
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge transfer dependence on CO2 hydrogenation activity to methanol in Cu nanoparticles covered with metal–organic framework systems†
Hirokazu
Kobayashi
*ab,
Jared M.
Taylor‡
a,
Yuko
Mitsuka
c,
Naoki
Ogiwara
a,
Tomokazu
Yamamoto
de,
Takaaki
Toriyama
e,
Syo
Matsumura
def and
Hiroshi
Kitagawa
 *afg
*afg
aDivision of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa-Oiwakecho, Sakyo-ku, Kyoto, 606-8502, Japan. E-mail: hkobayashi@kuchem.kyoto-u.ac.jp; kitagawa@kuchem.kyoto-u.ac.jp
bJST, PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
cShoei Chemical Inc., 5-3, Aza-wakazakura Fujinoki-machi, Tosu-shi, Saga 841-0048, Japan
dDepartment of Applied Quantum Physics and Nuclear Engineering, Graduate School of Engineering, Kyushu University, Motooka 744, Nishi-ku, Fukuoka, 819-0395, Japan
eThe Ultramicroscopy Research Center, Kyushu University, Motooka 744, Nishi-ku, Fukuoka, 819-0395, Japan
fInamori Frontier Research Center, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan
gInstitute for Integrated Cell-Material Sciences (iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto, 606-8501, Japan
First published on 6th February 2019
Abstract
We report the synthesis and characterization of highly active Cu nanoparticles covered with zirconium/hafnium-based metal–organic frameworks for CO2 hydrogenation to methanol. Compared to Cu/γ-Al2O3, Cu/ZIF-8, Cu/MIL-100 and Cu/UiO-66 composites, UiO-66 acts as the most active support, with Cu/Zr-UiO-66 producing methanol at a rate 70 times higher than that of Cu/γ-Al2O3. In addition, the replacement of Zr4+ with Hf4+ in UiO-66 tripled in the rate of methanol production. Furthermore, we describe a substituent effect on the catalytic activity, with Cu/Zr-UiO66-COOH providing a three-fold enhancement of methanol production, compared to that of Zr-UiO-66 or Zr-UiO66-NH2. The enhanced catalytic activity of Cu nanoparticles depends on the charge transfer degree from Cu nanoparticles to UiO-66 at the interface between Cu nanoparticles and UiO-66.
Introduction
Hybrid materials composed of metal nanoparticles and metal–organic frameworks (MOFs) have attracted much attention for their gas storage, magnetic, optical and catalytic applications due to the remarkable synergistic function between their constituent materials.1–9 In particular, these hybrid materials have been investigated as highly selective and/or active catalysts because, based on MOF selection, surface properties such as pore size, hydrophobicity/hydrophilicity, and Lewis or Brønsted acidity/basicity can be tuned to control the functionality of the composite.10 For example, Liyu Chen et al. reported Pd nanoparticles embedded in UiO-67 as a catalyst for selective olefin hydrogenation, with selectivity arising from molecular sieving by the pores of UiO-67.11 Kyungsu Na et al. reported the hydrogenation of methylcyclopentane to produce selective products depending on the pore size of the Zr-based MOF support.5 So far, the strategies on the design for hybrid catalysts of metal nanoparticles and MOFs have been mainly based on the combination of their characteristics. On the other hand, hybrid catalysts with electronic interactions between metal and MOF have received scant attention.12–14 Since catalytic properties such as activity or selectivity strongly depend on the electronic states of the metal catalysts, tuning the electronic properties of metal nanoparticles by modifying a MOF support is of significance for further development of hybrid catalysts.Carbon dioxide (CO2) conversion into basic chemicals such as formic acid, methanol and carbon monoxide is very important not only for the reduction of CO2 emissions, a greenhouse gas and major contributor to global warming, but also for the efficient use of CO2 to achieve a carbon neutral energy cycle. In particular, methanol is a good energy carrier as it is a stable liquid under ambient conditions, and it can act as a starting material for the production of olefins, formalin and so on. For these reasons, the development of efficient catalysts for methanol synthesis by CO2 hydrogenation has been extensively investigated, yet still remains a challenge as CO2 is a stable gas molecule which requires a 6-electron reduction for conversion into methanol. So far, most research on CO2 hydrogenation using MOF hybrid materials are related to CO (2-electron reduction) production,15,16 and there are few reports on CO2 hydrogenation to methanol.7,13 Herein we describe highly active Cu nanoparticle composites for CO2 hydrogenation to methanol, with activity controlled via charge transfer from Cu to UiO-66 by controlling the functional groups or metal species in the MOF for the first time.
For the MOF component of the composite material, we focused on Zr- and Hf-based UiO-66 (Zr-UiO-66 and Hf-UiO-66, respectively), which consist of hexanuclear M4+ oxy/hydroxy clusters linked into a cubic 3-dimensional network by terephthalate linkers. These MOFs were chosen because they have superior thermal, water and chemical stabilities, and because they are easily functionalized through linker substitution or defect formation.17–19 For functionalization of the terephthalate, we selected carboxylate and amino groups. Carboxylate and amino groups are known to work as electron withdrawing and donating groups to benzene ring, respectively. Therefore, the electronic states of the Zr6 cluster in UiO-66 are likely influenced by their substitution groups, which may lead to controlling the degree of charge transfer between Cu nanoparticles and UiO-66.
Results and discussion
Synthesis and characterization of Cu/UiO-66 composite catalysts
We synthesized Cu/UiO-66 composite catalysts by thermal decomposition of copper acetylacetonate, Cu(acac)2 as a Cu precursor in the presence of the pre-synthesized MOF (see experimental details, ESI†). In a typical synthesis, Zr-UiO-66 was firstly prepared by minor modifications to a solvothermal method reported previously.19 The obtained Zr-UiO-66 and Cu(acac)2 were then dispersed in an acetone solution, and the mixture was stirred at room temperature for 24 h. After impregnation, the solvent was removed by centrifugation. The collected solid, which included Zr-UiO-66 and Cu(acac)2, was heated at 350 °C for 1 h under vacuum to obtain the composite of Zr-UiO-66 and Cu nanoparticles (Cu/Zr-UiO-66) (see experimental details, ESI†). Cu/Hf-UiO-66, Cu/Zr-UiO-66-NH2, with a 2-aminoterephthalate linker, Cu/Zr-UiO-66-COOH with a 1,2,4-benzenetricarboxylate linker, and a γ-Al2O3 supported Cu catalyst (Cu/γ-Al2O3) as a standard control catalyst were also prepared by the same method to compare their catalytic activity with the non-functionalized Cu/Zr-UiO-66.The amounts of Cu loaded into Cu/Zr-UiO-66, Cu/Zr-UiO-66-NH2, Cu/Zr-UiO-66-COOH, Cu/Hf-UiO-66 and Cu/γ-Al2O3 were determined to be 14, 19, 19, 12 and 13 wt%, respectively, by inductively coupled plasma mass spectrometry. TEM images of the composites revealed that Cu nanoparticles are well dispersed and the mean diameters were estimated to be 13.1 ± 3.9, 18.6 ± 5.1, 19.6 ± 4.0, 15 ± 4.1 and 24 ± 4.1 nm for Cu/Zr-UiO-66, Cu/Zr-UiO-66-NH2, Cu/Zr-UiO-66-COOH, Cu/Hf-UiO-66 and Cu/γ-Al2O3, respectively (Fig. S1†).
Fig. 1a shows the powder X-ray diffraction (PXRD) patterns of Cu/Zr-UiO-66, Cu/Zr-UiO-66-NH2, Cu/Zr-UiO-66-COOH and Cu/Hf-UiO-66. The PXRD patterns of all composite materials consist of the diffraction from both Cu and the corresponding UiO-66. Cu/Zr-UiO-66-NH2 and Cu/Zr-UiO-66-COOH showed weak diffraction peaks from the MOF component (Fig. S2†), which suggests a decrease in crystallinity, likely arising from some instability of these MOFs at the high temperatures needed to decompose Cu(acac)2 (Fig. S3†).
 | ||
| Fig. 1 (a) PXRD patterns and (b) N2 adsorption/desorption isotherms at 77 K for Cu/Zr-UiO-66 (blue), Cu/Zr-UiO-66-NH2 (green), Cu/Zr-UiO-66-COOH (red) and Cu/Hf-UiO-66 (orange). | ||
In order to confirm the porosity of UiO-66 after hybridization with Cu nanoparticles, N2 sorption isotherms of the composites were measured at 77 K (Fig. 1b). Cu/Zr-UiO-66 and Cu/Hf-UiO-66 show typical type-I sorption behavior originating from the microporosity of the MOF, with a decrease in total uptake versus the Cu free MOF due to the presence of Cu nanoparticles. In contrast, a sharp decrease in the microporosity is observed in Cu/Zr-UiO-66-NH2 and Cu/Zr-UiO-66-COOH corresponding to the lower crystallinity of these samples, which is consistent with the PXRD results. The calculated BET surface areas for Cu/Zr-UiO-66, Cu/Hf-UiO-66, Cu/Zr-UiO-66-NH2 and Cu/Zr-UiO-66-COOH were 702, 207, 172 and 52 m2 g−1, respectively, in comparison to 1368, 1427, 1074 and 201 m2 g−1 for the Cu free MOFs (Fig. S4†). From the extended X-ray absorption fine structure analysis, we confirmed that the local structures of Z6-clusters in Cu/UiO-66 composite catalysts were maintained after hybridization with Cu nanoparticles (Fig. S5†).
In order to investigate the composite structure and elemental distribution in Cu/Zr-UiO-66, Cu/Hf-UiO-66 and Cu/Zr-UiO-66-COOH, we performed high-angle annular dark-field scanning TEM (HAADF-STEM) and energy-dispersive X-ray (EDX) elemental mapping of Cu and the constituent elements of UiO-66 (Fig. 2). HAADF–STEM images and STEM–EDX mappings of Cu/Zr-UiO-66, Cu/Zr-UiO-66-COOH and Cu/Hf-UiO-66 samples are shown in Fig. 2a–d, Fig. 2e–h and Fig. 2i–l, respectively. As shown in Fig. 2a, the HAADF–STEM image of Cu/Zr-UiO-66 revealed that the MOF covers the Cu nanoparticles. Fig. 2b and c show the corresponding Cu–K and Zr–L maps which are the constituent metal elements of Cu/Zr-UiO-66, and Fig. 2d presents an overlay map of Cu and Zr. These mapping data clearly show that the Zr elements of UiO-66 are distributed around the surface of the Cu nanoparticles. Similarly, HAADF-STEM images and STEM-EDX mappings of Cu/Zr-UiO-66-COOH and Cu/Hf-UiO-66 showed the same results as that of Cu/Zr-UiO-66. These results demonstrate that Cu/Zr-UiO-66 and the analogues are hybrid structures where Cu nanoparticles are covered with UiO-66.
 | ||
| Fig. 2 (a) HAADF image, (b) Cu–K and (c) Zr–L STEM-EDX maps of Cu/Zr-UiO-66. (d) Reconstructed overlay image of the maps shown in panels (b) and (c) (green, Cu; blue, Zr). (e) HAADF image, (f) Cu–K and (g) Zr–L STEM-EDX maps of Cu/Zr-UiO-66-COOH. (h) Reconstructed overlay image of the maps shown in panels (f) and (g) (green, Cu; blue, Zr). (i) HAADF image, (j) Cu–K and (k) Hf–L STEM-EDX maps of Cu/Hf-UiO-66. (l) Reconstructed overlay image of the maps shown in panels (j) and (k) (green, Cu; red, Hf). | ||
In order to address the possible formation mechanism on Cu nanoparticles covered with UiO-66, we performed the STEM-EDX mapping of the mixture of Zr-UiO-66 and Cu(acac)2 after impregnation. The STEM-EDX mapping revealed that the Cu precursor was located inside the pores of Zr-UiO-66 (Fig. S6†). The N2 sorption isotherm at 77 K indicated the decrease in the microporosity of Zr-UiO-66 due to incorporation of Cu(acac)2 (Fig. S7†). The PXRD patterns of the mixture of Zr-UiO-66 and Cu(acac)2 suggested that Cu(acac)2 exists in an isolated state (Fig. S8†). Considering that the mean diameter of Cu nanoparticles (13.1 nm) in Cu/Zr-UiO-66 is larger than that of the pore size of UiO-66 (ca. 1 nm), Cu(acac)2 loaded into the pores of Zr-UiO-66 is thermally decomposed to generate Cu atoms, and the Cu atoms migrate to form Cu nanoparticles by Ostwald ripening while partially eroding UiO-66. Consequently, Cu nanoparticles are preferentially present in the MOF particles.
CO2 hydrogenation activity to methanol in Cu/UiO-66 composite catalysts
To investigate the catalytic activities of the Cu/UiO-66 composites for CO2 hydrogenation to methanol, we performed the activity tests using a fixed bed flow reactor with a gas mixture of 20 sccm CO2, 100 sccm H2 and 20 sccm He, 2 atm at 220 °C (see experimental details, ESI†). The reaction temperature is lower than the thermally decomposed temperature of UiO-66 and the analogues (Fig. S3†). The amount of methanol synthesized by Cu/γ-Al2O3 and Cu/MOFs are shown in Fig. 3. The results of Cu composite catalysts with other well-known MOFs such as ZIF-8 and MIL-100 are also shown (Fig. S9 and 10†). It has been previously reported that Cu shows poor activity for the conversion of CO2 into methanol.20,21 In our experiment, Cu/γ-Al2O also produced extremely small amounts of methanol (1.9 μmol gCu−1 h−1) (Fig. 3 inset). On the other hand, Cu/Zr-UiO-66 exhibited a high catalytic activity, with a rate of methanol synthesis 70 times larger than that of Cu/γ-Al2O3 (114.2 μmol gCu−1 h−1). For comparison, pure Zr-UiO-66 does not show any catalytic activity for CO2 hydrogenation. Taking into consideration that Cu/ZIF-8 and Cu/MIL-100 have poor CO2 hydrogenation activities of 1.8 μmol gCu−1 h−1 and 16.4 μmol gCu−1h−1, respectively (Fig. 3 inset), this result indicates that Zr-UiO-66 demonstrates a highly effective support effect for the Cu catalyst.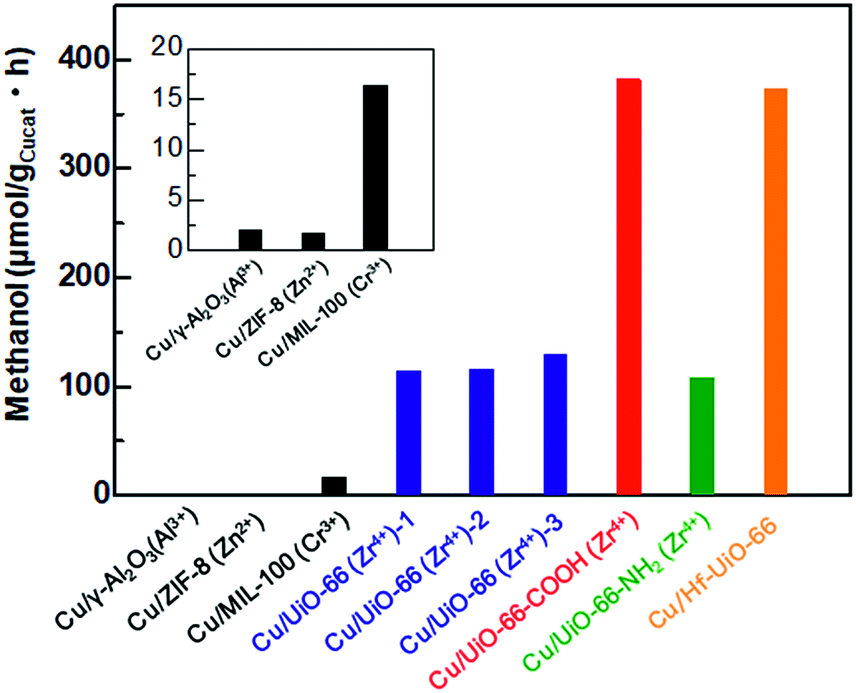 | ||
| Fig. 3 The amount of methanol synthesized from CO2 and H2 using Cu/γ-Al2O3 and Cu/MOF composite catalysts. From elemental analysis and TGA, the amount of defects were estimated to be [Zr6O4(OH)4.6(BDC)5.7], [Zr6O4(OH)4(BDC)4.5(AcO)3] and [Zr6O4(OH)4(BDC)3.6(AcO)4.8] for Zr-UiO-66-1, Zr-UiO-66-2 and Zr-UiO-66-3, respectively. AcO = acetate. | ||
It has recently been reported that the defects in MOF play an important role for controlling properties such as gas sorption,22 proton conductivity,23 optical properties24,25 and liquid phase catalytic activity.26–28 Furthermore, UiO-66 is well known to have widely controllable defect concentrations depending on the synthetic conditions.19 To investigate a defect effect on the catalytic activity of gas phase CO2 hydrogenation, we synthesized Cu/Zr-UiO-66-1, Cu/Zr-UiO-66-2 and Cu/Zr-UiO-66-3 with increasing amounts of acetic acid to introduce increasing amounts of defects into the Zr6-clusters (see experimental details and Fig. S11–S14†). Note: Cu/Zr-UiO-66 discussed above corresponds to Cu/Zr-UiO-66-2. As shown in Fig. 3, all of Cu/Zr-UiO-66-1, Cu/Zr-UiO-66-2 and Cu/Zr-UiO-66-3 exhibited high catalytic activities, compared to Cu/γ-Al2O3, but provided similar rates of methanol synthesis (114.2, 115.5 and 128. μmolgCu−1 h−1 for Cu/Zr-UiO-66-1, Cu/Zr-UiO-66-2 and Cu/Zr-UiO-66-3, respectively). These results indicate that the reactivity of CO2 is not strongly affected by the defects in Zr6-clusters of UiO-66. In addition, the N2 sorption isotherms of Cu/Zr-UiO-66-1, Cu/Zr-UiO-66-2 and Cu/Zr-UiO-66-3 (Fig. S14†) indicated that the porosity of UiO-66 also does not directly affect the catalytic activity.
Ligand substitution in Cu/Zr-UiO-66 seemed to have a major effect on the rate of methanol synthesis with a nearly 3-fold enhancement using carboxylate functionalized Cu/Zr-UiO-66-COOH (381.5 μmol gCu−1h−1vs. 114.2 μmol gCu−1 h−1), but little effect with the amine functionalized Cu/Zr-UiO-66-NH2 (107.7 μmol gCu−1 h−1). Metal substitution for Hf in Cu/Hf-UiO-66 also caused a nearly 3-fold enhancement compared to Cu/Zr-UiO-66 (373.2 μmol gCu−1 h−1vs. 114.2 μmol gCu−1 h−1). The catalytic activities of Cu/UiO-66-COOH and Cu/Hf-UiO-66 were higher than that of the commercially used Cu/ZnO/γ-Al2O3 system.29 In addition, all of the catalysts (Cu/Zr-UiO-66, Cu/Zr-UiO-66-NH2, Cu/Zr-UiO-66-COOH and Cu/Hf-UiO-66) gave very high selectivity of over 95% for methanol (Fig. S15†), which is in good agreement with the reported results.13
In general, the catalytic activity tends to increase with increasing BET surface area of the catalysts because the reactants can more easily access the surface of catalysts. The BET surface areas of Cu/Hf-UiO-66 and Cu/Zr-UiO-66-COOH in this report were much smaller than that of Cu/Zr-UiO-66, although they exhibited significantly enhanced catalytic activities. In addition, the mean diameters of Cu nanoparticles in Cu/Hf-UiO-66 and Cu/Zr-UiO-66-COOH are slightly larger than that of Cu/Zr-UiO-66. These results demonstrate that the surface area of UiO-66 does not directly affect the CO2 hydrogenation activity, but rather the functional group or metal species is a significant factor. In addition, considering that Cu nanoparticles deposited on UiO-66 produced a smaller amount of methanol (67.8 μmol gCu−1 h−1) (Fig. S16†), compared with that of Cu/Zr-UiO-66-2, the interface between Cu and UiO-66 is also critical to the enhanced catalytic activity.
It has been reported that charge transfer between metal nanoparticles and MOF plays an important role in altering the surface/bulk properties of nanoparticles.1,12–14 To investigate possible charge transfer between Cu and MOF, we performed X-ray photoelectron spectroscopy (XPS) of the MOF before and after the hybridization with Cu nanoparticles. As shown in Fig. 4a and b, for Cu/UiO-66 and Cu/UiO-66-COOH, the Zr 3d binding energies shifted to lower binding energy after hybridization with Cu nanoparticles, which indicates that Zr4+ is in a partially reduced state. In contrast, the Cu 2p binding energies of Cu/UiO-66 and Cu/UiO-66-COOH shifted to higher energies with UiO-66 coating, although the shifts became obscured due to the surface oxidation of Cu upon exposure to air (Fig. S17†). These results suggest that charge transfer from Cu nanoparticles to UiO-66 or Cu/UiO-66-COOH occurs in the composites. On the other hand, in the cases of Cu/γ-Al2O3 and Cu/ZIF-8 (Fig. 4c and d), the binding energy shifts of Al 2p and Zn 2p were not observable before and after the hybridization with Cu nanoparticles. The relationship between the produced methanol and the binding energy shift estimated by XPS analysis for Cu/γ-Al2O3 and Cu/MOF catalysts is shown in Fig. 5 (Fig. S18 and Table S1†). As shown in Fig. 5, we can clearly see a correlation between the charge transfer and the amount of the produced methanol – the negative shift of the MOF contributes to the catalytic activity for CO2 hydrogenation to methanol. In particular, among Zr-UiO-66 and the analogues such as Zr-UiO66-NH2 and Zr-UiO66-COOH, the largest negative shift is observed in Zr-UiO66-COOH, which provided the largest rate of methanol synthesis (314.3 μmol gCu−1h−1). These results are the first demonstration of altering the catalytic activity of Cu nanoparticles via charge transfer from Cu to UiO-66 by controlling the functional group or metal species in the MOF. Hf-UiO-66 showed a smaller binding energy shift (−0.17 eV), compared to Zr-UiO66-COOH (−0.32 eV) although the Cu/Hf-UiO-66 composite demonstrates a catalytic activity comparable with the most active Cu/UiO-66-COOH. This is because the binding energy shift of Hf 4f is considered to be relatively smaller than that of Zr 3d due to the valence change.30
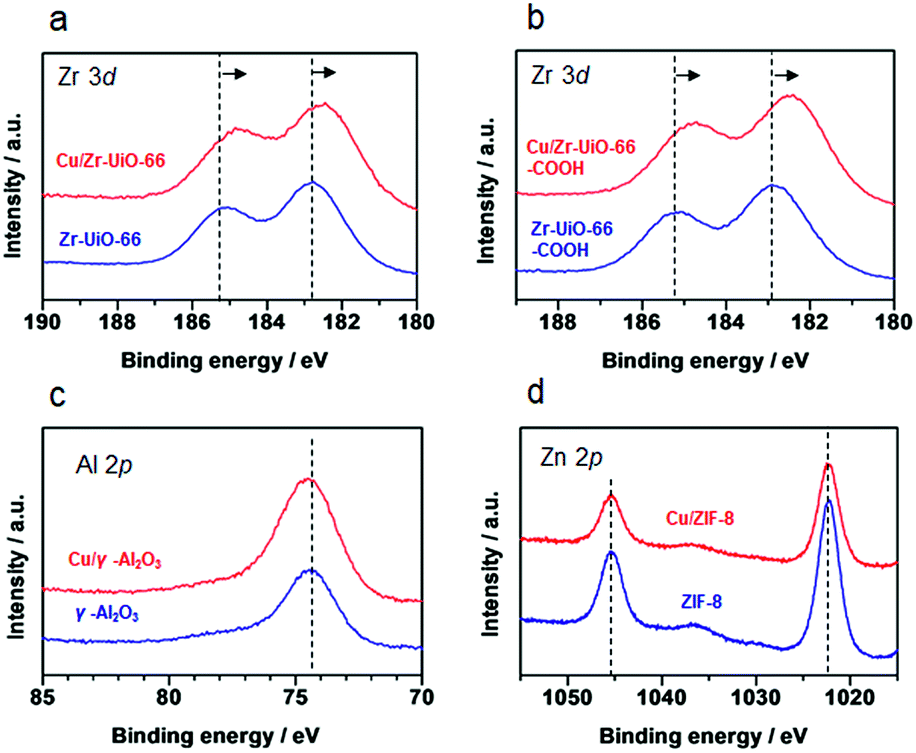 | ||
| Fig. 4 XPS spectra of γ-Al2O3 and MOF before and after the hybridization with Cu nanoparticles. (a) Cu/Zr-UiO-66, (b) Zr-UiO-66-COOH, (c) γ-Al2O3 and (d) ZIF-8. | ||
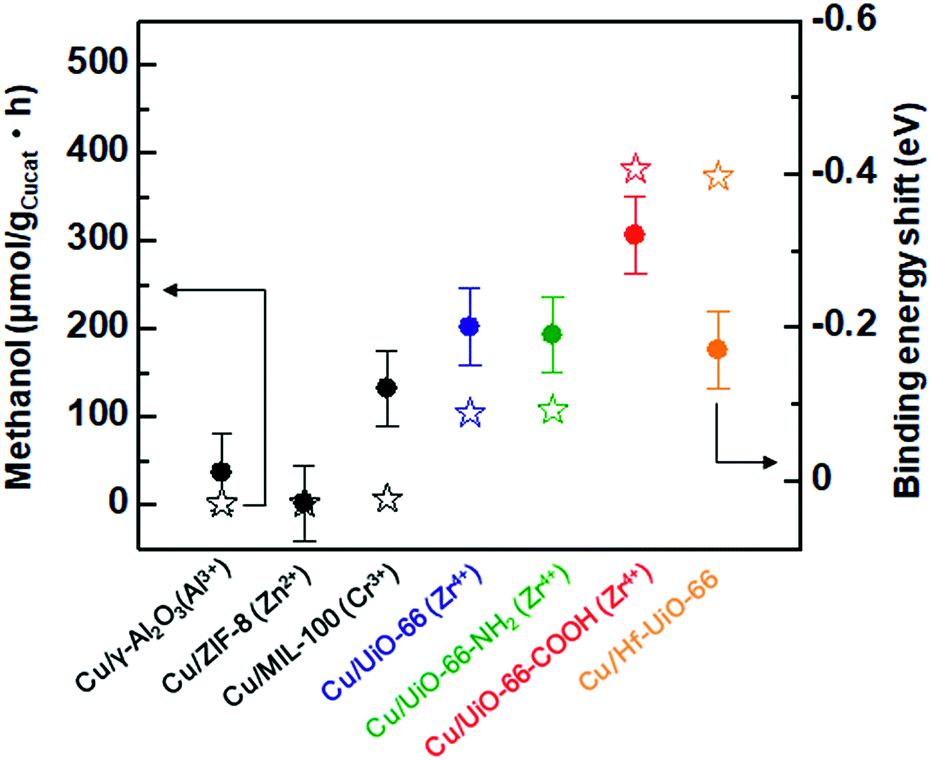 | ||
| Fig. 5 Relationship between the synthesized methanol and binding energy shift estimated by XPS analysis for Cu/γ-Al2O3 and Cu/MOF catalysts. Star and circle correspond to synthesized methanol and binding energy shift, respectively. | ||
The PXRD and TEM measurements revealed that the pristine structures of the Cu/UiO-66 catalysts were maintained after the CO2 hydrogenation test (Fig. S19 and S20†). Furthermore, after 5 cycles, the catalytic performance of Cu/Zr-UiO66-COOH had no obvious change (Fig. S21†), indicating stability of Cu/Zr-UiO66-COOH with excellent catalytic performance.
From theoretical calculations,31 the rate-determining step for CO2 conversion into methanol is hydrogenation of formate, so the stabilization of formate is key to produce methanol at a high rate. It is also known that cationic Cu species helps to stabilize the intermediates (formate).32 Therefore, in our system, cationic Cu species due to charge transfer from Cu to the metal components of UiO-66 is considered to promote the stabilization of formate, leading to the enhancement of catalytic activity for CO2 conversion into methanol.
Conclusions
In summary, we first demonstrated the charge transfer dependence on CO2 hydrogenation activity to methanol in Cu/MOF systems. Compared to Cu/γ-Al2O3, Cu/ZIF-8, Cu/MIL-100 and Cu/UiO-66 composites, UiO-66 is demonstrated to have the most highly effective support effect for CO2 hydrogenation to methanol, and Cu/Zr-UiO-66 produced methanol at a rate 70 times larger than that of Cu/γ-Al2O3. The activity of CO2 hydrogenation to methanol did not strongly correlate to the amount of defects in the Zr6-clusters of UiO-66. On the other hand, the replacement of Zr4+ with Hf4+ in UiO-66 tripled the rate of methanol production. Furthermore, we found a substituent effect to the catalytic activity, with Cu/Zr-UiO66-COOH providing a 3-fold increase in the rate of methanol produced compared to that of Zr-UiO-66 or Zr-UiO66-NH2. The charge transfer from Cu to UiO-66 is considered to play an important role in these enhancements of the catalytic activity of Cu. We hope that the results in this study will contribute to the further development of effective catalysts for CO2 hydrogenation to methanol, as well as to the future design of highly novel catalysts based on control of the charge transfer between metal nanoparticles and MOF by careful selection of the functional group and/or metal species.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NEDO, JST PRESTO (No. JPMJPR1514) and a Grant-in-Aid for Scientific Researches (B) (No. 17750056) from JSPS. The synchrotron radiation experiments were performed at the BL14B2 of SPring-8 with the approval of JASRI (Proposal No. 2018A1753). The STEM observations were performed as part of a program conducted by the Advanced Characterization Nanotechnology Platform sponsored by the MEXT, Japan.Notes and references
- G. Li, H. Kobayashi, J. M. Taylor, R. Ikeda, Y. Kubota, K. Kato, M. Takata, T. Yamamoto, S. Toh, S. Matsumura and H. Kitagawa, Nat. Mater., 2014, 13, 802–806 CrossRef CAS PubMed.
- C. M. Doherty, E. Knystautas, D. Buso, L. Villanova, K. Konstas, A. J. Hill, M. Takahashi and P. J. Falcaro, Mater. Chem., 2012, 22, 11470–11474 RSC.
- P. Falcaro, F. Normandin, M. Takahashi, P. Scopece, H. Amenitsch, S. Costacurta, C. M. Doherty, J. S. Laird, M. D. H. Lay, F. Lisi, A. J. Hill and D. Buso, Adv. Mater., 2011, 23, 3901–3906 CrossRef CAS PubMed.
- Y. Zhao, N. Kornienko, Z. Liu, C. Zhu, S. Asahina, T. R. Kuo, W. Bao, C. Xie, A. Hexemer, O. Terasaki, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 2199–2202 CrossRef CAS PubMed.
- K. Na, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2014, 14, 5979–5983 CrossRef CAS PubMed.
- K. M. Choi, K. Na, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 7810–7816 CrossRef CAS.
- B. An, J. Zhang, K. Cheng, P. Ji, C. Wang and W. Lin, J. Am. Chem. Soc., 2017, 139, 3834–3840 CrossRef CAS PubMed.
- M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef CAS.
- G. Li, S. Zhao, Y. Zhang and Z. Tang, Adv. Mater., 2018, 30, 1800702 CrossRef PubMed.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- L. Chen, H. Chen, R. Luque and Y. Li, Chem. Sci., 2014, 5, 3708–3714 RSC.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- B. Rungtaweevoranit, J. Baek, J. R. Araujo, B. S. Archanjo, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2016, 16, 7645–7649 CrossRef CAS PubMed.
- S. Yoshimasu, M. Sadakiyo, A. Staykov, K. Kato and M. Yamauchi, Chem. Commun., 2017, 53, 6720–6723 RSC.
- K. M. Choi, D. Kim, B. Rungtaweevoranit, C. A. Trickett, J. T. D. Barmanbek, A. S. Alshammari, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 356–362 CrossRef CAS PubMed.
- X. Zhao, H. Xu, X. Wang, Z. Zheng, Z. Xu and J. Ge, ACS Appl. Mater. Interfaces, 2018, 10, 15096–15103 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- S. Biswas and P. V. D. Voort, Eur. J. Inorg. Chem., 2013, 12, 2154–2160 CrossRef.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- Y. Yang, J. Evans, J. A. Rodriguez, M. G. White and P. Liu, Phys. Chem. Chem. Phys., 2010, 12, 9909–9917 RSC.
- Y. Yang, M. G. White and P. Liu, J. Phys. Chem. C, 2012, 116, 248–256 CrossRef CAS.
- O. Kozachuk, I. Luz, F. X. L. Xamena, H. Noei, M. Kauer, H. B. Albada, E. D. Bloch, B. Marler, Y. Wang, M. Muhler and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7058–7062 CrossRef CAS PubMed.
- J. Taylor, S. Dekura, R. Ikeda and H. Kitagawa, Chem. Mater., 2015, 27, 2286–2289 CrossRef CAS.
- Z. Fang, J. P. Dirholt, M. Kauer, W. Zhang, C. Lochenie, B. Jee, B. Albada, N. Metzler-Nolte, A. Pcppl, B. Weber, M. Muhler, Y. Wang, R. Schmid and R. A. Fischer, J. Am. Chem. Soc., 2014, 136, 9627–9636 CrossRef CAS PubMed.
- C. L. Whittington, L. Wojtas and R. W. Larsen, Inorg. Chem., 2014, 53, 160–166 CrossRef CAS PubMed.
- C. Chizallet, S. Lazare, D. B.-Bachi, F. Bonnier, V. Lecocq, E. Soyer, A.-A. Quoineaud and N. Bats, J. Am. Chem. Soc., 2010, 132, 12365–12377 CrossRef CAS PubMed.
- L. T. L. Nguyen, K. K. A. Le, H. X. Truong and N. T. S. Phan, Catal. Sci. Technol., 2012, 2, 521–528 RSC.
- K. Leus, I. Muylaert, M. Vandichel, G. B. Marin, M. Waroquier, V. V. Speybroeck and P. V. D. Voort, Chem. Commun., 2010, 46, 5085–5087 RSC.
- Manuscript in preparation..
- X-ray Photoelectron Spectroscopy Database, http://techdb.podzone.net/eindex.html Search PubMed.
- Y. Yang, C. A. Mims, D. H. Mei, C. Peden and C. T. Campbell, J. Catal., 2013, 298, 10–17 CrossRef CAS.
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. DeBartolo, B. v. Issendorff, P. Zapol, S. Vajda and L. A. Curtiss, J. Am. Chem. Soc., 2015, 137, 8676 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05441j |
| ‡ Present address: Department of Chemistry, University of Calgary, 2500 University Dr NW, Calgary, Alberta T2N 1N4 (Canada). |
| This journal is © The Royal Society of Chemistry 2019 |