
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA hypervalent and cubically coordinated molecular phase of IF8 predicted at high pressure†
Dongbao
Luo‡
a,
Jian
Lv‡
a,
Feng
Peng
d,
Yanchao
Wang
 a,
Guochun
Yang
*b,
Martin
Rahm
*c and
Yanming
Ma
*ae
a,
Guochun
Yang
*b,
Martin
Rahm
*c and
Yanming
Ma
*ae
aState Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun 130012, China. E-mail: mym@calypso.cn; mym@jlu.edu.cn
bCentre for Advanced Optoelectronic Functional Materials Research and Laboratory for UV Light-Emitting Materials and Technology of Ministry of Education, Northeast Normal University, Changchun 130024, China. E-mail: yanggc468@nenu.edu.cn
cDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, Gothenburg, 412 96, Sweden. E-mail: martin.rahm@chalmers.se
dCollege of Physics and Electronic Information, Luoyang Normal University, Luoyang 471022, China
eInternational Center of Future Science, Jilin University, Changchun 130012, China
First published on 3rd January 2019
Abstract
Up to now, the maximum coordination number of iodine is seven in neutral iodine heptafluoride (IF7) and eight in anionic octafluoride (IF8−). Here, we explore pressure as a method for realizing new hypercoordinated iodine compounds. First-principles swarm structure calculations have been used to predict the high-pressure and T → 0 K phase diagram of binary iodine fluorides. The investigated compounds are predicted to undergo complex structural phase transitions under high pressure, accompanied by various semiconductor to metal transitions. The pressure induced formation of a neutral octafluoride compound, IF8, consisting of eight-coordinated iodine is one of several unprecedented predicted structures. In sharp contrast to the square antiprismatic structure in IF8−, IF8, which is dynamically unstable under atmospheric conditions, is stable and adopts a quasi-cube molecular configuration with R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) symmetry at 300 GPa. The metallicity of IF8 originates from a hole in the fluorine 2p-bands that dominate the Fermi surface. The highly unusual coordination sphere in IF8 at 300 GPa is a consequence of the 5d levels of iodine coming down and becoming part of the valence, where they mix with iodine's 5s and 5p levels and engage in chemical bonding. The valence expansion of iodine under pressure effectively makes IF8 not only hypercoordinated, but also hypervalent.
symmetry at 300 GPa. The metallicity of IF8 originates from a hole in the fluorine 2p-bands that dominate the Fermi surface. The highly unusual coordination sphere in IF8 at 300 GPa is a consequence of the 5d levels of iodine coming down and becoming part of the valence, where they mix with iodine's 5s and 5p levels and engage in chemical bonding. The valence expansion of iodine under pressure effectively makes IF8 not only hypercoordinated, but also hypervalent.
1. Introduction
It is well known that the coordination number of atoms in compounds can greatly affect chemical properties1,2 and exploring new coordination motifs of elements is of importance to understand their chemical behavior and expand their applications.3–5 Iodine has, with the exception of astatine, the lowest electronegativity, the largest polarizability, and the largest atom size of the halogens.6–8 These properties and the ability to readily engage in hypercoordination, i.e. allowing for more atoms in its coordination sphere than what is predicted by the octet rule, combined with its low toxicity, make iodine distinct from the lighter p-block elements. The ability of iodine to engage in hypercoordination allows fabrication of compounds that are potential substitutes for transition metal-based catalysts.9,10 Commercially available hypercoordinated iodine compounds have shown promise for mild and highly selective oxidizing ability and environmentally benign catalysis.11–14The highest known coordination number in a neutral iodine fluoride compound is seven, in IF7.15 Isoelectronic XeF7+ and TeF7− have been synthesized.16 With the exception of some caged ions,17 higher coordination numbers in neutral main group compounds are, to the best of our knowledge, non-existent. XeF8 has been shown unstable even under pressures reaching 200 GPa.18 Chemically, the limit in the coordination number of iodine can be rationalized from the atom's seven valence electrons. Formally, the valence shell of iodine in IF7 can either be seen allowing for seven polar covalent bonds or, alternatively, the formation of closed-shell I(VII) surrounded by seven fluoride ligands. Because d-orbitals typically do not partake in the chemical bonding of main group elements, the classification as hypervalent is controversial, and the term hypercoordination is preferred.19
By this logic, octacoordination should be allowed by the addition of one electron. Indeed, anionic octafluoride (IF8−) has been synthesized as the [NO2]+[IF8]− salt.20 The coordination geometry of IF8− is square antiprismatic. This kind of arrangement is commonly observed in different transition metal octafluorides, for example in TaF83−, ReF8− and ZrF84−.21 Does this mean that the quest for higher coordination numbers in neutral iodine fluorides is over?
We do not believe so. Pressure is known to fundamentally change the chemistry and structure by, for example, overcoming reaction energy barriers, shortening interatomic distances, and modifying atomic orbital energy levels.22,23 There are many examples of the utility of pressure in allowing remarkable new hypercoordinated compounds, for example H5I,24,25 XeF6,18 HgF4,26 Li5Cs,27 CsF5,28 AuF6,29 Li5Au,30 AH6 (A = Sr, Ba),31,32 LaH10,33 and BaReH9.34
To explore the effects of pressure and search for higher coordination numbers in iodine fluorides, we have performed an extensive structure search of selected stoichiometries of IFx (x = 1/3, 1/2, 2/3, and 1–12) from 100 to 300 GPa using swarm-intelligence-based structural prediction calculations. Dynamical stability is an important criterion when predicting structural stability. The calculated phonon spectra of the predicted phases are shown in the ESI† and show no phonon modes with imaginary frequencies (Fig. S1†). The most notable of our predictions is the stabilization of a molecular IF8 phase of R symmetry at 300 GPa. The cubic coordination geometry of this molecular solid is distinct from the square antiprismatic structure in IF8−. We will return to discuss this unprecedented iodine coordination sphere and its underlying electronic structure.
2. Results and discussion
2.1 Stability and crystal structures
The relative stabilities of the different binary compositions were estimated by calculating the enthalpy of formation at T → 0 K and building the convex hull based on the most stable structures of the considered compositions (Fig. 1a). In Fig. 1a, the thermodynamically stable IFx structures are depicted as filled symbols, which resist decomposing into elemental solids or other stable compounds. Besides reproducing the known structures of IFx (x = 3, 5, 7) under ambient conditions,10 some unexpected compounds with stoichiometries of IFx (x = 8, 10, 11, 12) are predicted to become thermodynamically stable under high pressure. Note that these stoichiometries do not mean the same coordination number of fluorine around iodine. The maximal coordination number found is 8. The crystal structures of iodine fluorides that show stability in some part of the investigated pressure range are shown in Fig. 2. The stable structures will be discussed separately. All other considered stoichiometries, not shown in Fig. 2, have enthalpies that sit slightly above the solid line in Fig. 1a and are either unstable or metastable. Decomposition enthalpies with respect to stable IFx compounds and elemental solid I2 or F2 are shown in Table S1.†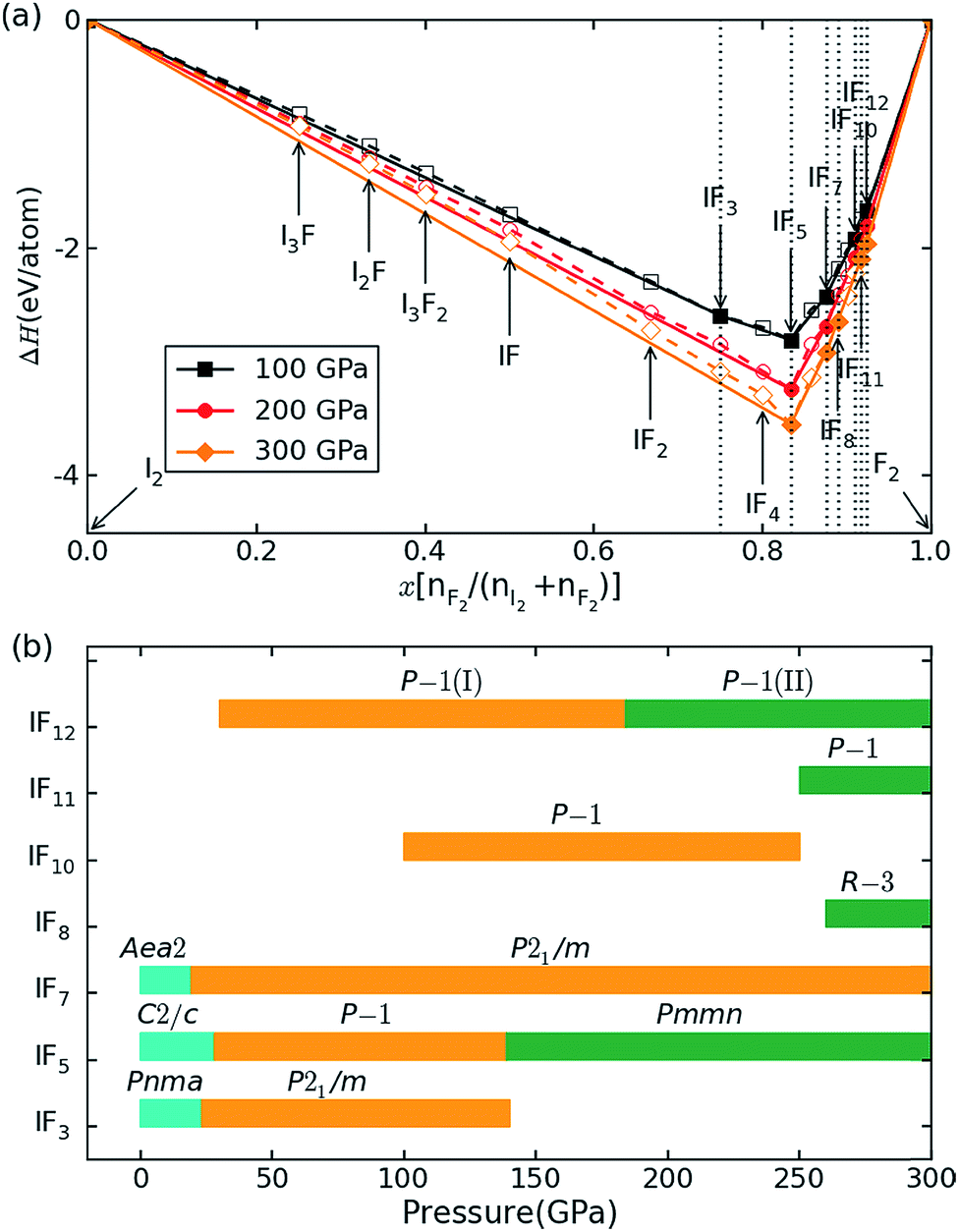 | ||
| Fig. 1 Relative stability of iodine fluorides. (a) Convex hull diagram for iodine fluoride compositions at 100, 200 and 300 GPa. The elemental reference structures used are I2 in the Fmm phase and F2 in the Cmca phase. These phases have previously been shown to be stable over the relevant pressure range.35,36 Dotted lines connect the lowest energy data points, and solid lines denote the convex hull. Stable compositions are indicated by filled symbols. (b) Pressure-composition phase diagram of iodine fluorides. | ||
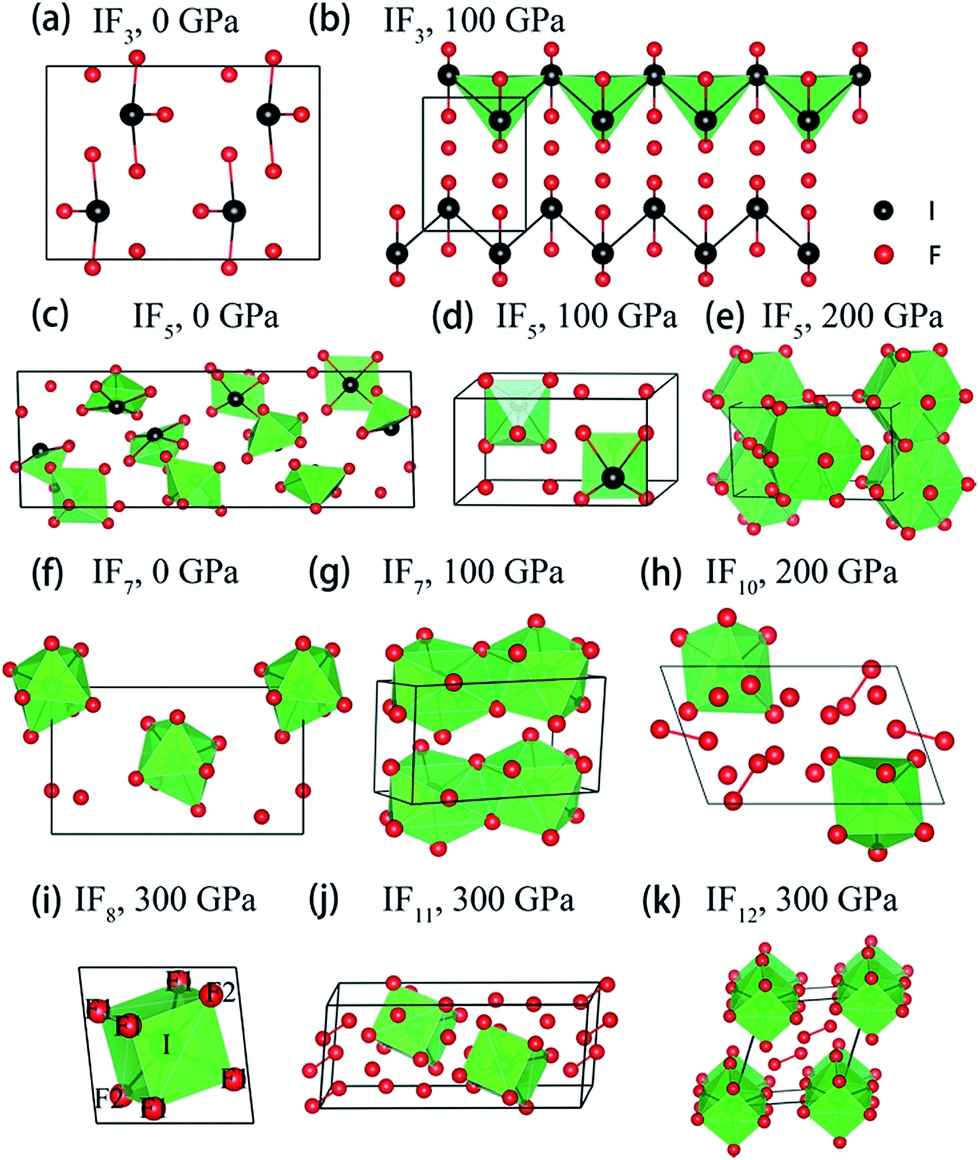 | ||
Fig. 2 Stable crystal structures of the considered I–F compounds. (a) Pnma phase of IF3 at 0 GPa. (b) P21/m phase of IF3 at 100 GPa. (c) C2/c phase of IF5 at 0 GPa (d) P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) phase of IF5 at 100 GPa. (e) Pmmn phase of IF5 at 200 GPa. (f) Aea2 phase of IF7 at 0 GPa. (g) P21/m phase of IF7 at 100 GPa. (h) P phase of IF10 at 200 GPa. (i) R phase of IF8 at 300 GPa. (j) P phase of IF11 at 300 GPa. (k) P phase of IF12 at 300 GPa. Black and red balls denote I and F atoms, respectively. The unit cell is drawn with black solid lines. Detail structural parameters of these I–F compounds are shown in Tables S2 and S3.† phase of IF5 at 100 GPa. (e) Pmmn phase of IF5 at 200 GPa. (f) Aea2 phase of IF7 at 0 GPa. (g) P21/m phase of IF7 at 100 GPa. (h) P phase of IF10 at 200 GPa. (i) R phase of IF8 at 300 GPa. (j) P phase of IF11 at 300 GPa. (k) P phase of IF12 at 300 GPa. Black and red balls denote I and F atoms, respectively. The unit cell is drawn with black solid lines. Detail structural parameters of these I–F compounds are shown in Tables S2 and S3.† | ||
A pressure-resolved phase diagram of iodine fluorides was built to facilitate experimental studies and is shown in Fig. 1b. At ambient pressure, the known molecular crystals of IFx (x = 3, 5 and 7) are reproduced (Fig. 2a, c, and f). Under compression, these molecular structures are predicted to undergo phase transitions and eventually transform into aggregated phases. For IF3, the ambient pressure orthorhombic Pnma structure transforms to a monoclinic phase (space group P21/m) at 23 GPa (Fig. 2b). The P21/m phase contains a zigzag iodine chain, in which an I–I distance of 2.7 Å is nearly the same as that in the I2 molecule at 1 atm. The IF3 molecular phase is predicted to become unstable with respect to IF5 and I2 above 140 GPa. Under compression, C2/c-structured IF5 is predicted to first transform to another molecular phase with P symmetry at 28 GPa (Fig. 2d) and subsequently into a non-molecular P21/m phase above 139 GPa (Fig. 2e). In contrast to IF5, IF7 directly transforms into a non-molecular P21/m phase at 20 GPa (Fig. 2g). Compared with the structures under near ambient conditions (Fig. 2c and f), the IF5 and IF7 molecular phases become gradually more distorted with compression. IF5 is predicted to transform into an orthorhombic structure with Pmmn symmetry, consisting of face-sharing 14-fold I–F polyhedra (Fig. 2e) at 200 GPa. P21/m-structured IF7 is similarly predicted to be structured as an edge-sharing 12-fold polyhedron (Fig. 2g) at 100 GPa.
2.2 Unexpected IF8
We arrive next to the IF8 stoichiometry. Here a new stable compound with R symmetry is predicted to be stable above 260 GPa (Fig. 2i). Whereas a cubic coordination geometry is common in extended materials, such as in the bcc lattice, a cubic ligand field has, to the best of our knowledge, never been experimentally found in a molecule. We will therefore focus most of the remainder of this paper discussing IF8. The unusual cubic coordination returns in the investigated IF10, IF11, and IF12 compositions, which all consist of F2 together with either IF8 or IF7 molecular structural units (Fig. 2h, j and k and S2†). Predicted IF10, IF11, and IF12 phases are all compositions with P symmetry in their respective stable pressure ranges.
The primitive cell of the R phase of IF8 contains one formula unit only, in which each I atom forms an eight-fold coordination with F atoms. That IF8 shows a clear molecular character is evident from the analysis of bond distances: six shorter I–F1 (1.789 Å) and two slightly longer I–F2 (1.802 Å), followed by a set of third nearest neighbors at 2.333 Å (Fig. 2i and S3†). The nearest intermolecular F–F contacts between IF8 units in the R crystal is 1.89 Å. In comparison, the nearest non-bonded contacts in a crystal of F2 optimized at 300 GPa is 1.75 Å. The molecular character is also supported by chemical bonding analyses that we will return to.
After examining the results of our structural searches, we found another IF8 phase with a square antiprismatic structure (space group Pnn). However, the Pnn-structured IF8 (Fig. S4†) is unstable with respect to the quasi-cubical IF8 (i.e. R-structured IF8) by 14.6 eV per formula unit. The large difference in enthalpy between the two structures can be attributed to a more favorable pV-term for the R structure and to the occupation of anti-bonding levels involving I 5d levels in the Pnn structure (details are provided in the ESI†)
2.3 Chemical bonding and electronic properties of IF8
Under normal ambient conditions molecular IF7 takes a pentagonal bipyramidal coordination and ionic IF8− a square antiprismatic coordination. The coordination sphere of these closed-shell molecules can be straightforwardly rationalized with simple valence shell electron-pair-repulsion (VSEPR) arguments, in which ligands maximize distances to their neighbors to minimize exchange, or Pauli repulsion.37 In contrast, molecular IF8 is an electron deficient radical, with one hole in its valence p-shell. Under ambient conditions we are correct to expect such a structure to dissociate into IF7 and an F atom. So how can IF8 represent a stable composition under high compression, and why does it take its peculiar cubic structure?We think that this can be explained in the following way: first, we know that the difference in electronegativity between F and I will not change meaningfully under high compression, and so we cannot expect a drastic change in the electronic structure for this reason.38 In atomic iodine, the 5d levels lie 8.2 eV above the 5p levels.39 Consequently, mixing, or hybridization, with 5d is not considered important for the chemical behavior of iodine under normal conditions.40 As we shall see, this changes under compression. To understand why, we take a molecular orbital (MO) perspective.
We know that for a cubic ligand field, the expected splitting of non-bonding d-orbitals is three (T2g) – over – two (Eg).41,42 Work on 8-coordination by Burdett, Hoffmann and Fay (BHF) compared different coordination geometries using group theory arguments and extended Hückel calculations.43 Among other things, it was demonstrated that the Eg levels (the dz2 and the dx2−y2 orbitals) should be non-bonding because they do not point along the bond axes of a cubic ligand field. However, the BHF work did not evaluate interactions that would occur with ligand p-levels in this specific coordination environment. The rarity of the cubic coordination sphere in nature is undoubtedly the reason for its near absence in the scientific literature.
To remedy the situation, we show in Fig. 3 the molecular orbital (MO) diagram for I 5s, 5p and 5d interacting with F 2p levels in a cubic (Oh symmetric) coordination geometry. This diagram is a sketch constructed from symmetry arguments applied to an isolated IF8 molecule. The energy orderings have been predicted using DFT calculations on the different fragments in vacuum. Two orbital combinations, of A1g and T1u symmetry, predict the specific interactions of I 5s and 5p with the corresponding symmetry adapted MOs of the F8 ligand cage. Assuming that the energy ordering shown in Fig. 3 is correct, five out of the six occupied MOs of these symmetries are I–F bonding. The T2g and Eg levels are noteworthy, as they predict the exclusive bonding interaction between the formally unoccupied I 5d levels and the F 2p ligand orbitals of the same symmetry. The designations of the MOs as bonding and antibonding are based on symmetry arguments, and this will not change under pressure. What might change is the relative importance of the MOs, their energy relative each other, and the degree of mixing of what are, under ambient conditions, unoccupied I 5d-levels. How can occupation of I 5d orbitals be possible under compression?
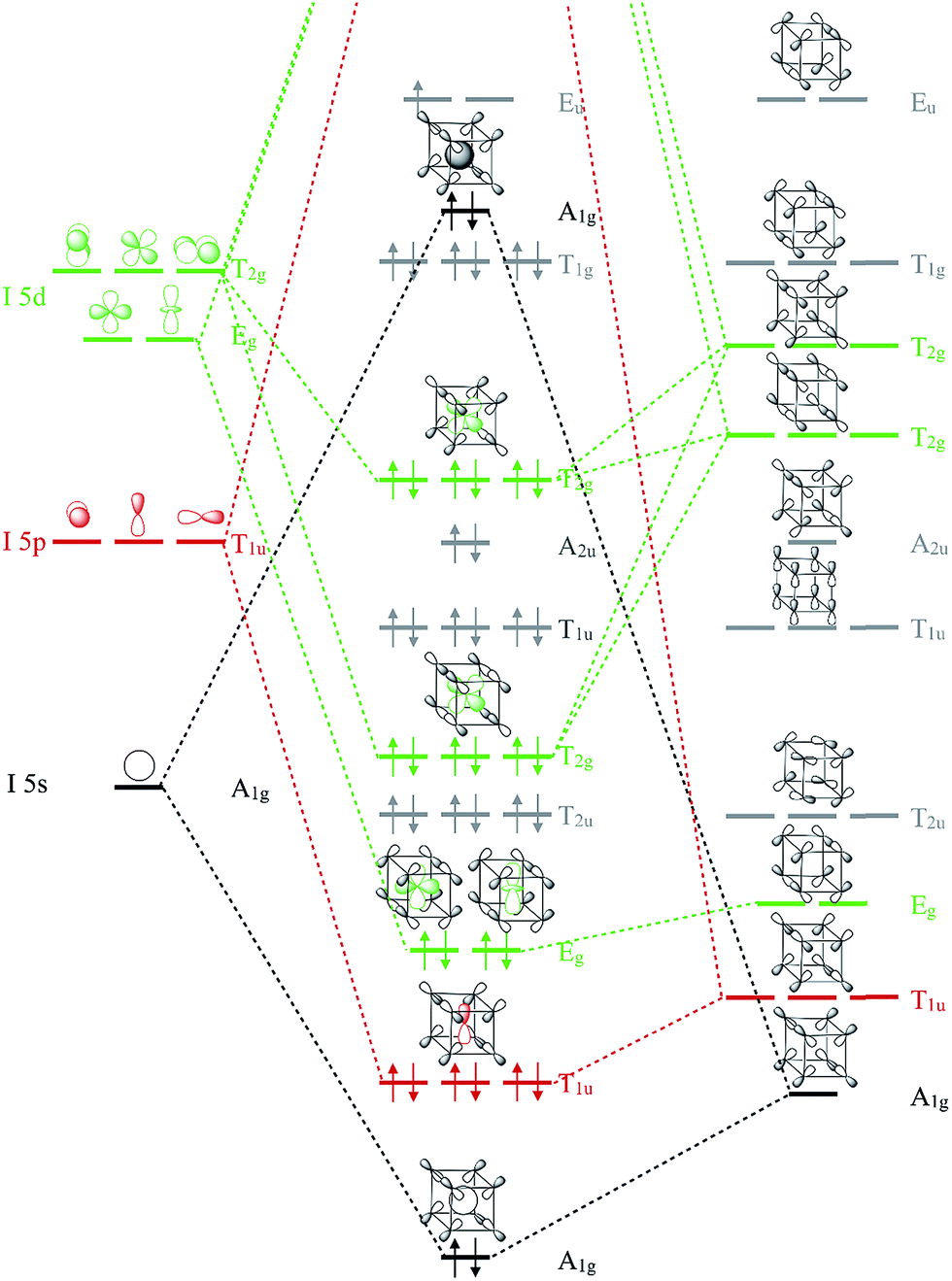 | ||
| Fig. 3 Molecular orbital diagram showing orbitals of iodine mixing with the symmetry adapted ligand orbitals of the F8 cage. Energies are not to scale. Approximate energy orderings were predicted using DFT calculations on frozen fragments of IF8 and F8 in vacuum. The energy levels of atomic I are known from experiment.37 Orbital symmetries responsible for I–F bonding are colored green for T2g and Eg (I 5d), red for T1u (I 5p) and black for A1g (I 5s). One bonding combination of A1g symmetry between I 5s and F 2s valence orbitals is omitted for clarity, as are the unoccupied orbitals. | ||
That orbitals of different kind can cross due to compression is well known.44,45 Mao-Sheng Miao and Roald Hoffmann have given a nice explanation as to why orbitals can cross in compressed atoms.46 In brief, orbitals that are smaller, and have fewer radial nodes, are less affected by compression. For a given principal quantum number, compression tend to increase level energies as s > p > d. In complex systems such as IF8, the purely physical effect of pressure is one factor affecting level ordering. A positive charge on iodine also helps to decrease the energy of I 5d levels relative to the ligand F orbitals, and we will return to address the oxidation state of iodine. As we shall see, chemical interactions with F atoms play a key role for bringing down I 5d levels in IF8.
We can use the MO diagram in Fig. 3 to understand that as the volume of the F8 cage decreases, as it will under compression, the overlap between I and F-based orbitals will naturally increase. Of course, iodine atoms and F8 are just fictional references here – what matters are the IF8 orbitals. Because the occupied T2g and Eg MOs of IF8 are all I–F bonding, these MOs will come down in energy relative to the other MOs as the pressure mounts and overlaps increase. The same goes for the I–F bonding A1g and T1u orbitals, which, together with the T2g and Eg set, are predicted to drive the stabilization of the high symmetry cubic coordination under high pressure. Put differently, participation of I 5d, and the cubic coordination geometry, can be rationalized by symmetry facilitated bonding interactions with the ligand framework, which become increasingly pronounced as the volume of the system decreases (Fig. S5†). The ligand coordination is essential for bringing down the d-orbitals of iodine. There is, for example, no participation of I 5d levels in similarly compressed elemental iodine (Fig. S6†). There are, as the MO diagram shows, also simultaneous F–F interactions of both bonding and antibonding character in IF8. However, as we shall see and quantify, I–F bonding interactions are significantly larger than F–F interactions, which largely cancel out under compression.
How does this analysis fit with the predicted R structure of IF8 at 300 GPa? Remarkably well, as it turns out. First, our crystal orbital Hamilton populations (COHPs) confirm the predominately molecular character of the electronic structure, already inferred by the predicted bond lengths. The integrated COHP up to the Fermi level between iodine and its 8 nearest neighbors (F1 & F2) is −6.4/−6.2 eV, which is indicative of strong bonding interactions. In contrast, iodine's interactions with its third nearest neighbor (F3) is estimated as −0.7 eV, which clearly suggests that R-structured IF8 can be viewed predominately as a molecular crystal. The integrated COHPs between F1–F1, F1–F2 and F1–F3 are 0.06, 0.07 and −0.14 eV, respectively (F3 refers to an F atom in the nearest neighboring IF8 unit). That F–F bonding and anti-bonding interactions within the IF8 molecule largely cancel out, but are destabilizing overall, is in qualitative agreement with the MO diagram in Fig. 3. Bonding between IF8 molecules (I–F3 and F1–F3) comes out as exclusively stabilizing in the COHP analysis (Fig. 4c and S4c†).
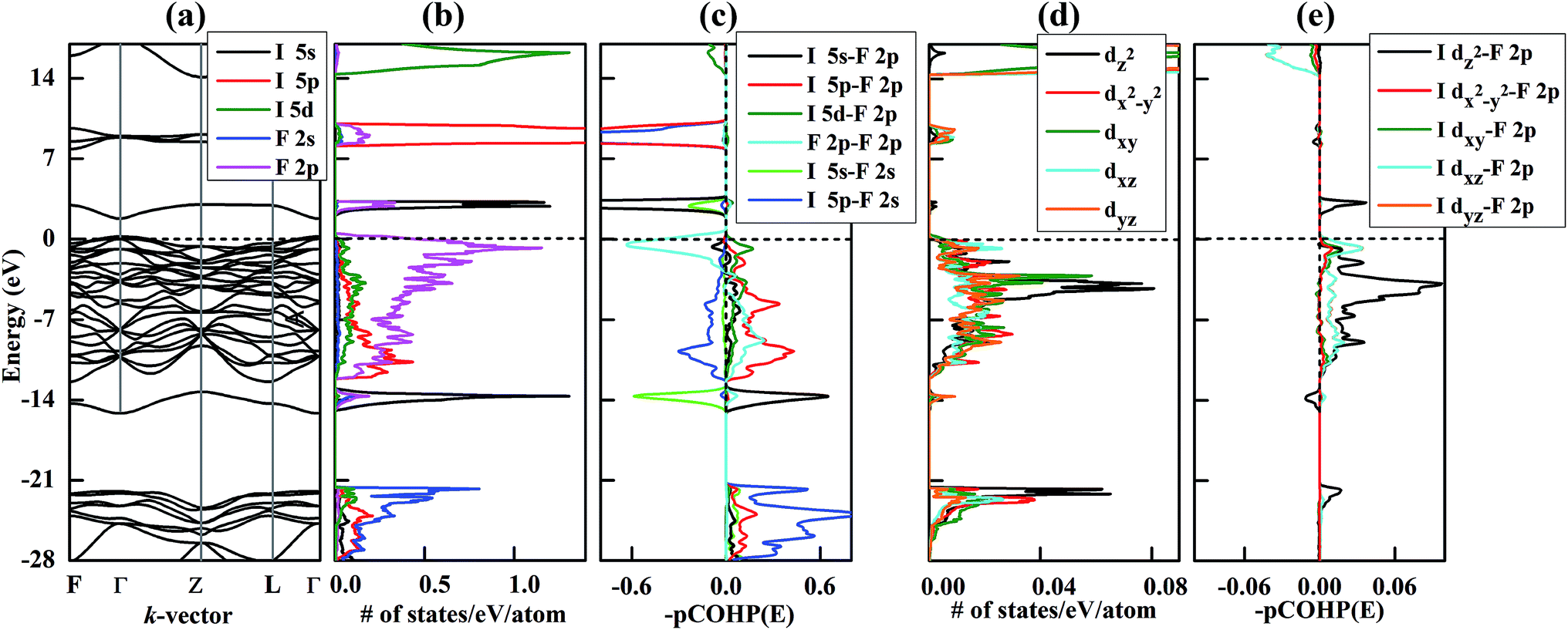 | ||
| Fig. 4 Electronic structure of IF8 in its R-phase at 300 GPa. (a) Band structure. (b) Partial density of states (PDOS). (c) Crystal orbital Hamilton populations (COHP) for I–F2 (1.802 Å). Positive and negative denote bonding and antibonding orbital interactions, respectively. I 5d–F 2s is omitted because the corresponding integrated COHP is very small below the Fermi level (−0.17 eV). (d) Calculated PDOS of I d-levels. (e) Calculated COHP of I 5d–F2 2p. Note that the lines corresponding to I dxy–F2 2p and I dyz–F2 2p are overlaid by I dx2−y2–F2 2p and I dxz–F2 2p in this figure. | ||
At 300 GPa, iodine's 5d band penetrates well into the valence region, where it undergoes a significant dispersion and mixes both with the 5s and 5p levels of iodine, and with the valence shell of fluorine (Fig. 4b). Our COHP analysis clearly shows significant bonding interactions involving d-levels below the Fermi level (Fig. 4c). The only anti-bonding interactions observed near the Fermi level are of I 5s–F 2p and F 2p–F 2p character, in good agreement with the schematic MO analysis of the IF8 molecule in Fig. 3. The only other anti-bonding states identified in the COHP are due to overlap with F 2s, which we omitted from Fig. 3 for clarity.
What about the metallicity of the material? That the R-structured IF8 shows metallicity (Fig. 4) is expected from the fact that it's a molecular solid of an open-shell molecule. In contrast, the P21/m-structured IF7, a molecular solid of a closed-shell molecule, is a semiconductor (Fig. S7†). We did perform an in silico experiment by artificially adding one electron to the IF8 unit cell (the negative charge being compensated by a smeared out positive background charge in our program). The result of the extra electron is a material with semiconducting character that exhibits a similar Projected Density of States (PDOS) distribution (Fig. S8†). In IF8, the Density of States (DOS) at the Fermi-level is dominated by the F 2p-bands. That the Fermi level should be dominated by F 2p is expected, and is also in good agreement with our MO analysis of the isolated IF8 molecule (Fig. 3), which predicts the highest occupied level to be a pure F 2p–F 2p antibonding level of Eu symmetry. Spin-polarized calculations on the R phase of IF8 shows that the material is not magnetic (Fig. S9†).
Because of significant mixing of s, p and d levels of iodine, we can conclude that a valence expansion of iodine has occurred. This implies that the IF8 molecule, predicted to exist in the crystalline state at 300 GPa, is not only hypercoordinated but also hypervalent. Expansion of iodine's valence space is here a consequence of additional energy levels being available for bonding, not a classification based on the fact that the number of ligands around iodine exceeds seven. Our bonding and density of states analyses are based on orbital projection methods, which differ slightly depending on the program used. Even though these methods are not exact, in that they do not recover all of the electron density, all approaches that we tested (see the Fig. S10†) do support the general conclusion that there is a valence expansion and that the I 5d levels are essentially equally populated. The dz2 orbital does appear to be slightly more populated on average, which may explain the small structural deviation from a perfect cube (Table S4†).
What about the oxidation state of iodine? Because the predicted R phase of IF8 is metallic, a formal ionic extrapolation is not possible.47 If we nonetheless consider each F ligand as F− and remove one electron from the ligands due to the radical character (the F 2p hole), the oxidation state of iodine in IF8 becomes +VII. Our best estimate of the total iodine orbital occupation in IF8 corresponds to an atomic charge of +3.8, when calculated using a Mulliken-type approach. The relative occupation of the I-based orbitals is s1p1.7d1. We must stress that these values are approximate and sensitive to the internal basis set used in the projection scheme. Nevertheless, the orbital-based charge is in strikingly good agreement with a separately calculated QTAIM charge of +4.0 on iodine (see the ESI†). Combined, the orbital and charge density analyses, and the Electron localization Function (ELF, Fig. S11†) suggest a rather large degree of covalency in IF8, and the presence of strong polar covalent and hypervalent bonds.
3. Conclusion
First-principles swarm structural search calculations have been employed to explore the phase stabilities and structures of I–F compounds under high pressure. Pressure favors the stabilization of fluoride-rich compounds (IFx = 3, 5, 7, 8, 10, 11, and 12) rather than iodine-rich compounds. Several I–F compounds are predicted to undergo pressure-induced molecular to extended phase transformations, accompanied by semiconductor to metal transitions. We have focused our analyses on a predicted R phase of IF8, where pressure stabilizes a unique cubical molecular structure. The predicted cubical coordination geometry is unique in main group chemistry, where square antiprismatic octa-coordination is common. The molecular crystal has an electron-deficient electronic structure that causes it to be metallic. Various molecular orbital and crystal orbital projection-based analyses support a conclusion that the electronic and geometric structure of IF8 is a consequence of valence expansion of iodine.
4. Computational details
Searches for I–F binary compounds were performed with the CALYPSO structure prediction method,48,49 while allowing for up to 4 formula units per unit cell. The predictive accuracy of this methodology has been repeatedly demonstrated on various systems, from elemental solids to binary and ternary compounds.50–55 The Vienna Ab initio Simulation Package (VASP) code56 and the Perdew–Burke–Ernzerhof57 functional in the generalized gradient approximation58 were adopted to perform structural relaxations within the framework of density-functional theory (DFT). The electron–ion interactions were represented by means of the all-electron projector augmented-wave method with 5s25p5 and 2s22p5 treated as the valence electrons of I and F atoms, respectively. A plane-wave basis set cutoff of 950 eV and the Monkhorst–Pack scheme59 with a dense k-point grid spacing of 2π × 0.032 Å−1 in the Brillouin zone were used to converge energies to less than 1 meV/atom. To ensure the validity of the pseudopotentials used, the equation of state of IF8 was also calculated using the full-potential WIEN2k code,60 with nearly identical results to the VASP calculations (Fig. S12†). Phonon calculations were performed using a supercell approach with the finite displacement method61 using Phonopy.62 The Electron Localization Function (ELF) was used to estimate the degree of electron localization.63 Crystal Orbital Hamilton Population (COHP) analyses were performed using the LOBSTER program.64,65 Van der Waals (vdW) interactions were included using the optB88-vdw approach.66,67 DFT calculations on molecular fragments were made using Gaussian 16, revision B.01.68Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by the National Key Research and Development Program of China (Grant No. 2016YFB0201200 and 2016YFB0201201), the National Natural Science Foundation of China (Grants No. 21573037, 21873017, 11534003, 11774127, and 11274136), the 2012 Changjiang Scholars Program of China, the Program for JLU Science and Technology Innovative Research Team, and the Science Challenge Project No. TZ2016001. Parts of the calculations were performed at the High-Performance Computing Center of Jilin University and Tianhe2-JK in the Beijing Computational Science Research Center. M. R. acknowledges financial support from Chalmers University of Technology and computational resources provided by the Swedish National Infrastructure for Computing (SNIC) at C3SE. Toby Zeng and Roald Hoffmann are acknowledged for valuable discussion and comments.References
- L. M. Yang, E. Ganz, Z. Chen, Z. X. WANG and P. V. R. Schleyer, Four Decades of the Chemistry of Planar Hypercoordinate Compounds, Angew. Chem., Int. Ed., 2015, 54, 9468–9501 CrossRef CAS PubMed
.
- H. Zhang, Y. Li, J. Hou, K. Tu and Z. Chen, FeB6 Monolayers: the Graphene-like Material with Hypercoordinate Transition Metal, J. Am. Chem. Soc., 2016, 138, 5644–5651 CrossRef CAS PubMed
- M. C. Lipke and T. D. Tilley, Hypercoordinate Ketone Adducts of Electrophilic η3-H2SiRR' Ligands on Ruthenium as Key Intermediates for Efficient and Robust Catalytic Hydrosilation, J. Am. Chem. Soc., 2014, 136, 16387–16398 CrossRef CAS
- Z. Wang and P. v. R. Schleyer, Planar Hypercoordinate Carbons Joined: Wheel-shaped Molecules with C–C axles, Angew. Chem., Int. Ed., 2002, 41, 4082–4085 CrossRef CAS
- A. Khan and D. Foucher, Hypercoordinate Compounds of the Group 14 Elements Containing κn-C, N-, C, O-, C, S-and C, P-Ligands, Coord. Chem. Rev., 2016, 312, 41–66 CrossRef CAS
-
N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier, 2012 Search PubMed
-
T. Kaiho, Iodine Chemistry and Applications, John Wiley and Sons, 2014 Search PubMed
- M. Rahm, R. Hoffmann and N. W. Ashcroft, Atomic and ionic radii of elements 1–96, Chem.–Eur. J., 2016, 22, 14625–14632 CrossRef CAS PubMed
- V. V. Zhdankin and J. D. Protasiewicz, Development of new hypervalent iodine reagents with improved properties and reactivity by redirecting secondary bonds at iodine center, Coord. Chem. Rev., 2014, 275, 54–62 CrossRef CAS
- S. Tang, Y. Wu, W. Liao, R. Bai, C. Liu and A. Lei, Revealing the Metal-like Behavior of Iodine: An Iodide-Catalysed Radical Oxidative Alkenylation, Chem. Commun., 2014, 50, 4496–4499 RSC
- R. D. Richardson and T. Wirth, Hypervalent Iodine Goes Catalytic, Angew. Chem., Int. Ed., 2006, 45, 4402–4404 CrossRef CAS
- H. Liang and M. A. Ciufolini, Chiral Hypervalent Iodine Reagents in Asymmetric Reactions, Angew. Chem., Int. Ed., 2011, 50, 11849–11851 CrossRef CAS PubMed
- A. Screenithya, C. Patel and C. Hadad, Hypercoordinate Iodine Catalysts in Enantioselective Transformation: The Role of Catalyst Folding in Stereoselectivity, ACS Catal., 2017, 7, 4189–4196 CrossRef
- A. Screenithya, K. Surya and R. B. Sunoj, Hypercoordinate Iodine (III) Promoted Reactions And Catalysis: An Update On Current Mechanistic Understanding, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 7, e1299 Search PubMed
-
V. V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds, John Wiley and Sons, 2013 Search PubMed
- K. O. Christe, D. A. Dixon, J. C. Sanders, G. J. Schrobilgen and W. W. Wilson, Heptacoordination: pentagonal bipyramidal heptafluoroxenon (1+) XeF7+ and heptafluorotellurate (1−) TeF7− ions, J. Am. Chem. Soc., 1993, 115, 9461–9467 CrossRef CAS
- M. G. Goesten, R. Hoffmann and F. M. Bickelhaupt, Eight-Coordinate Fluoride in A Sillicate Double-Four-Ring, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 828–833 CrossRef CAS
- F. Peng, J. Botana, Y. Wang, Y. Ma and M. Miao, Unexpected Trend in Stability of Xe-F Compounds Under Pressure Driven by Xe-Xe Covalent Bonds, J. Phys. Chem. Lett., 2016, 7, 4562–4567 CrossRef CAS PubMed
- E. Magnusson, Hypercoordinate molecules of second-row elements: d functions or d orbitals?, J. Am. Chem. Soc., 1990, 112, 7940–7951 CrossRef CAS
- K. O. Christe, J. C. Sanders, G. J. Schrobilgen and W. W. Wilson, High-Coordination Number Fluoro-and-Oxofluoro-Anions. IF6O−, TeF6O2−, TeF7−, IF8− and TeF82−, ChemInform, 1991, 13, 837–840 Search PubMed
- S. Riedel and M. Kaupp, The Highest Oxidation States of the Transition Metal Elements, Coord. Chem. Rev., 2009, 253, 606–624 CrossRef CAS
- L. Zhang, Y. Wang, J. Lv and Y. Ma, Materials Discovery at High Pressures, Nat. Rev. Mater., 2017, 2, 17005 CrossRef CAS
- W. Grochala, R. Hoffmann, J. Feng and N. W. Ashcroft, The Chemical Imagination at Work in Very Tight Places, Angew. Chem., Int. Ed., 2007, 46, 3620–3642 CrossRef CAS
- A. Shamp and E. Zurek, Superconducting High-pressure Phases Composed of Hydrogen and Iodine, J. Phys. Chem. Lett., 2015, 6, 4067–4072 CrossRef CAS
- D. Duan, F. Tian, Y. Liu, X. Huang, D. Li, H. Yu, Y. Ma, B. Liu and T. Cui, Enhancement of Tc in the Atomic Phase of Iodine-doped Hydrogen at High Pressures, Phys. Chem. Chem. Phys., 2015, 17, 32335–32340 RSC
- J. Botana, X. Wang, C. Hou, D. Yan, H. Lin, Y. Ma and M. Miao, Mercury under Pressure Acts as a Transition Metal: Calculated from First Principles, Angew. Chem., Int. Ed., 2015, 127, 9412–9415 CrossRef
- J. Botana and M. S. Miao, Pressure-Stabilized Lithium Caesides with Caesium Anions beyond the −1 State, Nat. Commun., 2014, 5, 1–8 Search PubMed
- M. Miao, Caesium in High Oxidation States and as a p-Block Element, Nat. Chem., 2013, 5, 846 CrossRef CAS PubMed
- J. Liu, S. Zhang, W. Guan, G. Yang and Y. Ma, Gold with +4 and +6 Oxidation States in AuF4 and AuF6, J. Am. Chem. Soc., 2018, 140, 9545–9550 CrossRef PubMed
- G. Yang, Y. Wang, F. Peng, A. Bergara and Y. Ma, Gold as a 6p-Element in Dense Lithium Aurides, J. Am. Chem. Soc., 2016, 138, 4046–4052 CrossRef CAS PubMed
- J. Hooper, T. Terpstra, A. Shamp and E. Zurek, Composition and constitution of compressed strontium polyhydrides, J. Phys. Chem. C, 2014, 118, 6433–6447 CrossRef CAS
- J. Hooper, B. Altintas, A. Shamp and E. Zurek, Polyhydrides of the alkaline earth metals: a look at the extremes under pressure, J. Phys. Chem. C, 2013, 117, 2982–2992 CrossRef CAS
- Z. M. Geballe, H. Liu, A. K. Mishra, M. Ahart, M. Somayazulu, Y. Meng and R. J. Hemley, Synthesis and stability of lanthanum superhydrides, Angew. Chem., Int. Ed., 2018, 57, 688–692 CrossRef CAS PubMed
- T. Muramatsu, W. K. Wanene, M. Somayazulu, E. Vinitsky, D. Chandra, T. A. Strobel, V. V. Struzhkin and R. J. Hemley, Metallization and superconductivity in the hydrogen-rich ionic salt bareh9, J. Phys. Chem. C, 2015, 119, 18007–18013 CrossRef CAS
- R. Reichlin, A. K. McMahan, M. Ross, S. Martin, J. Hu, R. J. Hemley, H.-K. Mao and Y. W. Wu, Optical, x-ray, and band-structure studies of iodine at pressures of several megabars, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 3725 CrossRef CAS
- Q. Lv, X. Jin, T. Cui, Q. Zhuang, Y. Li, Y. Wang and X. Meng, Crystal structures and electronic properties of solid fluorine under high pressure, Chin. Phys. B, 2017, 26, 076103 CrossRef
- R. J. Gillespie and E. A. Robinson, Electron domains and the VSEPR model of molecular geometry, Angew. Chem., Int. Ed. Engl., 1996, 35, 495–514 CrossRef CAS
-
M. Rahm, R. Cammi, N. Ashcroft and R. Hoffmann, Electron Configuration and Electronegativity of the Atoms Under Compression, 256th ACS National Meeting & Exposition, Boston, MA, United States, 2018, PHYS-366 Search PubMed
-
A. Kramida, Y. Ralchenko and J. Reader, NIST Atomic Spectra Database, version 5.1, 2013. See http://physics.nist.gov/asd Search PubMed
- W. Kutzelnigg, Chemical bonding in higher main group elements, Angew. Chem., Int. Ed. Engl., 1984, 23, 272–295 CrossRef
- M. Randić, Ligand field splitting of d orbitals in eight coordinated complexes of dodecahedral structure, J. Chem. Phys., 1962, 36, 2094–2097 CrossRef
- M. Randić, Ligand field splitting of d orbitals in eight coordinated complexes of square antiprism structure, Croat. Chem. Acta, 1960, 32, 189 Search PubMed
- J. K. Burdett, R. Hoffmann and R. C. Fay, Eight-Coordination, Inorg. Chem., 1978, 17, 2553–2568 CrossRef CAS
- J. P. Connerade, V. K. Dolmatov and P. A. Lakshmi, The filling of shells in compressed atoms, J. Phys. B: At., Mol. Opt. Phys., 2000, 33, 251 CrossRef CAS
- L. J. Parker and J. V. Badding, Transition Element-Like Chemistry for Potassium under Pressure, Science, 1996, 273, 95–97 CrossRef CAS PubMed
- M. S. Miao and R. Hoffmann, High Pressure Electrides: A Predictive Chemical and Physical Theory, Acc. Chem. Res., 2014, 47, 1311–1317 CrossRef CAS PubMed
- P. Karen, Oxidation State, A Long-Standing Issue!, Angew. Chem., Int. Ed., 2015, 54, 4716–4726 CrossRef CAS PubMed
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Crystal Structure Prediction via Particle-Swarm Optimization, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 94116 CrossRef
- Y. Wang, J. Lv, L. Zhu and Y. Ma, CALYPSO: A Method for Crystal Structure Prediction, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS
- H. Wang, S. T. John, K. Tanaka, T. Iitaka and Y. Ma, Superconductive Sodalite-like Clathrate Calcium Hydride at High Pressures, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6463–6466 CrossRef CAS PubMed
- L. Zhu, H. Liu, C. J. Pickard, G. Zou and Y. Ma, Reactions of Xenon with Iron and Nickel Are Predicted in the Earth's Inner Core, Nat. Chem., 2014, 6, 644 CrossRef CAS PubMed
- J. Lv, Y. Wang, L. Zhu and Y. Ma, Predicted Novel High-Pressure Phases of Lithium, Phys. Rev. Lett., 2011, 106, 15503 CrossRef PubMed
- L. Zhu, H. Wang, Y. Wang, J. Lv, Y. Ma, Q. Cui, Y. Ma and G. S. Zou, Substitutional Alloy of Bi and Te at High Pressure, Phys. Rev. Lett., 2011, 106, 145501 CrossRef PubMed
- Y. Li, J. Hao, H. Liu, Y. Li and Y. Ma, The Metallization and Superconductivity of Dense Hydrogen Sulfide, J. Chem. Phys., 2014, 140, 174712 CrossRef PubMed
- S. Zhang, L. Zhu, H. Liu and G. Yang, Structure and Electronic Properties of Fe2SH3 Compound under High Pressure, Inorg. Chem., 2016, 55, 11434–11439 CrossRef CAS PubMed
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671 CrossRef CAS
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin-Zone Integrations, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef
- P. Blaha, K. Schwarz, P. Sorantin and S. B. Trickey, Full-Potential, Linearized Augmented Plane Wave Programs for Crystalline Systems, Comput. Phys. Commun., 1990, 59, 399–415 CrossRef CAS
- K. Parlinski, Z. Q. Li and Y. Kawazoe, First-Principles Determination of the Soft Mode in Cubic ZrO2, Phys. Rev. Lett., 1997, 78, 4063 CrossRef CAS
- A. Togo, F. Oba and I. Tanaka, First-Principles Calculations of the Ferroelastic Transition between Rutile-Type and CaCl2-Type SiO2 at High Pressures, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef
- A. D. Becke and K. E. Edgecombe, A Simple Measure of Electron Localization in Atomic and Molecular Systems, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS
- R. Dronskowski and P. E. Bloechl, Crystal Orbital Hamilton Populations (COHP): Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Analytic Projection from Plane-wave and PAW Wavefunctions and Application to Chemical-bonding Analysis in Solids, J. Comput. Chem., 2013, 34, 2557–2567 CrossRef CAS PubMed
- K. Lee, É. D. Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Higher-Accuracy van Der Waals Density Functional, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 81101 CrossRef
- I. Hamada, Van Der Waals Density Functional Made Accurate, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 121103 CrossRef
-
M. J. Frisch, et al., Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed description of the calculation methods and structural predictions, main structural parameters, phonon band structures and density of states, various analyses of I2 and IFx (x = 7, 8, 12) structures, atomic charges on iodine in different structures, and equations of states. See DOI: 10.1039/c8sc04635b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |