
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect grafting of tetraaniline via perfluorophenylazide photochemistry to create antifouling, low bio-adhesion surfaces†
Cheng-Wei
Lin‡
 a,
Stephanie
Aguilar‡
a,
Ethan
Rao
ab,
Wai H.
Mak
a,
Xinwei
Huang
a,
Na
He
ab,
Dayong
Chen
a,
Dukwoo
Jun
c,
Paige A.
Curson
a,
Brian T.
McVerry
ab,
Eric M. V.
Hoek
d,
Shu-Chuan
Huang
*e and
Richard B.
Kaner
*af
a,
Stephanie
Aguilar‡
a,
Ethan
Rao
ab,
Wai H.
Mak
a,
Xinwei
Huang
a,
Na
He
ab,
Dayong
Chen
a,
Dukwoo
Jun
c,
Paige A.
Curson
a,
Brian T.
McVerry
ab,
Eric M. V.
Hoek
d,
Shu-Chuan
Huang
*e and
Richard B.
Kaner
*af
aDepartment of Chemistry and Biochemistry and California NanoSystems Institute, University of California, Los Angeles, Los Angeles, California 90095, USA
bHydrophilix, Inc., 12100 Wilshire Blvd, Suite 800, Los Angeles, CA 90025, USA
cGreen Technology Center, Jung-gu, Seoul, 04554, Republic of Korea
dDepartment of Civil and Environmental Engineering, University of California, Los Angeles, Los Angeles, California 90095, USA
eDepartment of Chemistry, National Dong Hwa University, Shoufeng, Hualien 97401, Taiwan. E-mail: schuang@gms.ndhu.edu.tw
fDepartment of Materials Science and Engineering and California NanoSystems Institute, University of California, Los Angeles, Los Angeles, California 90095, USA. E-mail: kaner@chem.ucla.edu
First published on 13th March 2019
Abstract
Conjugated polyaniline has shown anticorrosive, hydrophilic, antibacterial, pH-responsive, and pseudocapacitive properties making it of interest in many fields. However, in situ grafting of polyaniline without harsh chemical treatments is challenging. In this study, we report a simple, fast, and non-destructive surface modification method for grafting tetraaniline (TANI), the smallest conjugated repeat unit of polyaniline, onto several materials via perfluorophenylazide photochemistry. The new materials are characterized by nuclear magnetic resonance (NMR) and electrospray ionization (ESI) mass spectroscopy. TANI is shown to be covalently bonded to important carbon materials including graphite, carbon nanotubes (CNTs), and reduced graphene oxide (rGO), as confirmed by transmission electron microscopy (TEM). Furthermore, large area modifications on polyethylene terephthalate (PET) films through dip-coating or spray-coating demonstrate the potential applicability in biomedical applications where high transparency, patternability, and low bio-adhesion are needed. Another important application is preventing biofouling in membranes for water purification. Here we report the first oligoaniline grafted water filtration membranes by modifying commercially available polyethersulfone (PES) ultrafiltration (UF) membranes. The modified membranes are hydrophilic as demonstrated by captive bubble experiments and exhibit extraordinarily low bovine serum albumin (BSA) and Escherichia coli adhesions. Superior membrane performance in terms of flux, BSA rejection and flux recovery after biofouling are demonstrated using a cross-flow system and dead-end cells, showing excellent fouling resistance produced by the in situ modification.
Introduction
Polyaniline, a well-studied conjugated polymer known for its simple acid/base doping/de-doping chemistry and facile synthesis,1–5 has been widely applied to chemical gas sensors for ammonia detection,4,6 chemical and electrochemical actuators,7,8 non-volatile memory devices,9 supercapacitors,10 water filtration membranes,11 anticorrosive coatings,12 tissue engineering,13 among other uses.14,15 In the realm of coatings,16,17 and membranes,11,18–21 researchers often blend polyaniline with other matrix materials in order to obtain the hydrophilic and antibacterial characteristics of polyaniline at the surface.22–26 However, low solubility and gelation of polyaniline during processing, have hindered the development of improved materials.27,28 Therefore, grafting polyaniline onto other materials is crucial in order to achieve more robust coatings and functionalize surfaces for enhanced performance.29,30 Due to difficulties in processing polyaniline, chemically grafting polyaniline is often performed by functionalizing end groups with amines,31–33 amidation reactions with carboxylic groups,34–37 diazotization reactions with diazonium salts,38–40 and nitrogen doping41 followed by oxidative polymerization. However, such modification processes require chemically inert and mechanically strong materials in order to sustain the harsh pretreatments. In addition, the abovementioned methods are only suitable for materials within the micro- and nanoscale regime, which limit the feasibility of modifying large surfaces with such hydrophilic, pH-responsive, and antibacterial polyaniline.Herein, we report a newly synthesized molecule, 4-azidotetrafluorobenzoyl tetraaniline (ATFB-TANI), which can be used to couple tetraaniline (TANI) to other materials. TANI, the smallest representative repeat unit of polyaniline, is known to have similar properties to polyaniline, but can be readily dissolved in common organic solvents for facile processing.42–44 This bridging group is based on the solid foundation of perfluorophenylazide photochemistry,30 which can undergo addition and insertion reactions when exposed to UV light.45–49 Thus, with the assistance of perfluorophenylazide, TANI can now be covalently photografted onto carbon nanotubes (CNTs) and reduced graphene oxide (rGO) without pretreatments. Furthermore, large area modifications through dip-coating and spray-coating on polyethylene terephthalate (PET) films with high transparencies and low bio-adhesions show great potential to apply this material for biomedical uses.
Another application that could greatly benefit from a coating that prevents bio-fouling is ultrafiltration (UF) membranes. Typical commercial UF membranes are made of polyethersulfone (PES), polyvinylidene fluoride (PVDF), polysulfone (PSF), polypropylene (PP), or polyurethane (PU).50,51 The hydrophobic nature of these matrix polymers leads to protein adherence to their surfaces and the proliferation of bacteria.52,53 Additionally, the accumulation of both organic and inorganic foulants on membrane surfaces over time, i.e. fouling, can severely lower the permeation flux and the filtration efficiency, often damaging the membranes.54–58 In order to surmount these problems, scientists and engineers have already developed a couple rule-of-thumbs for designing antifouling membranes. First, hydrophilic membrane surfaces have been suggested as a method to form a few-molecule-thick hydration nanolayer, which may prevent foulant adhesions and cake formation. Second, stimuli-responsive polymers grafted onto membrane surfaces can undergo a coil-to-globule transition to “release” the accumulated foulants when exposed to a stimulus during washing cycles.50,51,59,60
To date, all the reported polyaniline-based filtration membranes are formed by blending polyaniline with base polymers. For instance, in 2008, Fan et al. reported a UF membrane made by blending polyaniline with PSF, doubling the pure water flux compared to a pristine PSF membrane and lessening the flux decline.11,18 Since then, other researchers have reported UF membranes composed of polyaniline blended with base polymer, including PES, PSF, PVDF, etc.21,61–65 Blending-in polyaniline enhances the membrane's hydrophilicity, thereby decreasing the flux decline and improving the flux recovery. Even more hydrophilic membranes were reported by further mixing CNTs into polyaniline-blended UF membranes; however, bovine serum albumin (BSA) rejection decreased due to the formation of larger porosity in these membranes.66–68 McVerry et al. and Zhao et al. incorporated self-doped sulfonated polyaniline (SPANI) into PSF and PVDF UF membranes, respectively, with superior hydrophilicity. The zwitterionic nature of SPANI resulted in a high flux recovery and high BSA rejection rate.20,69
The blending strategy mentioned above is straightforward but has several limitations: (1) the processability of polyaniline during membrane casting needs to be improved as polyaniline only appears dispersible in dimethyl sulfoxide (DMSO) and N-methyl-2-pyrrolidone (NMP). (2) The crucial pore size of the composite membranes that directly affects the water flux and rejection rate has to be tuned experimentally by trial and error. (3) The mechanical properties of the resultant composite membranes are strongly dependent on the intrinsic properties of polyaniline and the percentage blended in. As polyaniline possesses a more rigid backbone,70,71 the flexibility of its membranes are limited. (4) The confined polyaniline chains embedded in composite membranes cannot undergo a coil-to-globule transition when treated with acids in order to actively repel foulants. In order to graft polyaniline onto membranes or attach bridging groups to polyaniline through chemical reactions, dissolving polyaniline in DMSO or NMP is inevitable. However, DMSO and NMP are strong organic solvents that can readily dissolve and destroy the base UF membranes during the modification process. Hence, grafting polyaniline onto membrane surfaces without damaging the base UF membranes has so far been unsuccessful.
Here we report the first oligoaniline modified filtration membranes via photografting ATFB-TANI onto commercially available PES UF membranes. The modified membrane surfaces show enhanced hydrophilicity and improved performance in terms of antifouling ability, BSA rejection, and preventing bio-adhesions. This fast, facile, and non-destructive modification process offers a new route to graft conducting oligoanilines and can serve as the initiator for further polymerization as well.
Results and discussions
In order to graft TANI onto large scale membrane surfaces without ruining the base polymer membranes, perfluorophenylazide was applied due to its fast and non-destructive photochemistry. As shown in Fig. 1a, TANI and 4-azidotetrafluorobenzoic acid (ATFB) were stirred under basic conditions for 48 hours in order to complete the coupling reaction. Upon light activation, the phenylazides on ATFB-TANI undergo decomposition of nitrogen (N2) to form singlet phenylnitrenes, and the highly reactive phenylnitrene radicals can undergo C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C addition and C–H and N–H insertion reactions.72 The synthesized ATFB-TANI complex was confirmed by the peak shifts observed in 19F-NMR (Fig. 1c) when compared to the ATFB starting molecule (Fig. 1b), along with the electrospray ionization (ESI) spectrum with a mass observed within 0.7 ppm of the expected mass (Fig. 1d). As a small molecule, ATFB-TANI can be dissolved in a wide range of organic solvents, including toluene, tetrahydrofuran, chloroform, acetone, ethanol, acetonitrile, methanol, and NMP, as shown in Fig. 1e with the color differences being attributed to solvatochromism.42,73 The relatively high solubility of ATFB-TANI in common organic solvents compared to polyaniline indicates its advantageous processability. In this report, we chose ethanol as the solvent to carry out modifications, mainly due to its low toxicity, high solubility toward ATFB-TANI, low solubility toward polyethersulfone membranes, and high miscibility with water.
C addition and C–H and N–H insertion reactions.72 The synthesized ATFB-TANI complex was confirmed by the peak shifts observed in 19F-NMR (Fig. 1c) when compared to the ATFB starting molecule (Fig. 1b), along with the electrospray ionization (ESI) spectrum with a mass observed within 0.7 ppm of the expected mass (Fig. 1d). As a small molecule, ATFB-TANI can be dissolved in a wide range of organic solvents, including toluene, tetrahydrofuran, chloroform, acetone, ethanol, acetonitrile, methanol, and NMP, as shown in Fig. 1e with the color differences being attributed to solvatochromism.42,73 The relatively high solubility of ATFB-TANI in common organic solvents compared to polyaniline indicates its advantageous processability. In this report, we chose ethanol as the solvent to carry out modifications, mainly due to its low toxicity, high solubility toward ATFB-TANI, low solubility toward polyethersulfone membranes, and high miscibility with water.
 | ||
| Fig. 1 The synthesis procedure and characterizations of 4-azidotetrafluorobenzoyl-tetraaniline (ATFB-TANI). (a) ATFB-TANI was synthesized by coupling tetraaniline (TANI) with 4-azidotetrafluorobenzoic acid (ATFB). The ATFB-TANI can be covalently grafted onto the substrates by utilizing azide photochemistry. The 19F-NMR spectra of (b) ATFB and (c) ATFB-TANI. (d) Electrospray ionization (ESI) spectrum showing the molar mass of ATFB-TANI is 0.7 ppm away from the calculated value. (e) Photo showing ATFB-TANI can be dissolved in common organic solvents. | ||
Here we demonstrate that facile surface modification using ATFB-TANI can be applied to several important materials, which specifically undergo CC addition reactions, creating covalent bonds with phenylnitrenes after UV light exposure. In Fig. 2a and b, a lab grade Petri dish made of polystyrene becomes more hydrophilic after modification as indicated by water droplets spreading out and a very faint blue color. The water droplet contact angle as measured by a goniometer decreased from 63° to 42° (Fig. 2c and d). Analogously, a modified compressed graphite pellet also exhibits a decreased contact angle (Fig. 2f and g) indicating increased hydrophilicity (Fig. 2e). Furthermore, the ATFB-TANI molecule provides a simple way to graft TANI onto carbon nanotubes (CNTs) (Fig. 2h) and reduced graphene oxide (rGO) (Fig. 2k). Conventionally, grafting polyaniline onto CNTs or GO/rGO requires surface functional groups via harsh chemical pre-treatments in order to obtain active sites for chemical grafting or polymerization.35,41,74 Here, conducting TANI is successfully chemically grafted onto these materials via a non-destructive photochemical method (Fig. 2i, j and l).
 | ||
| Fig. 2 Images of water droplets on (a) an unmodified and (b) a modified polystyrene-based Petri dish. The contact angles of (c) unmodified and (d) modified Petri dish are 63° and 42°, respectively. The contact angle of (e) a compressed graphite pellet decreases from (f) 60° to (g) 35° after modification. The transmission electron microscopy (TEM) bright field images of (h) unmodified, (i and j) modified multi-walled carbon nanotubes (MWCNTs) and (k) unmodified and (l) modified reduced graphene oxide (rGO). | ||
Since ATFB-TANI can dissolve in common organic solvents, large-scale modifications are possible. Fig. 3a shows polyethylene terephthalate (PET) films subjected to an increasing number of dip-coatings. The coating process was performed by repeated dip-coating, UV light exposure, and ethanol washing. Both undoped (EB) and doped (ES) PET films show a gradual decrease in the transparency as the number of coating layers is increased. The percent transmittance of both EB and ES PET films is around 95% for a monolayer, and about 80% after 15 layers of modification (Fig. 3b and c). The sheet resistances of the doped conducting PET films decrease from around 1011 Ω sq−1 (Fig. S1†) to 109 Ω sq−1 (Fig. 3d). The high sheet resistances may be explained by the rather sparse grafting density, and short chains of TANI that do not greatly enhance carrier hopping.75 The modified films show similar sheet resistances, implying that the ATFB-TANI molecules grafted onto previous layers may react with those on pre-modified molecules, resulting in the loss of some conjugation for the pre-existing layers.
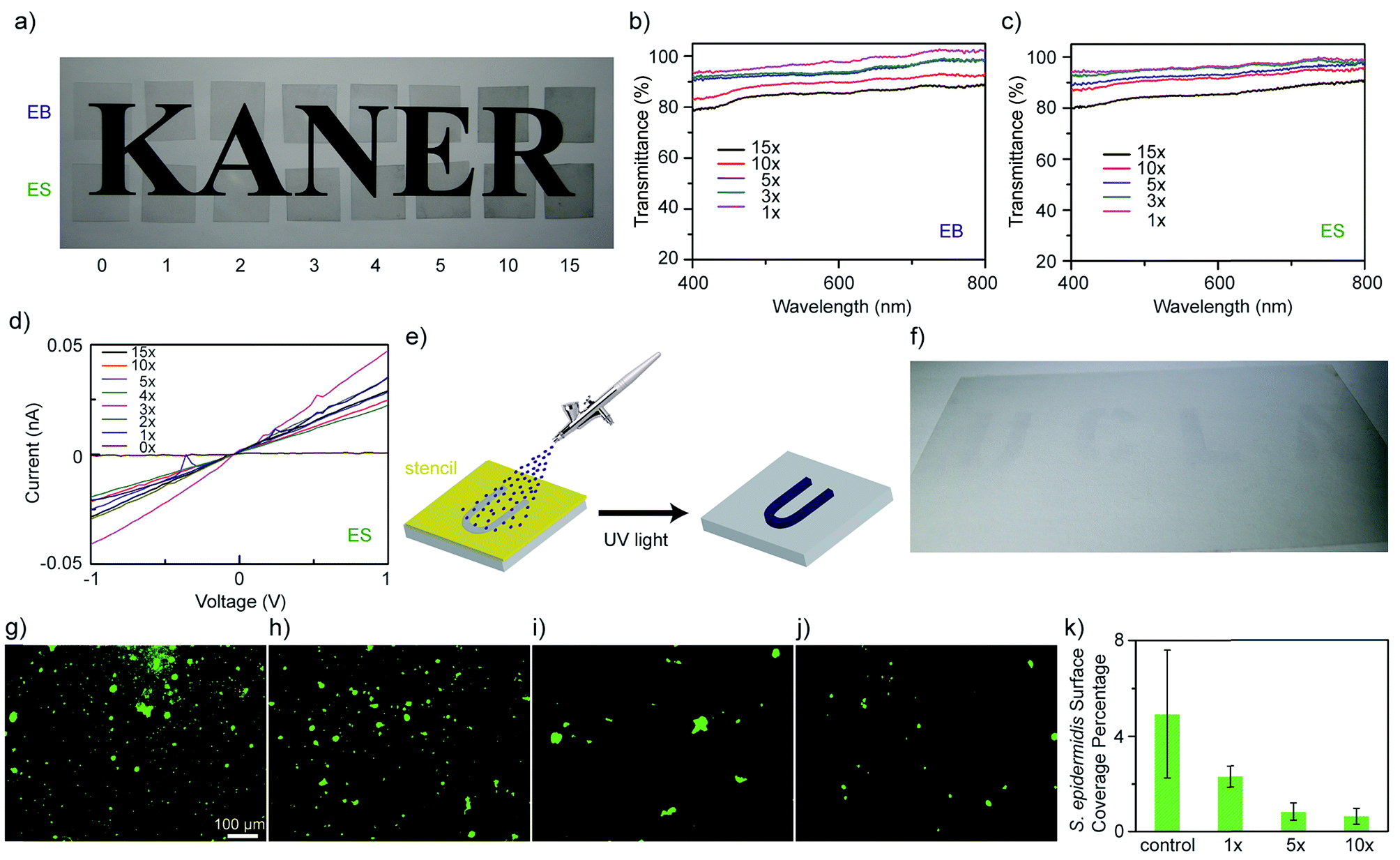 | ||
| Fig. 3 (a) Undoped (EB) and HCl doped (ES) polyethylene terephthalate (PET) films with different numbers of modifications showing their transparencies. UV-vis spectra show the transmittances of (b) undoped and (c) doped PET films with a pristine PET film as the reference. (d) The measured I–V curves of a pristine PET film and modified PET films after doping. (e) A schematic showing (f) a UCLA pattern by spray-coating ATFB-TANI solutions on top of a 13.5 cm × 8.0 cm PET film through stencil masks, followed by UV light exposure. Microscopic images showing the surface coverage of Staphylococcus epidermidis on the (g) unmodified, (h) 1, (i) 5, and (j) 10 times modified PET films, along with (k) a statistics bar graph ((g–j) are under the same magnification). | ||
Spray-coating combined with stencil masks is a method often used for large scale patterning. The schematic in Fig. 3e demonstrates how ATFB-TANI molecules dissolved in an ethanol solution can be airbrushed onto PET films using stencil masks to form a “UCLA” pattern. This achieves a highly transparent UCLA patterned coating after exposure to UV light and an ethanol rinse, as shown in Fig. 3f. Such a facile, fast, and non-destructive modification with ATFB-TANI to make highly transparent coatings could prove useful for biomedical applications such as artificial skin. The idea is to prevent common infections such as those caused by Staphylococcus epidermidis (S. epidermidis).76–78 Adhesion tests on both modified and unmodified PET films were carried out by exposure to S. epidermidis. The observed images with stained cells on unmodified (Fig. 3g), and one, five, and ten-fold modified (Fig. 3h–j) PET films show a readily noticeable drop in the number of cells adhered (Fig. S2†). A statistical bar graph reveals that the surface coverage percentage of S. epidermidis drops more than 50% from the unmodified to the more modified films (Fig. 3k). Note that the surface coverage of S. epidermidis for five-fold modified PET films is significantly lower than the one-fold ones, but similar with the ten-fold ones, indicating that the surface modification can be effective with no more than five times of treatment.
Next, we grafted TANI onto polyethersulfone (PES) UF membranes. The unmodified UF PES membranes appear white, while the modified membranes exhibit a lightblue color even after rinsing with DI water and ethanol (Fig. 4a). Unlike the modification of colored materials, one can directly observe the modification of these initially transparent membranes as they develop a blue hue. The colored modification also makes any surface imperfections on the membrane readily visible (Fig. 4b). Similar to aniline oligomers and polyaniline, ATFB-TANI can undergo a doping/de-doping process by treating with acids/bases. During the doping process, the imine nitrogens in the blue emeraldine base form become protonated, forming the green emeraldine salt form with the positively charged backbone surrounded by counter-anions to balance the charge (Fig. 4c).79 The modified membrane can be reversibly protonated with acid and deprotonated with base, switching from the blue emeraldine base form to the green emeraldine salt form (Fig. 4d). Note that the color change of the modified membranes takes a slightly longer time, about 3 to 5 seconds more, than protonating/deprotonating in solution. The reason is simply due to the slower diffusion rate of the dopants in solids when compared to liquids. Fig. 4e shows the UV-vis spectra of both the blue emeraldine base (EB) and the green emeraldine salt (ES) forms of ATFB-TANI dissolved in DMSO. Similar to TANI, the broad peak at ∼600 nm, i.e. the blue color, can be attributed to the benzenoid to quinoid excitation transition, while the sharper peak for emeraldine salt at ∼440 nm can be attributed to the allowed optical transition from the highest occupied molecular orbital (HOMO) to the higher bipolaron state.42,80–82
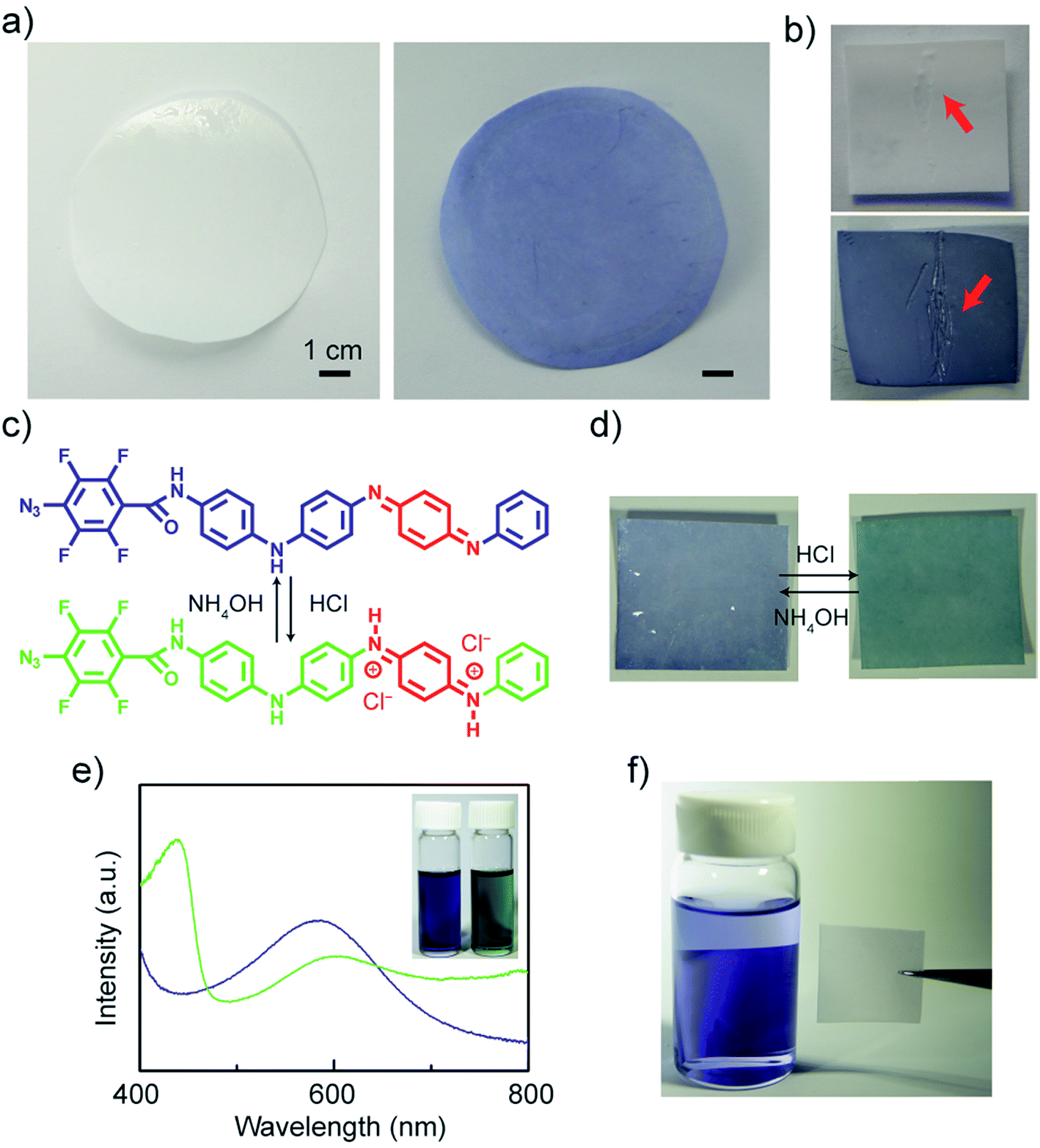 | ||
| Fig. 4 (a) Photographs of unmodified (left) and modified (right) polyethersulfone (PES) membranes. (b) Scratches are easily seen on the modified membrane (bottom) compared to the unmodified one (top). The (c) ATFB-TANI molecule and (d) the modified membrane can be protonated and deprotonated when treated with acids and bases. (e) The UV-vis spectra and photo (inset) of undoped (blue) and doped (green) ATFB-TANI dissolved in dimethyl sulfoxide (DMSO). (f) UV light treated TANI stained membranes appeared colorless after rinsing in a vial of ethanol. | ||
To confirm the chemical grafting of ATFB-TANI onto the PES membrane surfaces, we performed the same modification procedure with TANI instead of ATFB-TANI in an ethanol solution. Without the assistance of azides, the TANI modified membrane after the treatment of UV light maintained its unaltered white color after rinsing with ethanol (Fig. 4f). In contrast, the ATFB-TANI modified PES membrane remained blue after rinsing with ethanol, indicating that the blue color of ATFB-TANI modified membranes is not stained, and the ATFB-TANI is successfully grafted on by photochemistry. The degree of grafting (DG) of ATFB-TANI onto a membrane is 9.74 ± 1.30 μg cm−2, which is comparable to previous reports for the degree of grafting for other polymers (200 < DG < 1000 μg cm−2), in terms of the number of moles per surface area.83–85 Fig. S3† shows a modified membrane soaked in water for nearly one year, the blue color indicates the excellent stability of the grafting. Attempting a more careful examination using attenuated total reflectance infrared (ATR-IR) spectroscopy did not show the difference between the unmodified and modified membranes (Fig. 5a), likely due to the small amount of ATFB-TANI attached. Hence, a more sensitive and precise surface technique, such as X-ray photoelectron spectroscopy (XPS), was needed. The XPS spectra, as shown in Fig. 5b, cannot clearly differentiate the azide signal at 406.5 eV due to the existence of a high nitrogen signal from the commercially available unmodified membranes. However, the fluorine peak at 687.6 eV is evident, revealing that a small amount of ATFB-TANI has indeed been grafted onto the membrane.86–88
 | ||
| Fig. 5 (a) ATR-IR spectra of both modified and unmodified PES membranes. (b) X-ray photoelectron spectroscopy (XPS) spectra and F 1s spectrum (inset) of the modified and unmodified membrane surfaces. (c) Captive bubble images under deionized water, and (d) contact angles under different pH aqueous solutions of modified and unmodified membranes. | ||
Due to the high-water permeability of these PES membranes, their hydrophilicity was examined using a captive bubble contact angle goniometer. A previous study showed that TANI decorated polymeric films possess lower contact angles.89 The interfacial energy of TANI/water is comparatively smaller than PES/water, resulting in more rounded air bubbles, i.e. lower contact angles, for modified membranes (Fig. 5c). Although the interfacial energy of modified membranes is expected to change when exposed to acids because of the protonation of the imine nitrogens, the contact angles observed were consistently around 30 degrees different between unmodified and modified membranes at various pH values, as can be seen in Fig. 5d. This phenomenon can be explained by the insignificant differences in interfacial energy due to the very small amount of surface modification. The relatively more hydrophilic nature of the modified surfaces at different pH values indicates that these membranes are capable of being operated under harsh conditions. Polyaniline films may also possess increased roughness which can affect the contact angle measurement;90 TANI, as a small molecule, slightly increases the surface roughness, from 2.72 nm to 3.49 nm for the unmodified and modified membranes, respectively (Fig. S4†).
To evaluate their antifouling properties, the PES membranes were subjected to a 1.0 g L−1 of bovine serum albumin (BSA) solution with a pressure of 50 psi. The membranes were first compacted with deionized (DI) water until the permeation flux reached equilibrium. After exposure to the 1.0 g L−1 BSA feed solution, the permeation flux decreased dramatically due to the formation of a cake layer during the fouling process.51 As shown in Fig. 6a, the modified membrane exhibited a smaller flux decline after 45 minutes of BSA fouling compared to the unmodified one. The reason may be the more hydrophilic surfaces created after modification; however, the improvement is not significant likely due to the high pressure (50 psi) applied. As mentioned above, the hydrophilic surfaces of the modified membranes form a few-nanometer-thick hydration layer that prevents the adhesion of foulants directly onto the membrane surfaces. Thus, the foulant BSA will experience much lower adhesion forces from the modified membranes. Hence, the modified membranes were tested with cycles of fouling and water washes. The water wash should remove much of the accumulated BSA from the more hydrophilic membrane surfaces due to lower interaction forces and thus give a higher recovery rate. In Fig. 6b, the modified membranes were repeatedly water washed for 5 minutes after 45 minutes of BSA fouling. Note that the water wash duration on the graph shows about 10 minutes because of the extra time spent on cleaning and filling the water tank. The flux recovery percentages were found to be 73.30 ± 2.41%, 60.77 ± 2.41%, and 56.59 ± 0.15% for the first, second, and third fouling experiments, respectively, carried out on the modified membranes. Note that the average flux recovery of the modified membranes after three times fouling (56.59 ± 0.15%), is still comparable to the first flux recovery of the unmodified membranes (56.35 ± 6.45%) and other polyaniline blended membranes.91–94 The flux recovery percentages decrease with each cycle due to the irreversible fouling that occurrs from the BSA, preventing a full recovery on both the modified and unmodified membranes.
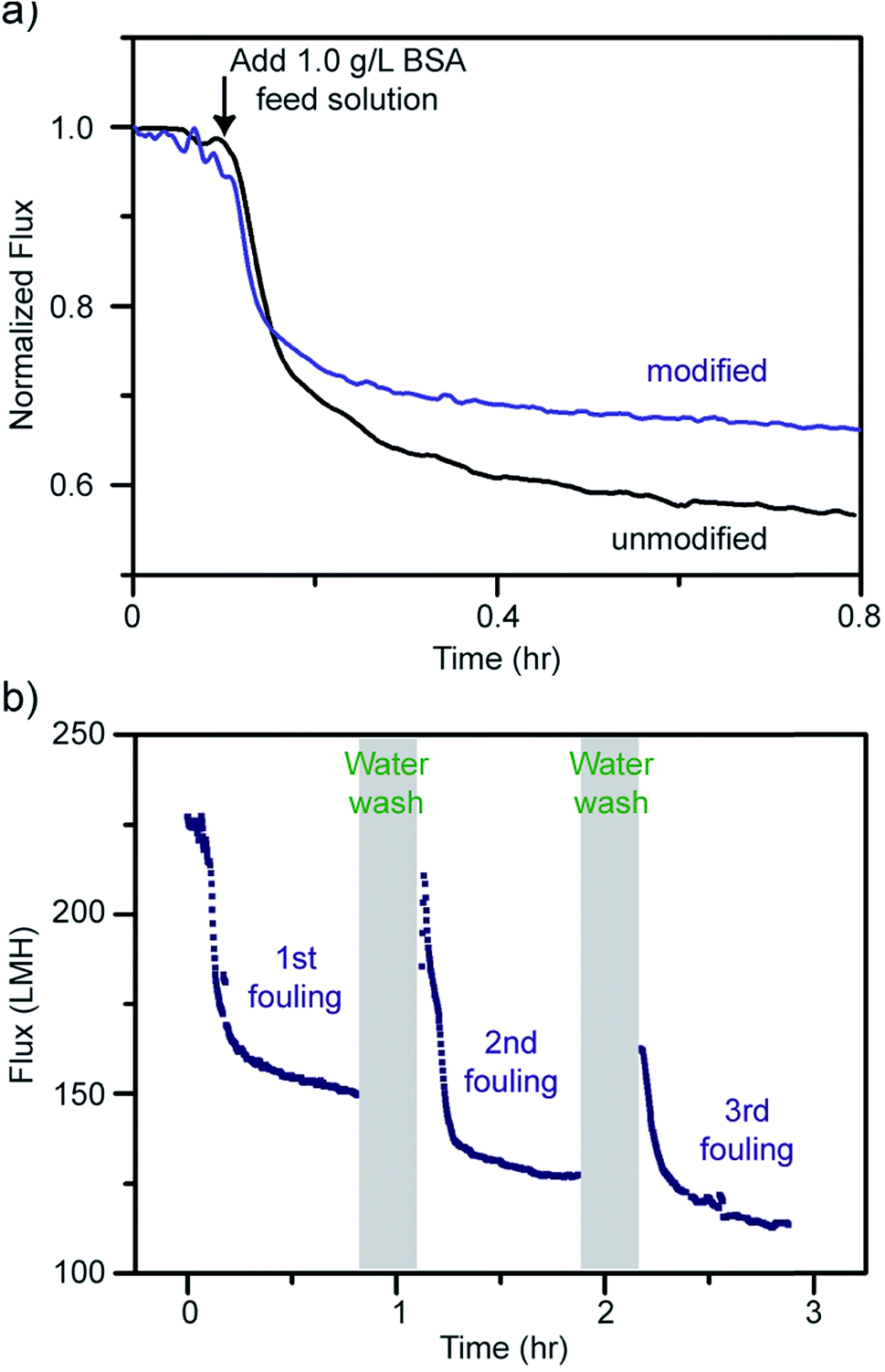 | ||
| Fig. 6 (a) Flux decline for unmodified and modified PES membranes after adding bovine serum albumin (BSA). (b) Flux declines and recoveries of a modified membrane after three cycles of fouling and water washing. | ||
In addition, the modified membranes possess a better BSA rejection rate of 97.53 ± 1.12%, comparable to polyaniline composite UF membranes,11,18,21,94,95 while the unmodified membranes possess a rejection rate of only 91.66 ± 0.85%. Furthermore, the modified membranes have slightly higher water permeation flux compared to the unmodified membranes.91 This counter-intuitive increase in the permeation flux is likely due to slight swelling of the PES membranes induced by their immersion in ethanol during the modification process.96 The performances of both unmodified and modified membranes are summarized in Table 1.
| Membrane | Pure water permeability at 50 psi (LMH) | BSA rejection (%) | Flux decline (%) | Flux recovery (%) | Contact angle at pH 7 (degree) | Root-mean-square roughness (nm) |
|---|---|---|---|---|---|---|
| Unmodified | 181 ± 21.0 | 91.06 ± 0.85 | 46.85 ± 5.22 | 56.35 ± 6.45 | 76.3 ± 1.6 | 2.72 ± 0.30 |
| Modified | 227 ± 4.9 | 97.53 ± 1.12 | 40.70 ± 8.95 | 73.30 ± 2.41 | 49.8 ± 0.4 | 3.49 ± 0.47 |
Conjugated aniline oligomers can be easily protonated and deprotonated under different pH conditions. We performed fouling on modified membranes, followed by washing with water at different pH values. However, washing at different pHs did not give superior results to washing at neutral pH, indicating that the doping/de-doping of TANI did not effectively expel BSA during the wash cycle.97 Based on the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory,98 polyaniline nanofibers are able to form stable colloids within a pH range from 2.2 to 3.5, while forming precipitates at pH values less than 1.5 and greater than ∼5.99 The pH responsive polyaniline nanofibers swell from a coiled state to a globule state when exposed to an appropriate pH environment. However, TANI only forms a stable dispersion in a solution of pH = 1 (zeta potential equal to 14.83 mV), as shown in Fig. 7a (15 hours standing), and it has been observed to last for at least 6 months. The moderately destabilized TANI under pH = 0.5 and more stabilized dispersion under pH = 2 after adding 0.1 M NaCl indicate that the TANI has a very narrow pH range for being stabilized in suspension (Fig. 7b). Therefore, for future research, in order for the grafted TANI molecules to be able to repel foulants merely via changes of solution pH, knowing the precise pH of the solution and the isoelectric point for the chosen foulants will be crucial.
 | ||
| Fig. 7 (a) Optical images of TANI dispersed in aqueous solutions (2 mg mL−1) at different pH and (b) with 0.1 M NaCl added after standing for 15 hours. | ||
Another key indicator for evaluating membranes is the adhesion of microorganisms and the proliferation of bacteria on the membrane surfaces. As such, we performed experiments in which both unmodified and modified PES membranes were soaked in BSA solutions, and E. coli cultured in Luria–Bertani broth, followed by observing the surface coverage of BSA and E. coli adherence under a fluorescent microscope. Intermediately modified membranes (designated 1.0 mM modified) were also used in these tests in order to examine the effects of the ATFB-TANI attachment on the UF membranes. In Fig. 8a, it can be seen that the modified membranes show significantly lower BSA adhesion compared to the unmodified membranes. The quantified BSA surface coverage is 4.10 ± 0.47%, 0.148 ± 0.077% and 0.033 ± 0.025% for the unmodified, 1.0 mM and 2.0 mM modified membranes, respectively (Fig. 8b). The extremely low adhesions are believed to be due to the hydration layer created at the surface of the hydrophilic ATFB-TANI modified membranes. The low BSA adhesion also provides an explanation for the higher flux recovery and BSA rejection observed with the modified membranes. Note that we used pH neutral water for both membrane fouling tests and BSA adhesion testing, so the TANI is not positively charged enough to have a strong interaction with negatively charged BSA. Severe flux decline and very low flux recovery were observed when using hydrochloric acid (pH = 1, 2) for membrane fouling testings.97 On the other hand, although polyaniline has been reported as an antibacterial material,22,65 we could not find any membrane filtration related studies with E. coli. As E. coli is known to have stronger interactions with hydrophobic materials,100 we expected that the hydrophilic modification would prevent E. coli adhesion. As can be seen in Fig. 8d, the live E. coli surface coverage for unmodified membranes is 7.66 ± 2.12%, which drops dramatically for the 1.0 mM and 2.0 mM modified membranes to 0.242 ± 0.216% and 0.057 ± 0.058%, respectively. The dead E. coli surface coverage was found to be 5.44 ± 2.04%, 0.396 ± 0.275%, and 0.187 ± 0.053%, respectively. While these results do not suggest that TANI possesses antibacterial properties, they do indicate that only a small amount of modification is needed to dramatically lessen the adherence of E. coli by up to two orders of magnitude.
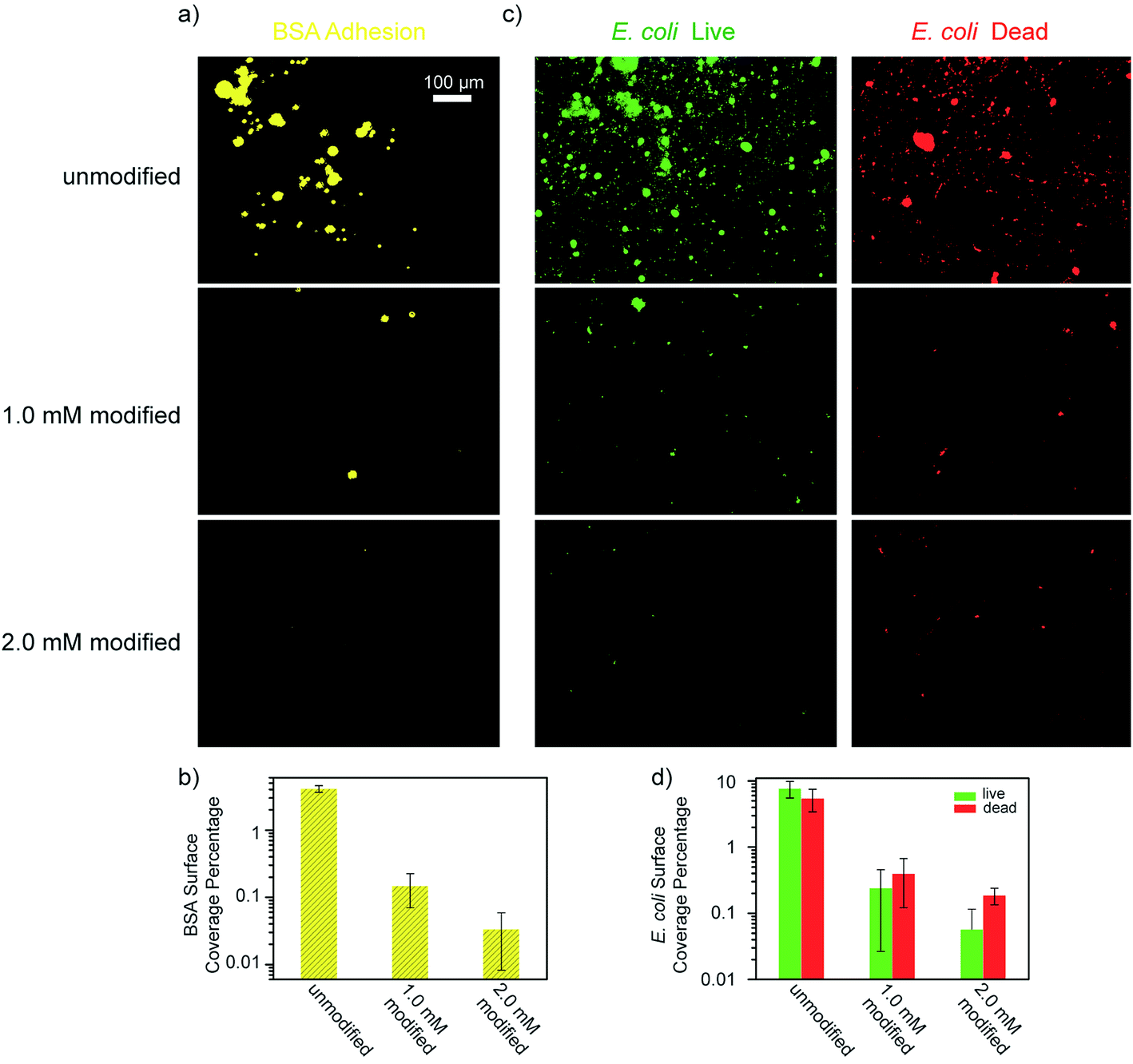 | ||
| Fig. 8 (a) BSA and (c) E. coli adhesion test microscopic images and (b and d) their surface coverage percentages of unmodified, 1.0 mM and 2.0 mM modified PES membranes. (All images share the same scale bar.) | ||
Conclusions
By synthesizing a novel ATFB-TANI molecule with a UV reactive azide group, a facile method for chemically grafting conjugated TANI onto important materials including graphite, carbon nanotubes, reduced graphite oxide and polymers is now available. Dip-coating or spray-coating with stencil masks leads to highly transparent, patterned, and low S. epidermidis adhesive PET films that show potential for low bio-adhesion coatings. Unlike conventional composite membranes, ATFB-TANI molecules can be grafted onto commercial polysulfone ultrafiltration membranes without complex pre-treatments. The TANI modified membranes exhibit increased hydrophilic surfaces, leading to low flux decline, high flux recovery, a high rate of BSA rejection, and low BSA and E. coli adhesions. The design concept of the ATFB-TANI molecule may inspire other modifications for grafting a variety of conjugated oligo/polymers to create hydrophilic and low bio-adhesion surfaces or to help with other materials suffering from processability issues.Experimental section
Materials
Polyether sulfone (PES) UF membranes were purchased from Synder Filtration (LX, 300 kDa). Aniline dimer, ferric chloride hexahydrate, bovine serum albumin (BSA) (heat shock fraction, pH = 7, ≥98%), sodium azide (NaN3), acetone, sodium hydroxide (NaOH), methanol, triethylamine (NEt3), 4-dimethylaminopyridine (DMAP), dichloromethane (DCM), ammonium hydroxide, and deuterated dimethyl sulfoxide (d6-DMSO) were purchased from Sigma Aldrich. Escherichia coli (E. coli), Luria–Bertani (LB) broth, and S. epidermidis were purchased from ATCC. Graphite powder (SP-1 325) for making graphite pellets was purchased from Bay Carbon Inc. The multi-walled carbon nanotubes (MWCNTs) (outer diameter >50 nm; length = 10–20 μm; purity > 95 wt%) were purchased from Cheap Tubes. Polystyrene Petri dishes were purchased from Fisher Scientific. Polyethylene terephthalate (PET) films (PP2950) were purchased from 3 M. All chemicals were used as received.ATFB-TANI synthesis
Modification
The commercial PES membranes were soaked in DI water, renewed every few hours, for at least two days in order to remove the chemicals added for transportation and storage. First, solutions of 1.0 mM and 2.0 mM ATFB-TANI in ethanol were prepared. The unmodified membranes were quickly rinsed with ethanol in order to remove any residual water that may lead to uneven modifications. The membranes were then dipped into the ATFB-TANI ethanol solution for one minute before irradiation with a handheld UV light (254 nm) for one minute. The UV-treated membranes were rinsed with ethanol and DI water, respectively. The grafted membranes were soaked in DI water for another 24 hours before use. In order to modify the Petri dishes, 0.5 mL of 2.0 mM ATFB-TANI solution was pipetted onto a Petri dish and gently swirled. The Petri dish was exposed to UV light for 90 seconds before rinsing with ethanol several times. The graphite pellets were made by compressing graphite powders with a hydraulic press at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lb; ∼0.2 mL of 2.0 mM ATFB-TANI solution was then drop-cast on top of the pellets followed by UV treatment for one minute, rinsing with ethanol and then DI water. The multi-walled carbon nanotubes (MWCNTs) were dispersed in a 2.0 mM ATFB-TANI ethanol solution (300 μg mL−1). The dispersion was ultrasonicated with a tip-ultrasonication processor (Ultrasonics FS-300N, 20% power) in an ice bath for 10 min. The well-dispersed solution (2.5 mL) was exposed to UV light for 3 minutes, followed by drop-casting onto a glass slide and gentle rinsing with ethanol in order to remove the unreacted ATFB-TANIs. The TEM samples were prepared by tapping TEM grids on the glass slide. The graphene oxide (GO) was synthesized through a modified Hummer's method, as reported elsewhere.102 As described previously,103 a GO aqueous solution was mixed with ascorbic acid and after vacuum filtration, the ascorbic acid was rinsed out and dried in a 100 °C oven overnight. The reduced GO (rGO) film was ultrasonicated with 2.0 mM ATFB-TANI ethanol solution for 1 minute, followed by UV treatment for 3 minutes. The PET films were dipped into ATFB-TANI solutions for 10 seconds and exposed to UV light for another minute, rinsed with ethanol and air dried. The spray-coatings on SWCNT films and the UCLA pattern on the PET film with stencil masks were carried out by spraying 2.0 mM solution with an Image® Dual Action airbrush. The airflow rate was 5 standard cubic feet per minute (SCFH) with approximately 1 mL of solution sprayed out per minute.
000 lb; ∼0.2 mL of 2.0 mM ATFB-TANI solution was then drop-cast on top of the pellets followed by UV treatment for one minute, rinsing with ethanol and then DI water. The multi-walled carbon nanotubes (MWCNTs) were dispersed in a 2.0 mM ATFB-TANI ethanol solution (300 μg mL−1). The dispersion was ultrasonicated with a tip-ultrasonication processor (Ultrasonics FS-300N, 20% power) in an ice bath for 10 min. The well-dispersed solution (2.5 mL) was exposed to UV light for 3 minutes, followed by drop-casting onto a glass slide and gentle rinsing with ethanol in order to remove the unreacted ATFB-TANIs. The TEM samples were prepared by tapping TEM grids on the glass slide. The graphene oxide (GO) was synthesized through a modified Hummer's method, as reported elsewhere.102 As described previously,103 a GO aqueous solution was mixed with ascorbic acid and after vacuum filtration, the ascorbic acid was rinsed out and dried in a 100 °C oven overnight. The reduced GO (rGO) film was ultrasonicated with 2.0 mM ATFB-TANI ethanol solution for 1 minute, followed by UV treatment for 3 minutes. The PET films were dipped into ATFB-TANI solutions for 10 seconds and exposed to UV light for another minute, rinsed with ethanol and air dried. The spray-coatings on SWCNT films and the UCLA pattern on the PET film with stencil masks were carried out by spraying 2.0 mM solution with an Image® Dual Action airbrush. The airflow rate was 5 standard cubic feet per minute (SCFH) with approximately 1 mL of solution sprayed out per minute.
Membrane performance testing
To measure fouling, membranes were placed in a stainless-steel holder. A feed tank was connected to a mechanical pump, which flowed feed solutions across the membrane with an effective area of 17 cm2. A pressure gauge was placed between the pump and the membrane holder to monitor the input pressure. The output after membrane filtration was monitored with a flow meter, and its readings were recorded by a connected computer. The membranes were first compacted with DI water at 50 psi until the flow reached a steady value. The fouling test began by introducing 1.0 g L−1 of BSA solution for 45 minutes. The flow decline percentage was calculated based on the equation:| Flow decline (%) = (1 – Jf/Ji) × 100% |
| Flow recovery (%) = JR/Ji × 100% |
The BSA rejection performance testing on the PES UF membranes was conducted in a stainless-steel dead-end stirred filtration cell (Sterlitech Corp., Kent, WA) with an active membrane area of approximately 110 cm2. The stirred cell was filled with DI water and pressurized. The water flow rate was recorded using a digital flow meter (FlowCal 5000, Tovatech LLC, South Orange, NJ). The membrane was then compacted at 50 psi until the flow rate was stable, approximately 1 hour for each membrane. BSA rejection of each membrane was characterized by filling the stirred cell with 1.0 g L−1 of BSA solution and pressurizing it at 50 psi. The BSA rejection rate was calculated through the equation:
| R = 1 – Ap/Af |
Bacterial and BSA adhesion testing
The antifouling properties of the ATFB-TANI modified PES membranes were investigated by a bacterial adhesion experiment using E. coli as a model organism. E. coli cell cultures were suspended in Luria–Bertani broth for 24 hours at 35 °C. 1 cm2 samples of modified and unmodified membranes were then soaked in the E. coli suspension (∼1 × 107 CFU mL−1) (CFU = colony forming unit) at 37 °C while shaken at 35 rpm for 24 h. The samples were then removed from the suspension and rinsed with a 1 M PBS buffer solution to remove any unbound cells. Membrane samples were then immersed and stained in a SYTO 9 dye solution (live/dead Baclight Bacterial Viability Kit) for 20 minutes. The samples were again rinsed with the 1 M PBS buffer solution. The samples were then immersed in a propidium iodide solution (PI) for 20 minutes and again rinsed with the 1 M PBS solution. SYTO 9 labels nucleic acids of both live and dead cells, whereas PI only labels nucleic acids of dead cells. When both dyes are present, PI exhibits a stronger affinity for nucleic acids than SYTO 9 and therefore SYTO 9 is displaced on the nucleic acids of dead cells. Images were then taken using a fluorescence microscope using different filters to view the SYTO9 and PI dyes. For BSA adhesion tests, the green fluorescence protein (GFP) BSA was dissolved in DI water (concentration equals to 50 μg mL−1), and samples of the membranes were immersed in that solution and placed in an incubator at 37 °C and 50 rpm. After 24 hours, the samples were removed, rinsed with DI water and imaged using a fluorescent microscope. Image analysis was performed using ImageJ. The surface coverage values were quantified by dividing the number of colored pixels by the total number of pixels.Characterization
The synthesized TANI was characterized by a Bruker UltraFlex Matrix Assisted Laser Desorption/Ionization (MALDI-TOF) spectrometer with 2,5-dihydroxybenzoic acid (DHB) as the matrix. NMR spectra were carried out on a Bruker AV300. Electrospray ionization mass spectrometry (ESI-MS) in methanol solvent was utilized to determine the composition of ATFB-TANI by comparison to Leucine Enkephalin. During ESI-MS characterization, a signal for ATFB-TANI (C31H19N7F4O)+ was observed at 583.1748 m/z and had an isotope pattern consistent with C, H, N, and O incorporation. The observed high-resolution ESI-MS for ATFB-TANI differed from the calculated masses by 0.7 ppm. UV-vis spectra were taken on a Shimadzu UV-3101 PC UV-vis-NIR scanning spectrometer with quartz cuvettes. The membrane topographies were investigated using a Bruker Dimension FastScan Probe Microscope (SPM) with silicon tips on nitride levers (Bruker Scanasyst-air) under the Tapping mode. The root-mean-square roughness and the image process were carried out by the software NanoScope Analysis. The conventional contact angles were measured through a First 10 Ångstroms Contact Angle Goniometer. The captive bubble contact angles were measured through a homemade setup where the membranes were clamped on glass substrates. The membrane was then faced down and immersed into a transparent acrylic box. Air bubbles were placed through a U-shape needle connected to a syringe. The transmission electron microscopy (TEM) images were collected on a Tecnai TF20 TEM (FEI Inc.) operated under low dose mode. The electrical performances of PET films were measured via a probe station HP 4155B using toothless alligator clips as the metal contacts. The measured Amperes were corrected to zero at zero voltage. The attenuated total reflectance infrared (ATR-IR) spectra were acquired on a PerkinElmer Spectrum One spectrometer equipped with a universal ATR sampling accessory. The XPS data were acquired using a Kratos Axis Ultra DLD spectrometer equipped with a monochromatic Al Kα X-ray source. The pH values of TANI dispersions (optical images) were calibrated by a pH meter (Mettler Toledo). For measuring the zeta potentials, around 4 mg of aniline tetramer and polyaniline nanofibers, synthesized by interfacial polymerization, were sonicated and vortexed until the powders were well-dispersed. 1.0 M HCl(aq) and 1.0 M NaOH(aq) (PanReact AppliChem) were used to adjust each solution to a total volume of about 15 mL with the desired pH value. The zeta potential values were measured via a Malvern Zetasizer Nano-ZS. For measuring the degree of grafting (DG), membranes were punched into circles with the diameter equal to 0.7 cm, and the weight (Wi) of the dried unmodified membrane measured using a microbalance (Mettler Toledo) recorded. The weights of dried modified membrane were then recorded as Wf. The degree of grafting is defined as:where A stands for the surface area of the membranes tested.
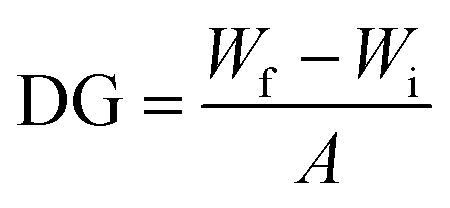
Author contributions
C.-W. L. and S. A. synthesized TANI, ATFB-TANI and measured the cross-flow membrane performances, BSA rejection, contact angles, transmittances, electrical performance and performed modifications. E. R. tested BSA, E. coli and S. epidermidis adhesions and analyzed them with ImageJ. W. H. M. made the AFM and XPS measurements. X. H. conceived the idea and obtained preliminary antifouling experiments and E. coli adhesion tests. N. H. helped measure the membrane fouling before and after modification. D. C. helped design the ATFB-TANI molecule. D. J. built the cross-flow system and the BSA rejection setup. P. C. helped make and modify the membranes. B. T. M. helped modify membranes and guide this work. All authors helped in writing the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Dr Martin Ignacio for help with UV-vis, contact angle, and XPS measurements, Dr Chris Turner for help making the acrylic box for captive bubble measurements, Jacqueline Yang for preliminary E. coli tests, Shuangmei Xue and Chen-Hao Ji for help with BSA rejection measurements, Matthew D. Kowal for making the graphite pellets and synthesizing the graphene oxide, Jakhangirkhodja Tulyagankhodjaev for preliminary contact angle measurements, Georgiy Akopov for acquiring SEM images, and Mackenzie Anderson for help with XPS measurements. The ESI instrumentation is maintained and made available through the support of Dr Gregory Khitrov (University of California, Los Angeles Molecular Instrumentation Center – Mass Spectrometry Facility in the Department of Chemistry). The authors thank Dr Robert S. Jordan for helpful discussions on designing the ATFB-TANI molecule. The authors also thank Prof. Jason Woo and Peng Lu for assistance measuring I–V electrical characteristics of PET films, Jeong Hoon Ko and Prof. Heather D. Maynard for help acquiring infrared spectra, and Alexandra Polasko and Prof. Shaily Mahendra for the bacterial tests. The authors would like to thank Chia-Liang Cheng's lab (Physics, NDHU), Yen-Peng Ho's lab (Chemistry, NDHU), Wenjea T. Tseng's lab (Materials Science and Engineering, NCHU) and Jacky Lin (Malvern in Taipei) for their help in the zeta potential measurements. The authors thank the National Science Foundation CBET Grant 1337065, the USA/China Clean Energy Research Center for Water-Energy Technologies (CERC-WET), and the UC Grand Challenge Program for financial support. C.-W. L. thanks the support from the UCLA Dissertation Year Fellowship. In addition, W. H. M. and S. A. would like to each acknowledge their UCLA Eugene V. Cota-Robles Fellowship and P. C. for Clare Booth Luce Fellowship. R. B. K. thanks the Dr Myung Ki Hong Endowed Chair in Materials Innovation at UCLA. This project was additionally supported by the National Center for Research Resources Grant Number S10RR025631. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.References
- A. J. Heeger, Angew. Chem., Int. Ed., 2001, 40, 2591–2611 CrossRef CAS PubMed.
- A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581–2590 CrossRef CAS PubMed.
- H. Shirakawa, Angew. Chem., Int. Ed., 2000, 40, 2574–2580 CrossRef.
- J. Huang, S. Virji, B. H. Weiller and R. B. Kaner, J. Am. Chem. Soc., 2003, 125, 314–315 CrossRef CAS PubMed.
- J. Huang and R. B. Kaner, J. Am. Chem. Soc., 2004, 126, 851–855 CrossRef CAS PubMed.
- S. Virji, J. Huang, R. B. Kaner and B. H. Weiller, Nano Lett., 2004, 4, 491–496 CrossRef CAS.
- C. O. Baker, B. Shedd, P. C. Innis, P. G. Whitten, G. M. Spinks, G. G. Wallace and R. B. Kaner, Adv. Mater., 2008, 20, 155–158 CrossRef CAS.
- J. Gao, J.-M. Sansinena and H.-L. Wang, Mater. Res. Soc. Symp. Proc., 2002, 698, 17–22 CAS.
- R. J. Tseng, J. Huang, J. Ouyang and R. B. Kaner, Nano Lett., 2005, 5, 1–4 CrossRef PubMed.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- Z. Fan, Z. Wang, M. Duan, J. Wang and S. Wang, J. Membr. Sci., 2008, 310, 402–408 CrossRef CAS.
- Z. Tian, H. Yu, L. Wang, M. Saleem, F. Ren, P. Ren, Y. Chen, R. Sun, Y. Sun and L. Huang, RSC Adv., 2014, 4, 28195–28208 RSC.
- N. K. Guimard, N. Gomez and C. E. Schmidt, Prog. Polym. Sci., 2007, 32, 876–921 CrossRef CAS.
- S. Bhadra, D. Khastgir, N. K. Singha and J. H. Lee, Prog. Polym. Sci., 2009, 34, 783–810 CrossRef CAS.
- D. Li, J. Huang and R. B. Kaner, Acc. Chem. Res., 2009, 42, 135–145 CrossRef CAS PubMed.
- A. Pud, N. Ogurtsov, A. Korzhenko and G. Shapoval, Prog. Polym. Sci., 2003, 28, 1701–1753 CrossRef CAS.
- P. K. Prabhakar, S. Raj, P. R. Anuradha, S. N. Sawant and M. Doble, Colloids Surf., B, 2011, 86, 146–153 CrossRef CAS PubMed.
- Z. Fan, Z. Wang, N. Sun, J. Wang and S. Wang, J. Membr. Sci., 2008, 320, 363–371 CrossRef CAS.
- X. Huang, B. T. McVerry, C. Marambio-Jones, M. C. Y. Wong, E. M. V. Hoek and R. B. Kaner, J. Mater. Chem. A, 2015, 3, 8725–8733 RSC.
- X. Zhao and C. He, ACS Appl. Mater. Interfaces, 2015, 7, 17947–17953 CrossRef CAS PubMed.
- S. Zhao, Z. Wang, J. Wang and S. Wang, Ind. Eng. Chem. Res., 2014, 53, 11468–11477 CrossRef CAS.
- M. R. Gizdavic-Nikolaidis, J. R. Bennett, S. Swift, A. J. Easteal and M. Ambrose, Acta Biomater., 2011, 7, 4204–4209 CrossRef CAS PubMed.
- M. S. Tamboli, M. V Kulkarni, R. H. Patil, W. N. Gade, S. C. Navale and B. B. Kale, Colloids Surf., B, 2012, 92, 35–41 CrossRef CAS PubMed.
- X. Liang, M. Sun, L. Li, R. Qiao, K. Chen, Q. Xiao and F. Xu, Dalton Trans., 2012, 41, 2804–2811 RSC.
- Z. Zhang, Z. Wei and M. Wan, Macromolecules, 2002, 35, 5937–5942 CrossRef CAS.
- D. S. Dhawale, R. R. Salunkhe, V. S. Jamadade, D. P. Dubal, S. M. Pawar and C. D. Lokhande, Curr. Appl. Phys., 2010, 10, 904–909 CrossRef.
- D. Yang and B. R. Mattes, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2702–2713 CrossRef CAS.
- D. Yang and B. R. Mattes, Synth. Met., 1999, 101, 746–749 CrossRef CAS.
- J. J. Orlando, J. B. Burkholder, S. A. McKeen and A. R. Ravishankara, J. Geophys. Res., 1991, 96, 5013–5023 CrossRef CAS.
- P. Roger, L. Renaudie, C. Le Narvor, B. Lepoittevin, L. Bech and M. Brogly, Eur. Polym. J., 2010, 46, 1594–1603 CrossRef CAS.
- J. Liu, J. An, Y. Zhou, Y. Ma, M. Li, M. Yu and S. Li, ACS Appl. Mater. Interfaces, 2012, 4, 2870–2876 CrossRef CAS PubMed.
- L. Lai, H. Yang, L. Wang, B. Teh, J. Zhong, H. Chou, L. Chen, W. Chen, Z. Shen and R. Ruoff, ACS Nano, 2012, 6, 5941–5951 CrossRef CAS PubMed.
- J. An, J. Liu, Y. Zhou, H. Zhao, Y. Ma, M. Li, M. Yu and S. Li, J. Phys. Chem. C, 2012, 116, 19699–19708 CrossRef CAS.
- M. Kotal, A. K. Thakur and A. K. Bhowmick, ACS Appl. Mater. Interfaces, 2013, 5, 8374–8386 CrossRef CAS PubMed.
- N. A. Kumar, H. Choi, Y. R. Shin, D. W. Chang and L. Dai, ACS Nano, 2012, 1715–1723 CrossRef CAS PubMed.
- J. An, J. Liu, Y. Zhou, H. Zhao, Y. Ma, M. Li, M. Yu and S. Li, J. Phys. Chem. C, 2012, 116, 19699–19708 CrossRef CAS.
- Z. F. Li, H. Zhang, Q. Liu, Y. Liu, L. Stanciu and J. Xie, Carbon, 2014, 71, 257–267 CrossRef CAS.
- X. Liu, P. Shang, Y. Zhang, X. Wang, Z. Fan, B. Wang and Y. Zheng, J. Mater. Chem. A, 2014, 2, 15273–15278 RSC.
- V. D. Filimonov, M. Trusova, P. Postnikov, E. A. Krasnokutskaya, Y. M. Lee, H. Y. Hwang, H. Kim and K. W. Chi, Org. Lett., 2008, 10, 3961–3964 CrossRef CAS PubMed.
- H. Ziani-Cherif, K. Imachi and T. Matsuda, Macromolecules, 1999, 32, 3438–3447 CrossRef CAS.
- A. U. Haq, J. Lim, J. M. Yun, W. J. Lee and T. H. Han, Small, 2013, 9, 3829–3833 CrossRef CAS PubMed.
- C. Lin, R. L. Li, S. Robbennolt, M. T. Yeung, G. Akopov and R. B. Kaner, Macromolecules, 2017, 50, 5892–5897 CrossRef CAS.
- Y. Wang, H. D. Tran and R. B. Kaner, Macromol. Rapid Commun., 2011, 32, 35–49 CrossRef CAS PubMed.
- Z. Wei and C. F. J. Faul, Macromol. Rapid Commun., 2008, 29, 280–292 CrossRef CAS.
- J. F. W. Keana and S. X. Cai, J. Org. Chem., 1990, 55, 3640–3647 CrossRef CAS.
- S. J. Pastine, D. Okawa, B. Kessler, M. Rolandi, M. Llorente, A. Zettl and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 4238–4239 CrossRef CAS PubMed.
- L.-H. Liu and M. Yan, Acc. Chem. Res., 2010, 43, 1434–1443 CrossRef CAS PubMed.
- S. Brase, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef CAS PubMed.
- Z. Li, A. Ajami, E. Stankevičius, W. Husinsky, G. Račiukaitis, J. Stampfl, R. Liska and A. Ovsianikov, Opt. Mater., 2013, 35, 1846–1851 CrossRef CAS.
- D. Rana and T. Matsuura, Chem. Rev., 2010, 110, 2448–2471 CrossRef CAS PubMed.
- R. Zhang, Y. Liu, M. He, Y. Su, X. Zhao, M. Elimelech and Z. Jiang, Chem. Soc. Rev., 2016, 45, 5888–5924 RSC.
- J. A. Koehler, M. Ulbricht, G. Belfort and N. York, Langmuir, 2000, 16, 10419–10427 CrossRef CAS.
- A. Fane, A. G. Fell and C. J. D. Suki, J. Membr. Sci., 1983, 16, 195–210 CrossRef CAS.
- F. Meng, S. Chae, A. Drews, M. Kraume and H. Shin, Water Res., 2009, 43, 1489–1512 CrossRef CAS PubMed.
- P. Le-clech, V. Chen and T. A. G. Fane, J. Membr. Sci., 2006, 284, 17–53 CrossRef CAS.
- W. Guo, H. Ngo and J. Li, Bioresour. Technol., 2012, 122, 27–34 CrossRef CAS PubMed.
- A. Drews, J. Membr. Sci., 2010, 363, 1–28 CrossRef CAS.
- K. J. Howe and M. M. Clark, Environ. Sci. Technol., 2002, 36, 3571–3576 CrossRef CAS PubMed.
- K. Dutta and S. De, J. Mater. Chem. A, 2017, 5, 22095–22112 RSC.
- C. M. Magin, S. P. Cooper and A. B. Brennan, Mater. Today, 2010, 13, 36–44 CrossRef CAS.
- S. Zhao, Z. Wang, X. Wei, B. Zhao and J. Wang, J. Membr. Sci., 2011, 385–386, 251–262 CrossRef CAS.
- S. B. Teli, S. Molina, E. G. Calvo, A. E. Lozano and J. de Abajo, Desalination, 2012, 299, 113–122 CrossRef CAS.
- S. Zhu, S. Zhao, Z. Wang, X. Tian and M. Shi, J. Membr. Sci., 2015, 493, 263–274 CrossRef CAS.
- B. Jiang, B. Wang, L. Zhang, Y. Sun, X. Xiao, N. Yang and H. Dou, J. Appl. Polym. Sci., 2017, 44452, 1–7 Search PubMed.
- S. Zhao, L. Huang, T. Tong, W. Zhang, Z. Wang, J. Wang and S. Wang, Environ. Sci.: Water Res. Technol., 2017, 3, 710–719 RSC.
- Y. Liao, D. Yu, X. Wang, W. Chain and X. Li, Nanoscale, 2013, 5, 3856–3862 RSC.
- W. Duan, A. Ronen, S. Walker and D. Jassby, ACS Appl. Mater. Interfaces, 2016, 8, 22574–22584 CrossRef CAS PubMed.
- B. Hudaib, V. Gomes, C. Zhou and Z. Liu, Sep. Purif. Technol., 2018, 190, 143–155 CrossRef CAS.
- B. T. McVerry, J. A. T. Temple, X. Huang, K. L. Marsh, E. M. V Hoek and R. B. Kaner, Chem. Mater., 2013, 25, 3597–3602 CrossRef CAS.
- S. Chen and H. Lee, Macromolecules, 1995, 28, 2858–2866 CrossRef CAS.
- M. Jaymand, Prog. Polym. Sci., 2013, 38, 1287–1306 CrossRef CAS.
- L. H. Liu and M. Yan, Acc. Chem. Res., 2010, 43, 1434–1443 CrossRef CAS PubMed.
- V. W. Liptay, Angew. Chem., 1969, 81, 195–206 CrossRef.
- N. Tsubokawa, Polym. J., 2005, 37, 637–655 CrossRef CAS.
- R. H. Baughman and L. W. Shacklette, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 5872–5886 CrossRef CAS.
- S. Satpathy, S. K. Sen, S. Pattanaik and S. Raut, Biocatal. Agric. Biotechnol., 2016, 7, 56–66 CrossRef.
- C. Vuong and M. Otto, Microbes Infect., 2002, 4, 481–489 CrossRef PubMed.
- M. Otto, Nat. Rev. Microbiol., 2009, 7, 555–567 CrossRef CAS PubMed.
- J. Jin-Chih and A. G. MacDiarmid, Synth. Met., 1986, 13, 193–205 CrossRef.
- J. L. Bredas and G. B. Street, Acc. Chem. Res., 1985, 18, 309–315 CrossRef CAS.
- Y. Cao, S. Li, Z. Xue and D. Guo, Synth. Met., 1986, 16, 305–315 CrossRef.
- A. Epstein, J. Ginder, F. Zuo, R. Bigelow, H. Woo, D. Tanner, A. Richter, W.-S. Huang and A. MacDiarmid, Synth. Met., 1987, 18, 303–309 CrossRef CAS.
- Q. Li, H. H. Lin and X. L. Wang, Membranes, 2014, 4, 181–199 CrossRef.
- T. A. Sergeyeva, H. Matuschewski, S. A. Piletsky, J. Bendig, U. Schedler and M. Ulbricht, J. Chromatogr. A, 2001, 907, 89–99 CrossRef CAS.
- H. M. Ulbricht and A. Matuschewski, J. Membr. Sci., 1996, 115, 31–47 CrossRef.
- B. T. McVerry, M. C. Y. Wong, K. L. Marsh, J. A. T. Temple, C. Marambio-jones, E. M. V Hoek and R. B. Kaner, Macromol. Rapid Commun., 2014, 35, 1528–1533 CrossRef CAS PubMed.
- K. Siegmann, J. Inauen, R. Sterchi and M. Winkler, Surf. Interface Anal., 2018, 50, 205–211 CrossRef CAS.
- K. Siegmann, J. Inauen, D. Villamaina and M. Winkler, Appl. Surf. Sci., 2017, 396, 672–680 CrossRef CAS.
- B. Guo, A. Finne-wistrand and A. Albertsson, Macromolecules, 2012, 45, 352–359 Search PubMed.
- P. Zhou, J. Li, W. Yang, L. Zhu and H. Tang, Langmuir, 2018, 34, 2841–2848 CrossRef CAS PubMed.
- Z. Fan, Z. Wang, N. Sun, J. Wang and S. Wang, J. Membr. Sci., 2008, 320, 363–371 CrossRef CAS.
- S. Zhao, Z. Wang, X. Wei, B. Zhao, J. Wang, S. Yang and S. Wang, J. Membr. Sci., 2011, 385–386, 251–262 CrossRef CAS.
- S. Zhao, Z. Wang, J. Wang and S. Wang, Ind. Eng. Chem. Res., 2014, 53, 11468–11477 CrossRef CAS.
- X.-G. Li, D.-G. Yu, W. Chain, E. M. V. Hoek, Y. Liao, X. Wang and R. B. Kaner, Nanoscale, 2013, 5, 3856–3862 RSC.
- Y. Liao, X. G. Li, E. M. V Hoek and R. B. Kaner, J. Mater. Chem. A, 2013, 1, 15390–15396 RSC.
- B. Kaeselev, P. Kingshottb and G. Jonsson, Desalination, 2002, 146, 265–271 CrossRef CAS.
- L. P. Wang, W. Wang, L. Di, Y. N. Lu and J. Y. Wang, Colloids Surf., B, 2010, 80, 72–78 CrossRef CAS PubMed.
- R. J. Hunter, Foundations of Colloid Science, Oxford University Press, New York, 1987 Search PubMed.
- D. Li and R. B. Kaner, Chem. Commun., 2005, 3286–3288 RSC.
- L. S. Dorobantu, S. Bhattacharjee, J. M. Foght and M. R. Gray, Langmuir, 2009, 25, 6968–6976 CrossRef CAS PubMed.
- Z. Shao, P. Rannou, S. Sadki, N. Fey, D. M. Lindsay and C. F. J. Faul, Chem.–Eur. J., 2011, 17, 12512–12521 CrossRef CAS PubMed.
- R. L. Li, C. W. Lin, Y. Shao, C. W. Chang, F. K. Yao, M. D. Kowal, H. Wang, M. T. Yeung, S. C. Huang and R. B. Kaner, Polymers, 2016, 8, 401–411 CrossRef.
- Y. Shao, M. F. El-Kady, C. W. Lin, G. Zhu, K. L. Marsh, J. Y. Hwang, Q. Zhang, Y. Li, H. Wang and R. B. Kaner, Adv. Mater., 2016, 6719–6726 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04832k |
| ‡ These two authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |