti-interference capability. However, turn-on sensing MOFs are still rare and most have some or other drawbacks, such as insufficiently weak original emission affecting sensitivity/selectivity, irreversible reactions disenabling material reuse and on-line detection, poor water stability limiting applications under aqueous or humid conditions, and poor manipulability of the mechanism hampering systematic material optimization. The rational design of sensing systems that make full use of the advantages of MOFs and turn-on mechanisms is still challenging.We propose a rational approach to fabricate turn-on sensors, utilizing the concept of indicator-displacement assays (IDAs) and the confined pore systems of MOFs. The IDA is a popular method to convert receptors into sensors making use of dynamic supramolecular interactions and has led to many sensing ensembles composed of various receptors and fluorescent or colorimetric indicators, targeting analytes of many classes.10 However, it has not yet been applied to MOFs. We propose that IDA can be performed within the pores of appropriate MOFs. Given the vast structural diversity and modifiability of MOFs, it should be possible to integrate both a receptor and fluorescent indicator in a MOF by framework functionalization and guest encapsulation. The receptor–indicator interaction could quench the emission from the indicator, giving an OFF state. The introduction of an analyte interacting with the receptor would separate the indicator from the receptor and thus evoke the fluorescence of the indicator. The crux for the turn-on sensing is the competition between the analyte and the indicator for interacting with the receptor. We envision that the host–guest interactions could be regulated or enhanced by the confined porous matrices resulting in high sensitivity and selectivity. In addition, the IDA inside a stable MOF should facilitate material regeneration and reuse (Scheme 1).
 | ||
| Scheme 1 (a) Fluorescence turn-off sensing of amine. (b) Turn-on sensing based on IDA. (c) The IDA in a MOF. CT = charge transfer, A = acceptor, and D = donor. | ||
For a proof-of-concept study, we demonstrate the IDA of amines inside an electron-deficient MOF, which proceeds through competing donor–acceptor charge-transfer (CT) interactions. Amines are important substances having wide applications but are also notorious pollutants harmful to human health and the environment.11 An increasing number of MOFs have been reported to be luminescently responsive to amines,12 of which only a few show a turn-on response.13 A judicious strategy toward amine-sensing MOFs is to incorporate electron-deficient units, with which electron-rich amines can form CT complexes to induce photophysical changes. Thus, some MOFs bearing a naphthalenediimide or 4,4′-bipyridinium unit can sense amines in the turn-off mode,14 and a dynamic coordination polymer shows a turn-on response to arylamines owing to emissive π–π CT complexation.5a However, highly sensitive MOFs for sensing amines in aqueous media are still rare. Recently, water-stable ionic MOFs furnished with electron-deficient 2,2′-bipyridinium and exchangeable anions have been prepared through postsynthetic N-alkylation of a 2,2′-bipyridyl-supported Zr(IV) MOF (UiO-67-bpy).15 It has been demonstrated that UiO-67-MQ (MQ2+ = N,N′-dimethyl-2,2′-bipyridinium) can sense alkylamines through reversible color and fluorescence changes induced by amine–bipyridinium CT complexation (Scheme 1a).15c However, the fluorescence sensitivity is low owing to the weak fluorescence of the MOF and the turn-off mode. We envision that electron-deficient MOFs should be nice candidates for turn-on IDAs through dynamic CT interactions. To implement this idea, an electron-rich and fluorescent anion (indicator) can be encapsulated into the MOF through anion exchange (Scheme 1b). The CT complexation between bipyridinium and the anion could quench the emission from the anion as well as that from the host. The amine–bipyridinium CT complexation could separate the anion from bipyridinium and thus the fluorescence of the anion could be turned on. With an appropriate anion, it should be possible to improve not only sensitivity but also selectivity. Here, we report the first example of the approach, using a MOF (UiO-67-DQ–PsO) integrated with diquat (DQ2+) units as receptor sites and 1-pyrenesulfonate (PsO−) as the indicator (Scheme 1c). The sensing properties of the precursor MOF (UiO-67-DQ–TfO with TfO− = triflate) are also presented first for comparison. As will be shown, the turn-on IDA of alkylamines in water using UiO-67-DQ–PsO shows dramatically improved sensitivity and anti-interference capability. Other benefits of implementing IDA within a MOF and the interactions underlying the IDA mechanism will be discussed. Notably, the space confinement effect is critical in enabling the IDA.
Results and discussion
Synthesis and characterization
UiO-67-DQ–TfO was prepared by postsynthetic N,N′-cycloalkylation of UiO-67-bpy with ethylene ditriflate (EDT) at room temperature, and UiO-67-DQ–PsO was obtained by anion exchange of UiO-67-DQ–TfO with sodium 1-pyrenesulfonate (NaPsO) in water. The reactions are confirmed by the IR spectra of the solids (Fig. S1†) and 1H NMR spectra of the solutions obtained by digesting the solids in HF (aq.)/d6-DMSO. The success of N-alkylation is indicated by the appearance of the characteristic IR bands of the ethylene group (3086 cm−1) and the triflate counteranion (1280, 1255, 1170 and 1030 cm−1).16 Anion exchange is indicated by the appearance of new multiple bands in the ranges of 1245–1160, 1090–1010, and 850–840 cm−1, which also appear in the spectra of NaPsO. 1H NMR spectra show new peaks characteristic of the DQ2+ cationic moiety (marked with Δ) after N-alkylation and new peaks characteristic of the PsO− anion (marked with ×) after anion exchange (Fig. 1). The N-alkylation ratio and anion exchange ratio were calculated according to integral analyses of the NMR spectra. A maximum N-alkylation ratio of about 65% can be achieved by prolonged reactions,15d but anion exchange of the highly N-alkylated MOF is incomplete (67%, Fig. S2†), perhaps because the pore size of the MOF prevents encapsulation of more PsO− anions, which are much bulkier than TfO−. The incomplete anion exchange has an adverse effect on the sensing properties (see below). Complete anion exchange (the molar ratio of the DQ2+ cation to the PsO− anion is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 as required by charge compensation) was achieved using UiO-67-DQ–TfO with a N-alkylation ratio of 42% (Fig. 1), which was obtained by reducing the duration time of the N-alkylation reaction. Hereafter unless otherwise specified, UiO-67-DQ–TfO refers to the MOF with the 42% modification ratio and UiO-67-DQ–PsO refers to the MOF after complete anion exchange. It is worth noting that the 1H NMR peaks of DQ are obviously shifted toward a high field after anion exchange, while the bpy peaks remain unshifted. On the other hand, some PsO− peaks for UiO-67-DQ–PsO are also obviously shifted compared with those for NaPsO.
:2 as required by charge compensation) was achieved using UiO-67-DQ–TfO with a N-alkylation ratio of 42% (Fig. 1), which was obtained by reducing the duration time of the N-alkylation reaction. Hereafter unless otherwise specified, UiO-67-DQ–TfO refers to the MOF with the 42% modification ratio and UiO-67-DQ–PsO refers to the MOF after complete anion exchange. It is worth noting that the 1H NMR peaks of DQ are obviously shifted toward a high field after anion exchange, while the bpy peaks remain unshifted. On the other hand, some PsO− peaks for UiO-67-DQ–PsO are also obviously shifted compared with those for NaPsO.
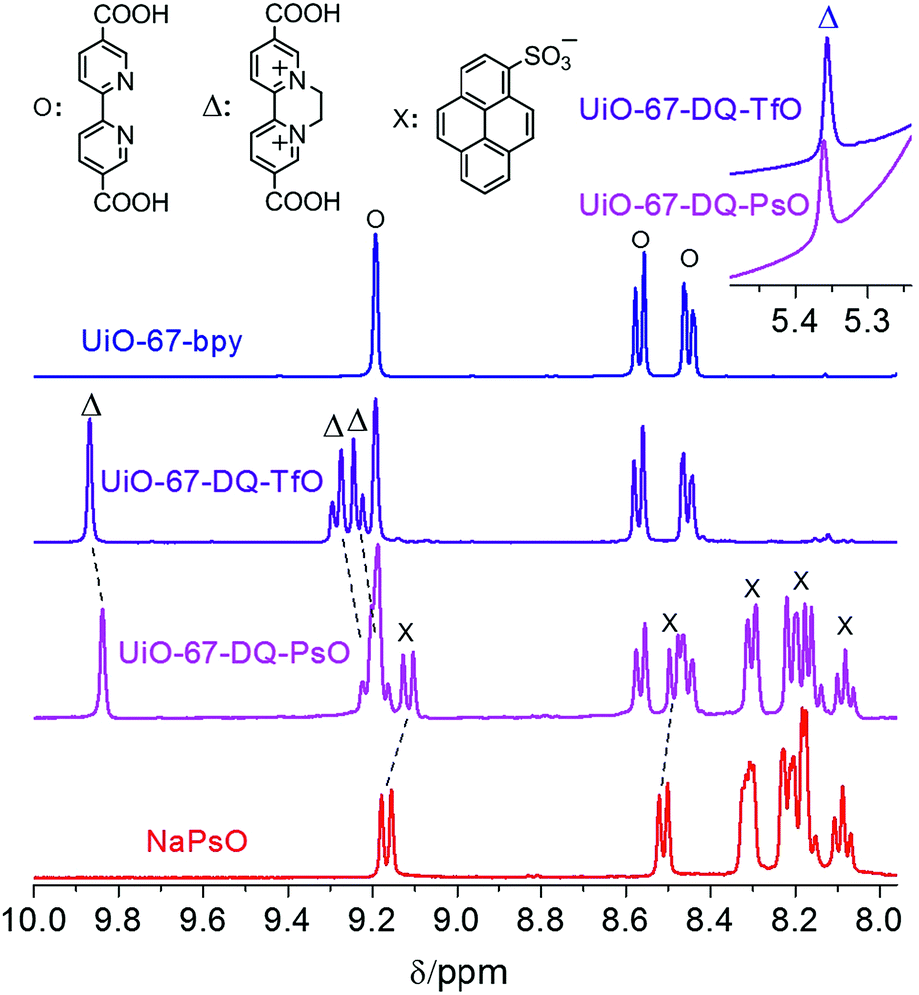 | ||
| Fig. 1 1H NMR spectra of the solutions obtained by digesting UiO-67-bpy, UiO-67-DQ–TfO, UiO-67-DQ–PsO and NaPsO with HF (aq.)/d6-DMSO. The dashed lines stress the shifts of some peaks. | ||
The phenomena are indicative of electronic interactions between PsO− and DQ− in solution.
Powder X-ray diffraction (PXRD) profiles of UiO-67-DQ–TfO and UiO-67-DQ–PsO are in good agreement with that of UiO-67-bpy (Fig. 2a), confirming that the crystalline framework is essentially retained after postsynthetic N-alkylation and anion exchange. The peak at 2θ = 9.4° (index [220]) increases in relative intensity on going from UiO-67-bpy to UiO-67-DQ–TfO and to UiO-67-DQ–PsO. This is consistent with the general phenomenon that the [220] peaks of UiO-66/67 MOFs (at 12.0° for UiO-66) decrease in intensity and even disappear upon guest removal.17 XRD simulation for UiO-66/67 reveals that the intensity of the peak is sensitive to the presence/absence of guest species in the pores of the MOFs, so the peak is mainly related to X-ray scattering of the guest. This justifies the change of the peak upon encapsulation of large anions. The XRD profile of UiO-67-DQ–PsO shows a small background hump between 20 and 30° (2θ), which could be tentatively ascribed to a slight structural degradation during anion exchange. The MOFs show a type-I nitrogen adsorption/desorption isotherm characteristic of microporosity (Fig. 2b). The Brunauer–Emmet–Teller (BET) surface area (977 m2 g−1) and the total pore volume (0.429 cm3 g−1) of UiO-67-DQ–TfO are significantly lower than the corresponding values (2047 m2 g−1 and 0.949 cm3 g−1) of UiO-67-bpy, owing to the occupation of the pores by the ethylene group and the TfO− anions. The values for UiO-67-DQ–PsO are further decreased (220 m2 g−1 and 0.136 cm3 g−1) because of the bulkiness of the PsO− anion. The MOFs show good thermal stability and retain the structural integrity after heating at 300 °C (UiO-67-DQ–TfO) and 200 °C (UiO-67-DQ–PsO), according to thermogravimetric and PXRD data (Fig. S3†).
 | ||
| Fig. 2 (a) PXRD patterns and (b) N2 adsorption/desorption isotherms of UiO-67-bpy, UiO-67-DQ–TfO and UiO-67-DQ–PsO. | ||
Response of UiO-67-DQ–TfO to amines
The MOF quickly changes from white to orange or red upon exposure to vapors of alkylamines such as butylamine, diethylamine, triethylamine and piperidine (Fig. 3a). By contrast, aniline vapor leads to a very faint color change only after prolonged exposure. Various non-amine VOCs (volatile organic compounds), including aliphatic/aromatic hydrocarbons, halohydrocarbons, alcohols, nitriles, amides, ketones, ethers, esters, and pyridines (only a few are shown in Fig. 3a), cannot cause any detectable color change. The selective chromogenic response to alkylamines is related to the appearance of the strong visible-light absorption band in the solid-state spectra of the amine-treated MOFs (Fig. 3c and S4†). The optical properties are reflective of CT complex formation between amines and the framework.15c,18 The precursor MOF UiO-67-bpy shows a negative color response to amines, so it is the DQ2+ chromophore that interacts with amine molecules. The deep colored UiO-67-DQ–TfO samples after amine treatment rapidly fade to their original color if treated with gaseous or aqueous HCl (Fig. S5a†), so it is the amine group that is responsible for the CT interaction. The fading also occurs when washed with water or just kept in open air. To our delight, the UiO-67-type framework remains intact during the coloration–decoloration cycle, as confirmed by PXRD (Fig. S5b†). | ||
| Fig. 3 (a) Photographs of UiO-67-DQ–TfO after being fumed with amines for few seconds. The negative response to some non-amine VOCs is also shown for comparison. (b) Relative fluorescence intensity (at 423 nm) of UiO-67-DQ–TfO/H2O dispersions in response to various VOCs (3 mM) in water. (c) Solid-state absorption (solid line) and emission spectra (dash line) of UiO-67-DQ–TfO before (red) and after exposure to butylamine (blue) and aniline (green). (d) The variation of In(I0/I − 1) upon incremental addition of butylamine to a UiO-67-DQ–TfO/H2O dispersion. | ||
The high-contrast, selective and reversible chromogenic properties imply potential applications for visual amine detection, food spoilage monitoring and invisible ink. For naked-eye and portable on-site detection, the test paper prepared by dip-coating filter paper with UiO-67-DQ–TfO dispersion shows an immediate color change when exposed to alkylamine vapors or solutions (Fig. S6†). Preliminary experiments indicate that the test paper can detect alkylamine vapors at a low concentration of 5 ppm, which is lower than the detection limits of most previous colorimetric sensors for alkylamine vapors (Table S1†).
UiO-67-DQ–TfO fluoresces in the solid state and aqueous dispersion. The emission intensity is significantly reduced after being treated with alkylamines (Fig. 3b and c). Consistent with the selective chromogenic properties, the aniline vapor or solution shows a much weaker quenching effect, and non-amine VOCs do not influence the emission. Considering that the emission band overlaps with the CT absorption band of the amine-treated MOF (Fig. 3c), the quenching effect could be due to Förster resonance energy transfer (FRET) from the framework to the non-emissive CT complex. To evaluate the performance of UiO-67-DQ–TfO for quantitative determination of alkylamines, the emission spectra of the MOF dispersed in water were monitored while butylamine (BA) was incrementally added. As expected, increasing the BA concentration leads to a gradual decrease in the emission intensity (Fig. S7†). The plot of I0/I versus BA concentration (C) can be fitted to the exponential function I0/I = AeKC + 1 (Fig. 3d), where I and I0 are the emission intensities at 423 nm in the presence and absence of BA, respectively. The limit of detection (LOD) was estimated to be 1.7 × 10−4 M by applying the 3σ/S convention (σ is the standard deviation of the blank measurements, and S is the slope of the I–C plot at a low concentration).
Fluorescence turn-on detection using UiO-67-DQ–PsO
The fluorescence spectrum of UiO-67-DQ–PsO dispersed in water is compared with those of UiO-67-DQ–TfO and NaPsO in Fig. 4. The PsO− solution shows strong emission bands at 376, 395 and 415 nm (λex = 346 nm), while the emission of UiO-67-DQ–PsO is very weak, even much weaker than that of UiO-67-DQ–TfO. It is clear that encapsulation of PsO− into the MOF efficiently quenches not only the emission from the framework but also that from the organic anion itself. Therefore, UiO-67-DQ–PsO provides an excellent OFF state for potential turn-on sensing. When BA (10 μM) was added to UiO-67-DQ–PsO, the PsO− emission was triggered, so the turn-on sensing was achieved. The enhancement ratio (I/I0) at 395 nm is as high as 25, suggesting a high signal-to-background ratio. The fluorescence turn-on is clearly visible to the naked eye under a UV lamp (254 nm) (Fig. 4, inset).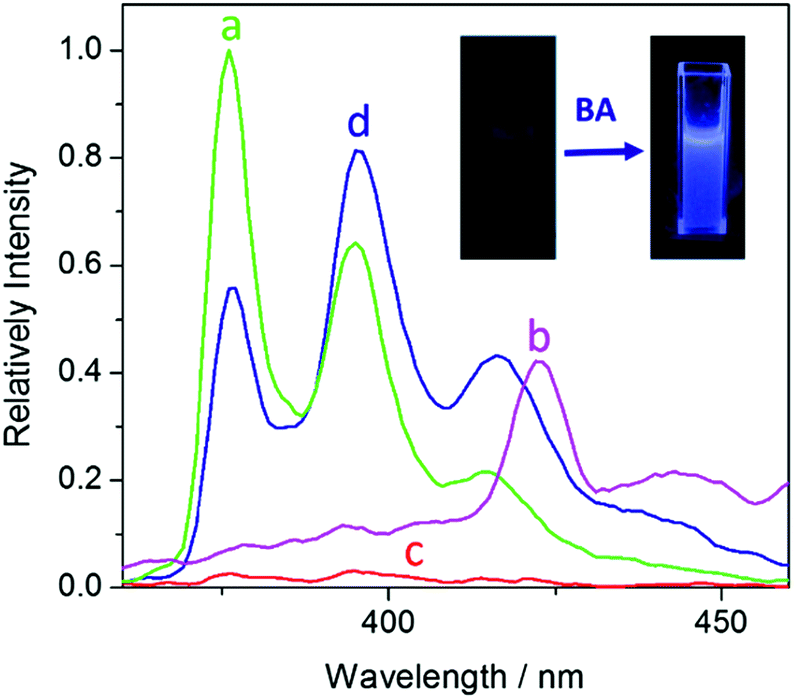 | ||
| Fig. 4 Emission spectra of NaPsO (a), UiO-67-DQ–TfO (b), and UiO-67-DQ–PsO ((c) original; (d) after addition of butylamine (BA)) dispersed in water. Inset: the visual change under UV light (254 nm) upon addition of BA. | ||
To evaluate the sensitivity of the turn-on detection system for amine, fluorescence titrations were performed by stepwise addition of BA to the aqueous dispersion of UiO-67-DQ (Fig. 5a). An ultratrace amount of BA (2 nM) causes a high enhancement ratio of 4.3, and the ratio increases linearly with logCBA in the range of 10−9 to 10−5 M (I/I0 = 5.42logCBA + 52.29, Fig. 5b). The LOD is estimated to be 1.7 × 10−10 M. This implies that UiO-67-DQ–PsO can be used as an ultrasensitive luminescent sensor for quantitative detection of alkylamines in the micro- to subnanomolar range. The LOD value is lower by six orders of magnitude compared to that of the turn-off detection using UiO-67-DQ–TfO, emphasizing the superiority of the IDA method.
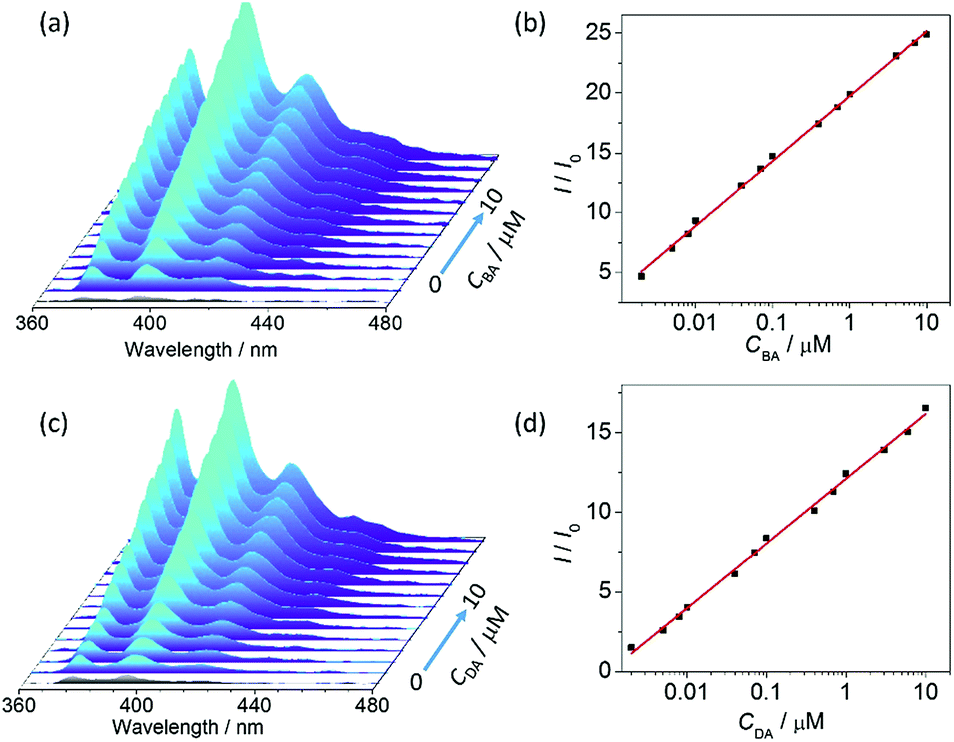 | ||
| Fig. 5 Variation of the emission spectrum of UiO-67-DQ–PsO dispersed in water upon incremental addition of butylamine (BA, a) and dopamine (DA, c) and the corresponding plots (b and d) of I/I0 at 395 nm versus the amine concentration. | ||
The alkylamine-responsive fluorescence turn-on is also highly selective. Negligible changes of emissions were observed when the aqueous dispersion of UiO-67-DQ–PsO was treated with aniline and a variety of other organic compounds (Fig. 6a). To determine the interference of excessive aniline with alkylamine sensing, the emission of the UiO-67-DQ–PsO suspension was monitored upon successive addition of BA and incremental amounts of aniline. We found that the emission is only responsive to BA addition and nearly independent of aniline addition, even a large excess of aniline causing no significant change (Fig. 6b). The interference rate measured using (I′ − I)/(I − I0) × 100% is only 1.0% for 1000 equivalents of aniline (I′ is the intensity in the copresence of BA and aniline). The trivially low interference rates suggest that the turn-on system has excellent anti-interference capability. For comparison, the turn-off detection of BA using UiO-67-DQ–TfO is strongly interfered with by aniline (Fig. 6b), the interference rate being 18% at 1 equivalent of aniline and increasing rapidly to 127% at 50 equivalents. The results show that the turn-on IDA using UiO-67-DQ–PsO not only dramatically enhances the sensitivity but also significantly improves the anti-jamming performance.
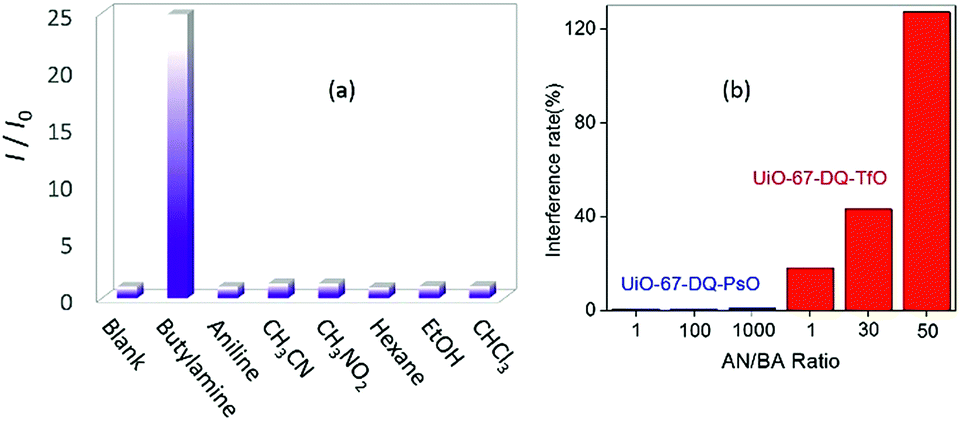 | ||
| Fig. 6 (a) Relative emission intensity of aqueous UiO-67-DQ–PsO dispersions in response to various organic compounds. (b) The interference rate of different amounts of aniline (AN) for detection of butylamine (BA) using UiO-67-DQ–PsO (CBA = 4 × 10−7 M) and UiO-67-DQ–TfO (CBA = 1 × 10−3 M). | ||
Dopamine (DA) is an important neurotransmitter and medication. Abnormal concentrations of DA have been correlated with various diseases in the mammalian central nervous system, so accurate determination of DA is important for disease diagnosis and monitoring.19 To further demonstrate the fluorescence turn-on system, UiO-67-DQ–PsO was tested as a sensor for determination of DA in water. The results obtained with DA are similar to those with BA (Fig. 5c and d). The enhancement ratio is linear with logCDA, and the LOD is 3.4 × 10−10 M. The subnanomolar LOD makes UiO-67-DQ–PsO stands out in sensitivity not only among the few MOFs with available LODs13c,d,20 but also among materials of various classes21 reported as luminescence sensors for DA (Table S1†).
Furthermore, from the economic and environmental viewpoints, it is highly desired that a sensory material can be reused through easy regeneration procedures. However, the reusability of MOF-based sensors is often limited by their poor stability against solvents (especially water) and analytes. To test the recyclability of UiO-67-DQ–PsO for DA detection, the used solid was isolated from the suspension by centrifugation, washed with water to remove adsorbed DA, dried and then reused for the next run of detection. As shown in Fig. 7a, the OFF state is recovered in the used MOF after the simple treatments, and the ON state evoked by DA shows no significant degradation in emission intensity compared with that of the fresh sample. Therefore, the sensor is recyclable after simple separation and washing. The reusability is endowed by the high structural stability of the ionic MOF against water and amines, which is evidenced by the PXRD measurements before and after the recycle tests (Fig. 7b). The facile recoverability and recyclability are some of the advantages of the IDA within a solid porous matrix. The homogeneous nature of traditional IDA processes complicates removal and recovery of receptors/indicators from solutions. UiO-67-DQ–PsO is a two-in-one sensor combining both the receptor and indicator in one stable solid, facilitating recovery. It is notable that for the sake of electroneutrality, the anionic indicator (PsO−) would not leave the cationic framework during amine-sensing.
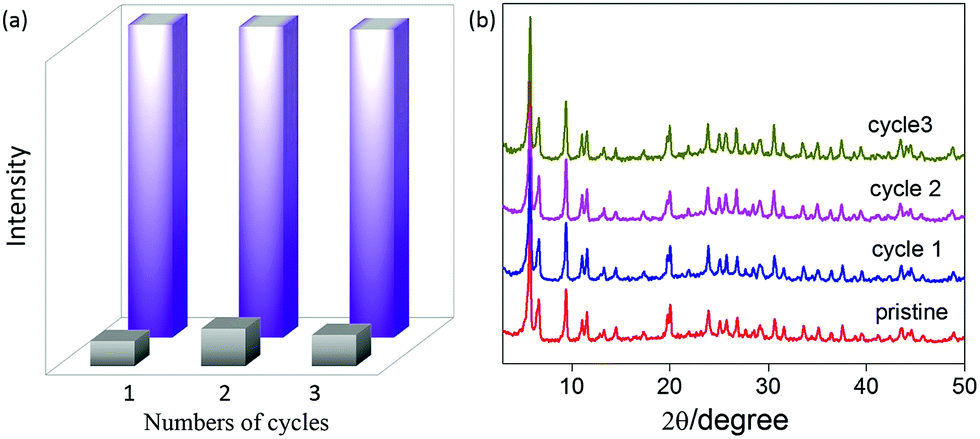 | ||
| Fig. 7 (a) Reproducibility of the fluorescence off (gray) and on (violet) states of UiO-67-DQ–PsO during three cycles of dopamine sensing (0.01 mM). (b) PXRD patterns of UiO-67-DQ–PsO before and after the cycles. | ||
Mechanism
To probe the interactions underlying the IDA, the UV-vis spectra of UiO-67-DQ–PsO its precursors (UiO-67-DQ–TfO and NaPsO) (Fig. 8) are compared. UiO-67-DQ–PsO shows a shoulder visible-light absorption that is absent in the precursors, consistent with the color change (Fig. S8†). The differences indicate ground-state PsO–DQ CT complexation. As observed for amine–DQ CT complexation in UiO-67-DQ–TfO, the PsO–DQ CT complexation serves to quench the emission of PsO− and the framework, leading to the OFF state of UiO-67-DQ–PsO. The PsO–DQ interactions also occur in solution: as marked in Fig. 1, some DQ2+ and PsO− NMR peaks for the digested UiO-67-DQ–PsO are shifted compared with the corresponding peaks for the precursors. After being treated with BA, both UiO-67-DQ–PsO and UiO-67-DQ–TfO turn red brown and show significantly increased visible-light absorbance, suggesting the formation of CT complexes between the amine and DQ2+ in UiO-67-DQ–PsO. CT interactions between the amine and PsO− can be precluded because NaPsO shows no color and spectral response to amines. The amine–DQ CT complexation serves to separate PsO− from DQ2+ (but PsO− still remains in the MOF), and the fluorescence of the indicator is turned on, completing the IDA mechanism.10a The high sensitivity implies that the CT interaction of DQ2+ with alkylamine is stronger than that with PsO− in UiO-67-DQ–PsO. Consistent with the mechanism, the excellent anti-interference capability of the alkylamine-sensing system toward aniline can be well justified. Aniline can form weak CT complexes with DQ2+ in UiO-67-DQ–TfO, interfering with alkylamine detection. However, the amino group of aniline has weaker Lewis basicity (electron-donor strength) and polarity. It can be assumed that the interactions between aniline and DQ2+ are not sufficiently strong to effectively interfere with the CT complexes between PsO− and DQ2+, so aniline cannot induce indicator displacement and does not interfere in the IDA of alkylamines. | ||
| Fig. 8 UV-vis spectra of NaPsO, UiO-67-DQ–TfO and UiO-67-DQ–PsO before (solid lines) and after (dashed lines) exposure to butylamine (BA) vapor. | ||
The mechanism is also supported by some results obtained with different MOF-indicator systems. The mixed-anion MOF obtained by incomplete anion exchange (UiO-67-DQ–PsO–TfO with PsO/TfO = 2, see above) has been tested for BA detection (Fig. S9†). No significant fluorescence turn-on was observed at BA concentrations below 1 μM. The LOD is 1.5 × 10−5 M, so the detection sensitivity of the mixed-anion MOF is much lower than that of the completely exchanged MOF, which can be justified as follows. In the mixed-anion MOF, not all of the DQ2+ sites form CT complexes with PsO−. The “free” receptor sites that do not form complexes with the anionic indicators should have priority in interacting with incoming amine molecules. Therefore, the amine cannot separate PsO− from DQ2+ and the fluorescence of PsO− is not turned on until the “free” DQ2+ sites are used up at a higher amine concentration. Similar phenomena have been observed in our initial studies using 2-hydroxy-1,3,6-pyrenetrisulfonate (HPTS3−) as the fluorescent indicator. UiO-67-DQ–HPTS, prepared by complete anion exchange of UiO-67-DQ–TfO with Na3HPTS, also shows a fluorescence turn-on response to alkylamine (Fig. S10†), but the sensitivity is also much lower compared with that of UiO-67-DQ–PsO: as shown in Fig. S11,† the addition of 10 μM BA to the UiO-67-DQ–HPTS dispersion causes an enhancement ratio of 1.4, which is much lower than that (25) for UiO-67-DQ–PsO under identical conditions. The low sensitivity of UiO-67-DQ–HPTS can be explained as follows. The charge balance dictates that the amount of the trianionic indicator in the MOF is less than that of the dicationic receptor site, and therefore the “free” DQ2+ sites delay the indicator displacement. The adverse effects of incomplete anion exchange and a higher-charged indicator provides corroborative evidence for the IDA mechanism.
To gain clues as to the possible effects of the confined space in the MOF on the IDA, the interactions of DQ2+ with PsO− and amine in an unconfined environment (i.e., in solution) were probed by monitoring the emission of an NaPsO solution (0.8 mM, equivalent to the amount of UiO-67-DQ–PsO used for sensing experiments) upon successive addition of DQ2+ and BA (Fig. S12†). The emission can be quenched by DQ2+, but the quenching efficiency is only 37% at CPsO:CDQ = 1:0.5, which corresponds to the ratio in UiO-67-DQ–PsO. For comparison, the PsO− emission is almost completely quenched in UiO-67-DQ–PsO. This suggests that the PsO–DQ interaction in solution is dramatically weakened by solvation and cannot afford a good OFF state for IDA. In addition, the addition of excessive BA hardly influenced the emission of the solution mixture of PsO− and DQ2+, suggesting that the IDA cannot be performed using the DQ2+–PsO− ensemble in homogeneous solutions. To gain further insight, similar studies were performed using aqueous [Me2DQdc]2+ (dimethyl diquat-5,5′-dicarboxylate, the ester of the very linker for UiO-67-DQ) (Fig. S13†). We chose the ester instead of the carboxylic acid (H2DQdc) because the acid–base reaction with amine would influence the interactions between bipyridinium and amine. Although the quenching efficiency (53%) of [Me2DQdc]2+ at CPsO:CMe2DQdc = 1:0.5 is higher than that (37%) of DQ2+, it is still far from the complete quenching observed for UiO-67-DQ–PsO. The addition of 0.8 mM BA (BA:Me2DQdc molar ratio = 2:1) to the [Me2DQdc]2+–PsO− mixture enhances the emission by 30%, indicating a weak indicator displacement effect. However, the enhancement is much lower than that for the UiO-67-DQ–PsO dispersion, where 0.01 mM BA enhances the emission by as much as 24 times (vide supra). That is, the turn-on IDA within the MOF is much more efficient than that in solution. We propose that the confined space provided by the MOF should significantly enhance both receptor–indicator and receptor–analyte interactions. Therefore, integrating a receptor–indicator ensemble within a MOF can lead to efficient and sensitive IDA that is insensitive or impossible without space confinement. By the way, the different behaviors between DQ2+ and [Me2DQdc]2+ could be due to the electron-withdrawing nature of the carboxylate groups, which makes the bipyridinium moiety more electron-deficient to the advantage of CT interactions with both PsO− and amine.
Conclusions
In summary, we proposed and demonstrated the IDA-in-MOF approach for the design of fluorescence turn-on sensors. The MOF integrated with electron-deficient receptor sites and an electron-rich fluorescent indicator can efficiently sense alkylamines in water because of indicator displacement through competing CT interactions of the receptor with analytes/indicators. We demonstrated that implementing IDA within the MOF affords several appreciable benefits. The turn-on assay shows dramatically enhanced sensitivity and anti-interference capability compared to the turn-off detection with the indicator-free precursor MOF. The integration of the receptor and indicator into the stable porous solid facilitates the regeneration and recyclability of the IDA ensemble. Furthermore, a strong and positive space-confinement effect was demonstrated. The confined space provided by the MOF can regulate the competing supramolecular interactions for an efficient IDA, which may be inefficient and even impossible in solution. Considering the great diversity and modifiability of MOFs and the various interactions applicable to IDA, the IDA-in-MOF approach should be of general interest for the rational design of superior fluorescence turn-on sensors for various analytes. It can also be extended to other porous materials such as porous organic frameworks. This work also has important implications for space-confined supramolecular chemistry in porous solids.Experimental section
Synthesis of UiO-67-DQ–TfO
UiO-67-bpy and EDT were synthesized according to literature methods.22 EDT (750 μL) was added to UiO-67-bpy (100 mg, ∼0.046 mmol) in CHCl3 (2.5 mL). After stirring for 2.5 h at room temperature, the solid was harvested by filtration, washed with ethanol, and then dried in air.Preparation of UiO-67-DQ–PsO
A freshly prepared sample (100 mg) of UiO-67-DQ–TfO was stirred in the aqueous solution (75 mL) of NaPsO (200 mg) for 5 h. The resulting gray-green solid was isolated, washed with water several times to remove any NaPsO and then dried at room temperature.Luminescence sensing measurements
UiO-67-DQ–TfO or UiO-67-DQ–PsO (6.0 mg) was dispersed in distilled water (10 mL) and sonicated for 30 min to form a stable milk white suspension. After recording the fluorescence using 2.5 mL of the dispersion, an aqueous solution containing the analyte with a given concentration was added to the cuvette and stirred for 30 min, and then fluorescence was measured.Preparation of test paper
The filter paper was dipped into a dispersion of UiO-67-DQ–TfO in EtOH (10 mg mL−1, uniformly dispersed by ultrasonication) for a few seconds and then left to dry at room temperature. The dip-coating and drying procedure was repeated three times.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 21773070 and 21471057).Notes and references
- (a) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (b) Y. Zhang, S. Yuan, G. Day, X. Wang, X. Yang and H.-C. Zhou, Coord. Chem. Rev., 2018, 354, 28 CrossRef CAS; (c) W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242 RSC.
- (a) Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483 CrossRef CAS PubMed; (b) R. W. Huang, Y. S. Wei, X. Y. Dong, X. H. Wu, C. X. Du, S. Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689 CrossRef CAS PubMed.
- (a) A. Karmakar, P. Samanta, A. V. Desai and S. K. Ghosh, Acc. Chem. Res., 2017, 50, 2457 CrossRef CAS PubMed; (b) S. M. Cohen, Chem. Rev., 2012, 112, 970 CrossRef CAS PubMed.
- (a) B. Yan, Acc. Chem. Res., 2017, 50, 2789 CrossRef CAS PubMed; (b) F. Wang, W. Liu, S. J. Teat, F. Xu, H. Wang, X. Wang, L. An and J. Li, Chem. Commun., 2016, 52, 10249 RSC; (c) Z. Ding, J. Tan, G. Feng, Z. Yuan, C. Wu and X. Zhang, Chem. Sci., 2017, 8, 5101 RSC.
- (a) R. Haldar, R. Matsuda, S. Kitagawa, S. J. George and T. K. Maji, Angew. Chem., Int. Ed., 2014, 53, 11772 CrossRef CAS PubMed; (b) Y. Takashima, V. M. Martinez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef PubMed.
- (a) Y. Li, S. Zhang and D. Song, Angew. Chem., Int. Ed., 2013, 52, 710 CrossRef CAS PubMed; (b) J.-N. Haoand and B. Yan, Chem. Commun., 2015, 51, 7737 RSC; (c) B. Zhao, X.-Y. Chen, P. Cheng, D.-Z. Liao, S.-P. Yan and Z.-H. Jiang, J. Am. Chem. Soc., 2004, 126, 15394 CrossRef CAS PubMed.
- V. Martínez-Martínez, S. Furukawa, Y. Takashima, I. López Arbeloa and S. Kitagawa, J. Phys. Chem. C, 2012, 116, 26084 CrossRef.
- (a) H. Wang, W. P. Lustig and J. Li, Chem. Soc. Rev., 2018, 47, 4729 RSC; (b) F. Y. Yi, D. X. Chen, M. K. Wu, L. Han and H. L. Jiang, Chempluschem, 2016, 81, 675 CrossRef CAS; (c) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC; (d) R.-B. Lin, S.-Y. Liu, J.-W. Ye, X.-Y. Li and J.-P. Zhang, Adv. Sci., 2016, 3, 1500434 CrossRef PubMed; (e) H.-R. Fu, L.-B. Yan, N.-T. Wu, L.-F. Ma and S.-Q. Zang, J. Mater. Chem. A, 2018, 6, 9183 RSC; (f) P. Kumar, A. Deep and K.-H. Kim, Trends Anal. Chem., 2015, 73, 39 CrossRef CAS; (g) B. Wang, X.-L. Lv, D. Feng, L.-H. Xie, J. Zhang, M. Li, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204 CrossRef CAS PubMed; (h) S. Wu, Y. Lin, J. Liu, W. Shi, G. Yang and P. Cheng, Adv. Funct. Mater., 2018, 28, 1707169 CrossRef.
- (a) S. S. Nagarkar, A. V. Desai and S. K. Ghosh, Chem.–Eur. J., 2015, 21, 9994 CrossRef CAS PubMed; (b) Y.-Z. Chenand and H.-L. Jiang, Chem. Mater., 2016, 28, 6698 CrossRef; (c) B. Chen, L. Wang, F. Zapata, G. Qian and E. B. Lobkovsky, J. Am. Chem. Soc., 2008, 130, 6718 CrossRef CAS PubMed; (d) N. B. Shustova, A. F. Cozzolino, S. Reineke, M. Baldo and M. Dinca, J. Am. Chem. Soc., 2013, 135, 13326 CrossRef CAS PubMed; (e) Y. Guo, X. Feng, T. Han, S. Wang, Z. Lin, Y. Dong and B. Wang, J. Am. Chem. Soc., 2014, 136, 15485 CrossRef CAS PubMed; (f) R. N. Das, M. Debnath, A. Gaurav and J. Dash, Chem.–Eur. J., 2014, 20, 16688 CrossRef CAS PubMed; (g) K. S. Lim, S. Y. Jeong, D. W. Kang, J. H. Song, H. Jo, W. R. Lee, W. J. Phang, D. Moon and C. S. Hong, Chem.–Eur. J., 2017, 23, 4803 CrossRef CAS PubMed; (h) M. Zhang, G. Feng, Z. Song, Y.-P. Zhou, H.-Y. Chao, D. Yuan, T. T. Y. Tan, Z. Guo, Z. Hu, B. Z. Tang, B. Liu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 7241 CrossRef CAS PubMed; (i) N. B. Shustova, A. F. Cozzolino and M. Dinca, J. Am. Chem. Soc., 2012, 134, 19596 CrossRef CAS PubMed; (j) B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1693 CrossRef CAS; (k) H. Xu, X. Rao, J. Gao, J. Yu, Z. Wang, Z. Dou, Y. Cui, Y. Yang, B. Chen and G. Qian, Chem. Commun., 2012, 48, 7377 RSC; (l) D. Yue, D. Zhao, J. Zhang, L. Zhang, K. Jiang, X. Zhang, Y. Cui, Y. Yang, B. Chen and G. Qian, Chem. Commun., 2017, 53, 11221 RSC.
- (a) L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840 CrossRef CAS PubMed; (b) B. T. Nguyenand and E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118 CrossRef; (c) A. Norouzy, Z. Azizi and W. M. Nau, Angew. Chem., Int. Ed., 2015, 54, 792 CrossRef CAS PubMed; (d) P. Koutnik, E. G. Shcherbakova, S. Gozem, M. G. Caglayan, T. Minami and P. Anzenbacher Jr, Chem, 2017, 2, 271 CrossRef CAS; (e) Z. Zheng, W.-C. Geng, J. Gao, Y.-Y. Wang, H. Sun and D.-S. Guo, Chem. Sci., 2018, 9, 2087 RSC; (f) M. A. Beatty, J. Borges-Gonzalez, N. J. Sinclair, A. T. Pye and F. Hof, J. Am. Chem. Soc., 2018, 140, 3500 CrossRef CAS PubMed; (g) B. M. Chapin, P. Metola, S. L. Vankayala, H. L. Woodcock, T. J. Mooibroek, V. M. Lynch, J. D. Larkin and E. V. Anslyn, J. Am. Chem. Soc., 2017, 139, 5568 CrossRef CAS PubMed.
- (a) S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, 2004 Search PubMed; (b) M. Papageorgiou, D. Lambropoulou, C. Morrison, E. Klodzinska, J. Namiesnik and J. Plotka-Wasylka, Trends Anal. Chem., 2018, 98, 128 CrossRef CAS.
- (a) S.-S. Zhao, J. Yang, Y.-Y. Liu and J.-F. Ma, Inorg. Chem., 2016, 55, 2261 CrossRef CAS PubMed; (b) X. Y. Xu, X. Lian, J. N. Hao, C. Zhang and B. Yan, Adv. Mater., 2017, 29, 1702298 CrossRef PubMed; (c) J. Chen, F.-Y. Yi, H. Yu, S. Jiao, G. Pang and Z.-M. Sun, Chem. Commun., 2014, 50, 10506 RSC; (d) S. Khatua, S. Goswami, S. Biswas, K. Tomar, H. S. Jena and S. Konar, Chem. Mater., 2015, 27, 5349 CrossRef CAS; (e) P. Rouschmeyer, N. Guillou, C. Serre, G. Clavier, C. Martineau, P. Audebert, E. Elkaim, C. Allain and T. Devic, Inorg. Chem., 2017, 56, 8423 CrossRef CAS PubMed.
- (a) P. Mani, A. A. Ojha, V. S. Reddy and S. Mandal, Inorg. Chem., 2017, 56, 6772 CrossRef CAS PubMed; (b) S. W. Jaros, J. Sokolnicki, A. Woloszyn, M. Haukka, A. M. Kirillov and P. Smolenski, J. Mater. Chem. C, 2018, 6, 1670 RSC; (c) C. Zhao, Z. Jiang, R. Mu and Y. Li, Talanta, 2016, 159, 365 CrossRef CAS PubMed; (d) Y. Cheng, J. Wu, C. Guo, X.-G. Li, B. Ding and Y. Li, J. Mater. Chem. B, 2017, 5, 2524 RSC.
- (a) A. Mallick, B. Garai, M. A. Addicoat, P. S. Petkov, T. Heine and R. Banerjee, Chem. Sci., 2015, 6, 1420 RSC; (b) T. Gong, P. Li, Q. Sui, J. Chen, J. Xu and E.-Q. Gao, J. Mater. Chem. A, 2018, 6, 9236 RSC; (c) J.-J. Liu, Y.-B. Shan, C.-R. Fan, M.-J. Lin, C.-C. Huang and W.-X. Dai, Inorg. Chem., 2016, 55, 3680 CrossRef CAS PubMed; (d) J.-J. Liu, Y.-F. Guan, M.-J. Lin, C.-C. Huang and W.-X. Dai, Cryst. Growth Des., 2015, 15, 5040 CrossRef CAS.
- (a) Q. Zhang, J. Yu, J. Cai, L. Zhang, Y. Cui, Y. Yang, B. Chen and G. Qian, Chem. Commun., 2015, 51, 14732 RSC; (b) L. Xu, Y. Luo, L. Sun, S. Pu, M. Fang, R. X. Yuan and H. B. Du, Dalton Trans., 2016, 45, 8614 RSC; (c) N. N. Yang, W. Sun, F. G. Xi, Q. Sui, L. J. Chen and E. Q. Gao, Chem. Commun., 2017, 53, 1747 RSC; (d) N. N. Yang, J. J. Fang, Q. Sui and E. Q. Gao, ACS Appl. Mater. Interfaces, 2018, 10, 2735 CrossRef CAS PubMed.
- S. J. Angus-Dunne, L. E. P. Lee Chin, R. C. Burns and G. A. Lawrance, Transition Met. Chem., 2006, 31, 268 CrossRef CAS.
- (a) C. J. Hafizovic, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed; (b) A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643 CrossRef CAS PubMed; (c) F.-G. Xi, Y. Yang, H. Liu, H.-F. Yao and E.-Q. Gao, RSC Adv., 2015, 5, 79216 RSC.
- C. J. Bender, Chem. Soc. Rev., 1986, 15, 475 RSC.
- (a) X. Liu, T. Xiao, F. Wu, M.-Y. Shen, M. Zhang, H.-h. Yu and L. Mao, Angew. Chem., Int. Ed., 2017, 56, 11802 CrossRef CAS PubMed; (b) A. Pandikumar, G. T. Soon How, T. P. See, F. S. Omar, S. Jayabal, K. Z. Kamali, N. Yusoff, A. Jamil, R. Ramaraj, S. A. John, H. N. Lim and N. M. Huang, RSC Adv., 2014, 4, 63296 RSC; (c) M. Sajid, M. K. Nazal, M. Mansha, A. Alsharaa, S. M. S. Jillani and C. Basheer, Trends Anal. Chem., 2016, 76, 15 CrossRef CAS; (d) J.-J. Fang, N.-N. Yang and E.-Q. Gao, Electrochem. Commun., 2018, 89, 32 CrossRef CAS.
- (a) C. Zhao, Y. Liu and Y. Li, Anal. Sci., 2015, 31, 1035 CrossRef CAS PubMed; (b) Q. Zhu, Y. Chen, W. Wang, H. Zhang, C. Ren, H. Chen and X. Chen, Sens. Actuators, B, 2015, 210, 500 CrossRef CAS.
- (a) Y.-K. Linand and Y.-C. Yeh, Anal. Chem., 2017, 89, 11178 CrossRef PubMed; (b) N. Pallikkarathodi Mani, M. Ganiga and J. Cyriac, Analyst, 2018, 143, 1691 RSC; (c) D. Bharathi, B. Siddlingeshwar, R. H. Krishna, V. Singh, N. Kottam, D. D. Divakar and A. AbdullahAlkheraif, J. Fluoresc., 2018, 28, 573 CrossRef CAS PubMed; (d) C. Sun, F. Yuan, H. Li and X. Wu, Microchim. Acta, 2018, 185, 1 CrossRef CAS PubMed; (e) M. I. Halawa, F. Wu, T. H. Fereja, B. Lou and G. Xu, Sens. Actuators, B, 2018, 254, 1017 CrossRef CAS; (f) Y.-S. He, C.-G. Pan, H.-X. Cao, M.-Z. Yue, L. Wang and G.-X. Liang, Sens. Actuators, B, 2018, 265, 371 CrossRef CAS.
- (a) G. Nickerl, M. Leistner, S. Helten, V. Bon, I. Senkovska and S. Kaskel, Inorg. Chem. Front., 2014, 1, 325 RSC; (b) M. Kuroboshi, T. Kondo and H. Tanaka, Heterocycles, 2015, 90, 723 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary graphics, Fig. S1–S13. See DOI: 10.1039/c9sc00032a |
| This journal is © The Royal Society of Chemistry 2019 |