
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA double helix of opposite charges to form channels with unique CO2 selectivity and dynamics†
Guolong
Xing
 a,
Irene
Bassanetti
b,
Silvia
Bracco
b,
Mattia
Negroni
b,
Charl
Bezuidenhout
b,
Teng
Ben
*a,
Piero
Sozzani
*b and
Angiolina
Comotti
*b
a,
Irene
Bassanetti
b,
Silvia
Bracco
b,
Mattia
Negroni
b,
Charl
Bezuidenhout
b,
Teng
Ben
*a,
Piero
Sozzani
*b and
Angiolina
Comotti
*b
aDepartment of Chemistry, Jilin University, Changchun 130012, People's Republic of China
bDepartment of Materials Science, University of Milano Bicocca, Via R. Cozzi 55, Milan, Italy. E-mail: angiolina.comotti@unimib.it
First published on 24th October 2018
Abstract
Porous molecular materials represent a new front in the endeavor to achieve high-performance sorptive properties and gas transport. Self-assembly of polyfunctional molecules containing multiple charges, namely, tetrahedral tetra-sulfonate anions and bifunctional linear cations, resulted in a permanently porous crystalline material exhibiting tailored sub-nanometer channels with double helices of electrostatic charges that governed the association and transport of CO2 molecules. The charged channels were consolidated by robust hydrogen bonds. Guest recognition by electrostatic interactions remind us of the role played by the dipolar helical channels in regulatory biological membranes. The systematic electrostatic sites provided the perfectly fitting loci of complementary charges in the channels that proved to be extremely selective with respect to N2 (S = 690), a benchmark in the field of porous molecular materials. The unique screwing dynamics of CO2 travelling along the ultramicropores with a step-wise reorientation mechanism was driven by specific host–guest interactions encountered along the helical track. The unusual dynamics with a single-file transport rate of more than 106 steps per second and an energy barrier for the jump to the next site as low as 2.9 kcal mol−1 was revealed unconventionally by complementing in situ13C NMR anisotropic line-shape analysis with DFT modelling of CO2 diffusing in the crystal channels. The peculiar sorption performances and the extraordinary thermal stability up to 450 °C, combined with the ease of preparation and regeneration, highlight the perspective of applying these materials for selective removal of CO2 from other gases.
Introduction
The manufacture of materials with permanent porosity is one of the major challenges in current research. This goal can be achieved through various strategies that promote the formation of covalent and metal–organic frameworks as well as molecular architectures.1–8 They are diversified by their greater or lesser tendency to form stable structures and their degree of crystalline periodicity. Highly crystalline porous molecular materials can be constructed easily by spontaneous assembly of precursor molecules, provided that their shapes and functionalities are adequately designed.9–11 Indeed, fully organic porous materials can be assembled by means of soft and reversible interactions enabling the highest modularity, processability and large single-crystal formation. Their excellent performances in gas capture and interaction with chemical and physical stimuli have been explored in recent years.12–29Organic functions of opposite charges on complementary molecules promote intense attractive interactions, and reciprocal recognition such as positively charged ammonium groups with carboxylic and sulfonic anions; thus, electrostatic interactions can be exploited to sustain robust molecular frameworks.30–35 Additionally, electrostatic charges can be exposed to the pores, making it possible to expand research towards unexplored developments.
In our approach, multiple-charged functional groups protruding in the space produce directional interactions, favourable to the design of porous molecular architectures, forming programmable charged channels. In general, the shape and symmetry of the building blocks to construct porous frameworks must be taken into account: specifically, highly symmetric and rigid tetrahedral struts (S4 point group) constitute a primary element, which sustained permanently porous materials and yielded very high surface areas.36–44
Biological systems are promising sources for the formation of sub-nanometer channels for the transport of molecules and ions, wherein the electrostatic distribution of charges on the channel walls and steric factors play a strategic role for building useful functions.45 By analogy, we have engineered a permanently porous 3D molecular architecture containing sub-nm channels formed by a double helix with two intertwined and alternatively charged ribbons, characteristic of transmembrane channels. It was generated by the assembly of tetrahedral and tetrafunctional 4,4′,4′′,4′′′-methane tetrabenzenesulfonate (TBS) anions and a bifunctional cation 1,4-diamidiniumbenzene (DAB) and was found to be highly stable. The porous structure was capable of reversibly absorbing relevant gases such as CO2 and CH4 with an exceptional CO2/N2 selectivity of 690 owing to the tight fitting and the complementary electrostatic pattern of the sub-nm channels. The direct detection of CO2 pervading the porous molecular framework was provided by multinuclear 2D MAS NMR spectra, depicting the exceptional CO2-matrix association in the properly fitting pockets. Furthermore, the anisotropic 13C NMR variable-temperature lineshapes unveiled the unique coordinated rotation and translation dynamics of CO2 while it jumps from one site to the next, following the helical charge patterning of the channel. Intriguingly, CO2 molecules receive the orientational encoding from the nanochannel sites at each diffusion turn of the screwing motion. Thus, by this unconventional means, we could establish the transport rate of a single gas molecule that is as fast as a million steps per second, corresponding to guest diffusion rates observed in biological channels. DFT calculations and Molecular Dynamics (MD) supported the NMR determination of the preferred arrangement and the energy landscape perceived by CO2 exploring the channels.
Results and discussion
Fabrication of charged sub-nanometer channels
The organotetrasulfonate TBS self-assembled with organodiamidinium DAB to form needlelike single crystals suitable for X-ray analysis (ESI†). After guest removal by thermal treatment under vacuum at 150 °C, the 3D architecture is maintained, yielding a new crystalline porous organosulfonate–amidinium salt (CPOS-5) (Fig. 1). It crystallizes in the tetragonal I41a space group, which contains a quarter of TBS (a phenyl-SO3− group) and half of DAB in the asymmetric unit (molecular formula equal to TBS(DAB)2). A 4-fold rotoinversion axis crosses the central carbon (C1), which orients four sulphonate groups along the vertices of a tetrahedron. Hence, organosulfonate anions and organoamidinium cations, driven by hydrogen bonds, assemble in a double helix of electrically charged ribbons (Fig. 1C). Both negatively and positively charged moieties draw alternatively charged helices that lead to the formation of 1D-channels (Fig. 1D), running parallel to the c-axis. The empty channels with a cross-section of 5.3 × 6.8 Å2 generate a porosity of 743.03 Å3 per cell corresponding to 14.6% of the cell volume (radius of the rolling sphere = 1.2 Å). The 13C MAS NMR spectrum confirmed the absence of guests in the cavities (ESI†). The porous compound exhibits high stability to water and temperature (up to 400 °C) owing to the connection of four organosulfonate anions to DAB monomers through charge-assisted hydrogen bonds in all directions.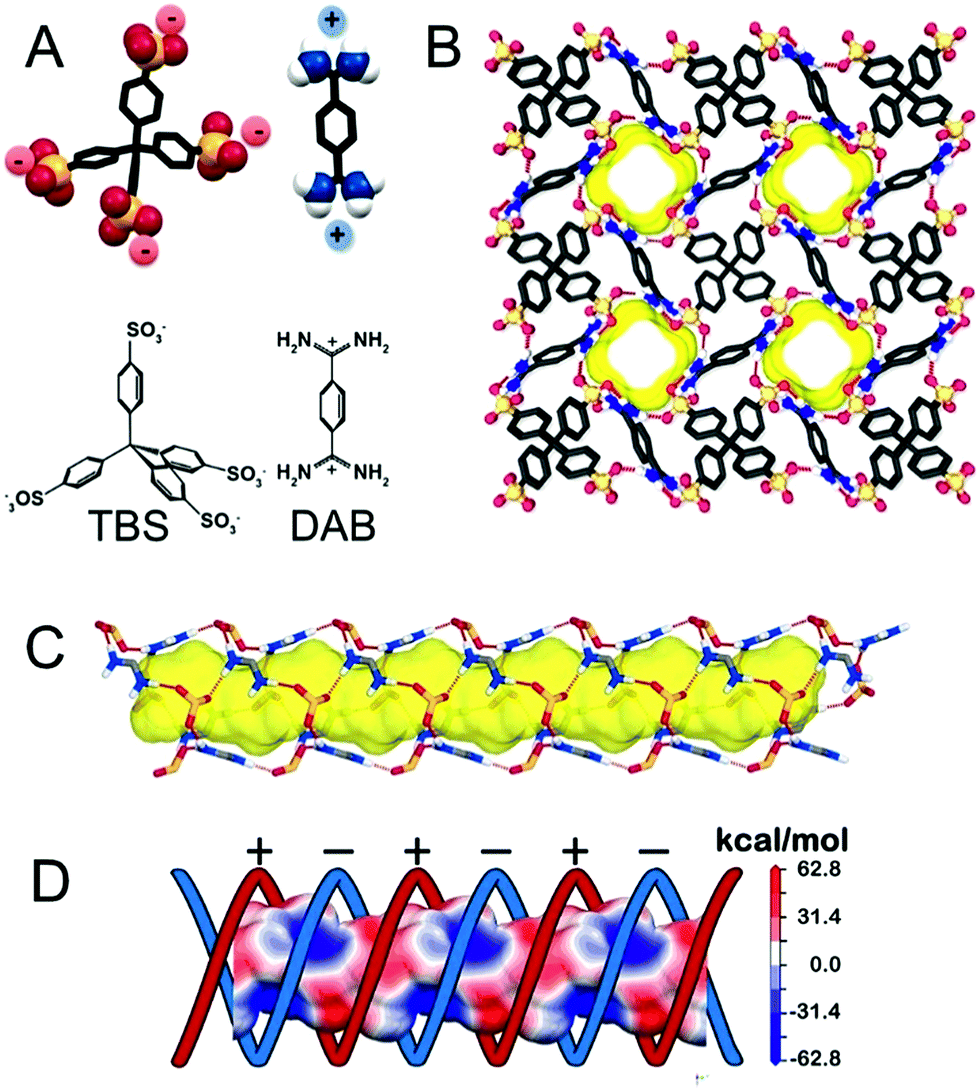 | ||
| Fig. 1 (A) Schematic representation of anionic TBS and cationic DAB synthons of the porous organic framework. (B) View of the porous framework along the c-axis; yellow represents the empty channels (a rolling sphere of 1.4 Å was used). The channel running parallel to the c-axis: (C) the synthons are connected through a H-bonding network in a helical fashion; (D) electrostatic map showing the positively and negatively charged helical ribbons. | ||
Gas capture and CO2 selectivity
CO2 adsorption isotherms were collected on CPOS-5 to demonstrate its permanent porosity (Fig. 2). Interestingly, at 273 K the CO2 adsorption isotherm displayed a Langmuir profile, which at 1 bar reached the value of 48 cm3 g−1, representing 8 CO2 molecules per unit cell (Fig. 2A). Notably, CO2 uptake was already extremely high at low pressure and exhibited a loading as high as 59 mg g−1 (273 K) at 0.1 bar, suggesting excellent affinity of the quadrupolar gas molecules with charge-decorated pore surfaces. At 0.1 bar and at room temperature, typical conditions of CO2 pressure in flue gas from power plants, CPOS-5 adsorbs 39 mg g−1, outperforming many well-known MOFs with greater surface areas such as HKUST-1, UMCM-150(N2), MIL-47, IRMOF-3 and ZIF-8,46 porous hydrogen-bonds and supramolecular organic frameworks such as SOF-1,23 SOF-7,47 HOF-3 (ref. 27) and CB6 (ref. 17) and its absorption value is comparable to that of the best performing HOF-5.24 | ||
| Fig. 2 (A) CO2 and N2 isotherms of CPOS-5 collected at 273 K (blue circles and diamonds, respectively) and 298 K (light-blue circles and diamonds, respectively). (B) CO2/N2 selectivity values of CPOS-5 versus pressure calculated starting from a 15/85 mixture and applying IAST calculation. (C) Isosteric heats of adsorption (Qst) versus CO2 and CH4 loading. | ||
The isosteric heat of adsorption Qst calculated using the van't Hoff equation accounted for 34.5 kJ mol−1, which is in agreement with the binding energy determined by DFT calculations ΔE = 33.8 kJ mol−1 (ESI†). The Qst value is comparable to those of some high-performance porous crystalline materials such as SOF-1,23 MIL-53,48 HKUST-1 (ref. 49) and NaX zeolites,50 which contain channels decorated with charged moieties.
The prominent difference in the CO2 uptake with respect to N2 under the same pressure and temperature conditions prompted us to calculate the CO2/N2 selectivity using Ideal Adsorbed Solution Theory (IAST) applied to single-component isotherms starting from a 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 (CO2:N2) mixture in order to simulate the flue gas conditions (Fig. 2B). The CO2/N2 selectivity at 273 K exhibited values in the 320–690 range at low pressures (<0.1 bar) and 110 at 1 bar. The CO2/N2 selectivity at room temperature was 540, as calculated using the ratio of Henry's constants (ESI†). This is one of the highest selectivities shown to date in porous molecular materials.19,20,24,47
:85 (CO2:N2) mixture in order to simulate the flue gas conditions (Fig. 2B). The CO2/N2 selectivity at 273 K exhibited values in the 320–690 range at low pressures (<0.1 bar) and 110 at 1 bar. The CO2/N2 selectivity at room temperature was 540, as calculated using the ratio of Henry's constants (ESI†). This is one of the highest selectivities shown to date in porous molecular materials.19,20,24,47
Furthermore, the isosteric heat of adsorption calculated for methane is 21.0 kJ mol−1, which is remarkably high compared to those of typical 3D frameworks with open metal sites or higher surface areas (Fig. 2C).9 It is comparable to SOF-1 (ΔH = 20.8 kJ mol−1) and one of the highest reported values in microporous polymers and crystals.23,51–53
Screwing dynamics and diffusion rate of CO2 in the channels
The remarkable CO2-matrix association allowed direct observation of the gas exploring the nanochannels by NMR spectroscopy. The 13C MAS NMR spectrum showed two dominant signals at δc = 125.5 and 125.8 ppm for 13C-enriched CO2: the former identifying free CO2 and the latter CO2 housed in the confined space of the crystal (Fig. 3A). The downfield signal is generated by the magnetic susceptibility effect experienced by CO2 molecules embedded in the polar environment of the crystal. In the cross-polarization (CP) spectrum, the δc = 125.8 ppm signal is largely dominating, for the fast magnetization transfer even at room temperature, from the matrix hydrogens to the CO2 carbons lying a short distance away (<1 nm),54 proving that the gas is exceptionally well-retained (ESI†). This is a remarkable result given the well-known difficulty of transferring nuclear polarization in conventional systems from a solid towards a gas, because of the short gas residence time, especially at room temperature.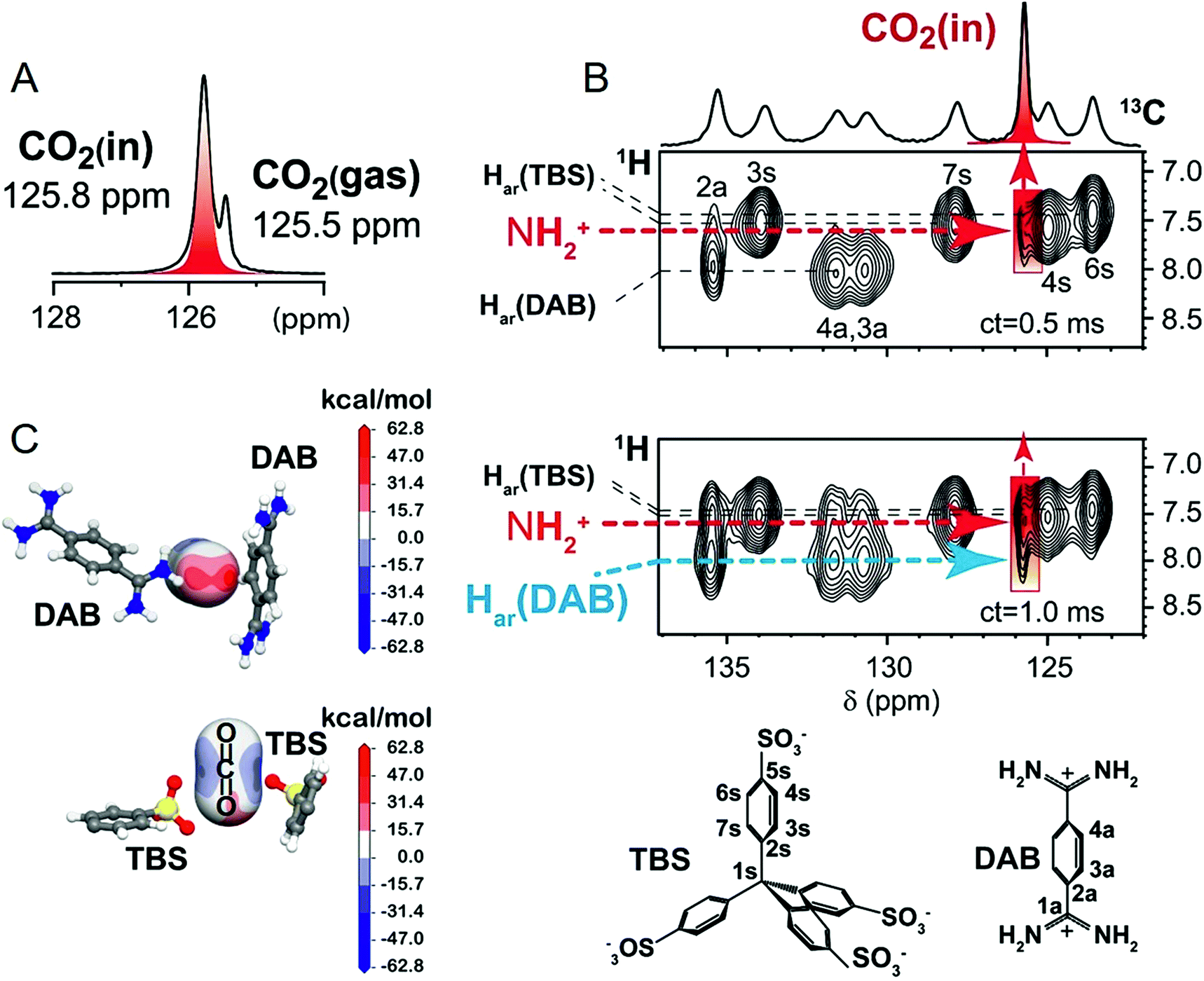 | ||
| Fig. 3 CPOS-5 loaded with 13C-enriched CO2 (0.75 bar at RT): (A) 13C MAS NMR spectrum; (B) 2D 1H–13C HETCOR MAS NMR spectra collected at room temperature and at two distinct contact times (ct). The host–CO2 cross peaks are highlighted in red. Chemical structures of TBS and DAB with labels (below). (C) Electrostatic potential projected on CO2 molecules by the DAB and TBS host moieties. The CO2 molecules were localized by DFT calculations. | ||
The surprising efficiency of gas–solid CP was exploited for the success of 2D 1H–13C CP NMR, which allowed for both in situ spectroscopic detection of CO2 and the identification of the specific interaction sites (Fig. 3B).33,55,56 Even at a contact time as short as 0.5 ms and room temperature, CO2 carbon nuclei receive magnetization from the amidinium hydrogens (at δH = 7.4 ppm), showing the short distances of the gas from these species lining the channel walls. The short internuclear distances were confirmed by ab initio DFT calculations, which pointed out the CO2 location and a distance of 3.24 Å between the N–H hydrogen and CO2 carbon. The close proximity of the CO2 carbon with the N–H group is imposed by the interaction of one of the CO2 oxygens with the amidinium cation. Also the aromatic C–H group of DAB undergoes favorable interactions with CO2 oxygens as demonstrated by the highly positive electrostatic potentials (Fig. 3C: the red regions on the oxygen atom of CO2). Actually, at a longer contact time of 1 ms the 2D NMR spectrum highlights the correlation between the aromatic C–H of DAB and CO2 carbon (distance of 4.18 Å). With regard to the organosulfonate anions, DFT calculations show CO2 molecules clamped in between the oxygens of two TBS sulfonate groups (Fig. 3C: the blue regions on the carbon atom of CO2), keeping the C–H hydrogens of TBS far apart from the CO2 carbon (distance of 5.75 Å), consistent with the absence of NMR cross-correlations. From the combined NMR and modelling approach, we can conclude that the charge distribution of the host complements the charges on the CO2 molecule and determines the stabilization of the gas in the channels.
Remarkably, multiple aspects of the dynamics of the CO2 molecules in the channels came to light from variable temperature 13C NMR spectra recorded in the static mode (Fig. 4A). Actually, the spectra depicted axial chemical shift anisotropy (CSA) restricted to a fraction of the total spectral width of solid CO2 in the bulk.57,58 From the NMR lineshape simulation we were able to determine the mechanism of motion, the dynamics and the inclination of CO2 molecules in the channels. The lineshape at room temperature describes a rapid 4-site re-orientation of CO2 molecules by a 90° jump mechanism (Fig. 4B). Nonetheless, the CO2 main axis maintains an inclination angle equal to θ = 56.7° with respect to the rotation axis. By lowering the temperature to 215 K, the inclination increases to 58.1° due to the increased loading and the larger contribution of CO2–CO2 interactions (ESI†). Notably, by the spectral analysis at variable temperature, it was possible to measure the 90° jump rates (k) that are higher than 106 Hz at room temperature and still over 105 Hz at 215 K.
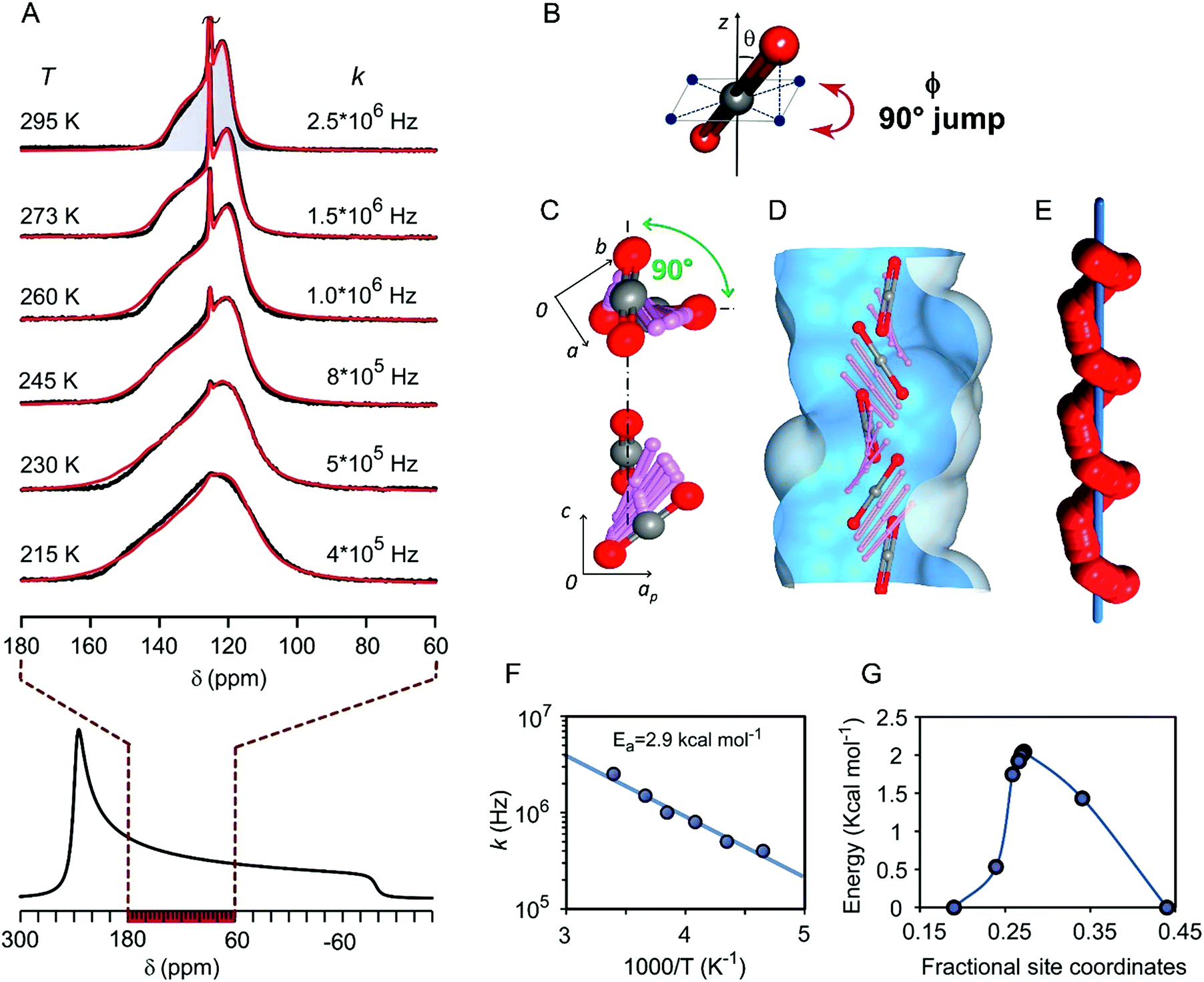 | ||
| Fig. 4 (A) Variable temperature 13C NMR spectra of the CPOS-5/13CO2 sample under static conditions. The chemical shift anisotropic lineshape of solid CO2, simulated starting from the main tensor components, is reported below for comparison. (B) Inclination angle (θ) of the CO2 main-axis with respect to the reorientation-axis z about which the 90° (ϕ angle) jump occurs. (C) Screwing mechanism for CO2 rototranslational dynamics in the channel: the 90° rotation angle of CO2 projection onto the ab-plane and along the channel c-axis for CO2 reorientation from one site to the next. (D) Overlay of the CO2 positions along the channel as calculated by the DFT transition state method. (E) Helical trajectory of one CO2 oxygen atom (red spheres) about the channel axis (blue bar). (F) Arrhenius plot of reorientational rates (k) versus the inverse of temperature. (G) Calculated energy profile for the rototranslation of CO2 along the channel. | ||
To understand in-depth the NMR determinations of the gas diffusing in the tight-fitting channels, molecular dynamics combined with ab initio calculations provided a model for the energy profile and interaction sites perceived by CO2 travelling in the channels. Each site accommodates a CO2 molecule at a time and imparts to the CO2 molecule an average inclination angle θ of 57°–58°, in perfect agreement (±0.7°) with the NMR data (ESI†). Regarding the fast 90° re-orientation, it cannot occur within the same site without a very high energy cost, for steric and electrostatic reasons. In contrast, the next site along the channel (symmetry related by a four-fold screw axis, 41) can host CO2 after its 90° turn (Fig. 4C). Thus, CO2 reorientation, observed by NMR, implies rotation correlated with sliding motion to the next site with the rate of 106 steps per second (Fig. 4D). Indeed, the unique feature of this diffusion phenomenon is the concerted roto-translation mechanism (i.e. simultaneous rotation and translation along the c axis) occurring with extremely rapid dynamics of the confined CO2, which follows a helical trajectory by a screwing path along a single file (Fig. 4E). From the NMR jump rates (k) vs. temperature, an Arrhenius plot can be derived, establishing a small activation energy of 2.9 kcal mol−1 (Fig. 4F), in excellent agreement with the energy barrier of 2.1 kcal mol−1 for a diffusion step of CO2 molecules, during the helical transport, as calculated by DFT analysis (Fig. 4G).
Conclusions
Multifunctional tetra-anionic and di-cationic molecules combine together to form frameworks with the property of being permanently porous. The crystalline framework was designed by the use of intrinsically rigid molecules as constructive elements and ionic interactions between sulfonate anions and amidinium cations. The ionic patterning of the crystalline channels allows the establishment of high interactions of 35 kJ mol−1, ideal for CO2 capture/release cycles. This high energy value stably retains the quadrupolar molecules of CO2. Such CO2 sequestration was so efficient that the CO2/N2 selectivity values at low pressures (<0.1 bar) range from 320 to 690.The interaction strength of the CO2 with the host was recognized by the striking capability of CO2 to receive nuclear polarization from the framework at room temperature and to develop the anisotropic patterns in the static 13C NMR spectra. A unique screwing mechanism of CO2, which follows the helical track of the charges on the channel walls, was observed. The rate of a single step (>106 steps per second) in the diffusion trajectory along the tunnel could be unconventionally established by NMR. The orientational and dynamical features are enforced by the computational models of gas–framework interactions. The contribution of the charges patterning the channel walls is of paramount importance for directing the association of a gas, such as CO2, with the host. These pieces of evidence encourage the use of multiple charge molecules of opposite polarity to spontaneously self-organize into functional materials endowed with high thermal stability and exploit the rich modularity of polyanions and polycations to design structures tailored to a specific gas for capture and sequestration.
Moreover, the fabrication of artificial charged channels in crystalline materials with a high degree of order and uniformity as well as a controllable structure promotes the understanding of the role of electrostatic interactions in the transport properties of transmembrane channels of biological systems.45,59 Indeed, the CO2 diffusion in helical narrow channels, such as in aquaporins, plays a strategic role in the selection and concentration of gases in plant cells.59 Our newly fabricated crystalline channels share helical and sub-nanometric structural features, and transport properties in a single file through the polar environment, thus achieving high selectivity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. C. would like to thank the Cariplo Foundation (Balance Project), INSTM Consortium/Lombardy Region and PRIN 2016. The “Dipartimenti di Eccellenza 2018–2022” project is acknowledged for the financial support. This work is supported by the NSFC (Grant no. 21390394, 21471065) and the “111” project (B07016).Notes and references
- S. Kitagawa, R. Kitaura and S.-I. Noro, Functional Porous Coordination Polymers, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- A. Schoedel, Z. Li and O. M. Yaghi, The role of metal–organic frameworks in a carbon-neutral energy cycle, Nat. Energy, 2016, 1, 1–13 Search PubMed.
- H. C. Zhou and S. Kitagawa, Metal–Organic Frameworks (MOFs), Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- S. Das, P. Heasman, T. Ben and S. Qiu, Porous Organic Materials, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- Z. Zhang and M. J. Zaworotko, Template-directed synthesis of metal–organic materials, Chem. Soc. Rev., 2014, 43, 5444–5445 RSC.
- A. Thomas, Functional Materials: From Hard to Soft Porous Frameworks, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed.
- P. Ramaswamy, N. E. Wong and G. H. K. Shimizu, MOFs as proton conductors – challenges and opportunities, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- M. Simard, D. Su and J. D. Wuest, Use of hydrogen bonds to control molecular aggregation. Self-assembly of three-dimensional networks with large chambers, J. Am. Chem. Soc., 1991, 113, 4696–4698 CrossRef CAS.
- G. Zhang and M. Mastalerz, Organic cage compounds – From shape-persistency to function, Chem. Soc. Rev., 2014, 43, 1934–1947 RSC.
- T. Hasell and A. I. Cooper, Porous organic cages: soluble, modular and molecular pores, Nat. Rev. Mater., 2016, 1, 1–14 Search PubMed.
- N. B. J. McKeown, Nanoporous molecular crystals, J. Mater. Chem., 2010, 20, 10588–10597 RSC.
- A. G. Skaterand and A. I. Cooper, Porous materials. Function-led design of new porous materials, Science, 2015, 348, aaa8075 CrossRef PubMed.
- P. Sozzani, S. Bracco, A. Comotti, L. Ferrettia and R. Simonutti, Methane and carbon dioxide storage in a porous van der Waals crystal, Angew. Chem., Int. Ed., 2005, 44, 1816–1820 CrossRef CAS PubMed.
- L. J. Barbour, Crystal porosity and the burden of proof, Chem. Commun., 2006, 1163–1168 RSC.
- K. J. Msayib, D. Book, P. M. Budd, N. Chaukura, K. D. M. Harris, M. Helliwell, S. Tedds, A. Walton, J. E. Warren, M. Xu and N. B. McKeown, Nitrogen and Hydrogen Adsorption by an Organic Microporous Crystal, Angew. Chem., Int. Ed., 2009, 48, 3273–3277 CrossRef CAS PubMed.
- A. Comotti, S. Bracco, G. Distefano and P. Sozzani, Methane, carbon dioxide and hydrogen storage in nanoporous dipeptide-based materials, Chem. Commun., 2009, 284–286 RSC.
- H. Kim, H. Kim, Y. Kim, M. Yoon, S. Lim, S. M. Park, G. Seo and K. Kim, Highly Selective Carbon Dioxide Sorption in an Organic Molecular Porous Material, J. Am. Chem. Soc., 2010, 132, 12200–12202 CrossRef CAS PubMed.
- M. Mastalerz, M. W. Schneider, I. M. Oppel and O. Presly, A Salicylbisimine Cage Compound with High Surface Area and Selective CO2/CH4 Adsorption, Angew. Chem., Int. Ed., 2011, 50, 1046–1051 CrossRef CAS PubMed.
- V. N. Yadav, A. Comotti, P. Sozzani, S. Bracco, T. Bonge-Hansen, M. Hennum and C. H. Gorbitz, Microporous Molecular Materials from Dipeptides Containing Non-proteinogenic Residues, Angew. Chem., Int. Ed., 2015, 54, 15684–15688 CrossRef PubMed.
- I. Bassanetti, S. Bracco, A. Comotti, M. Negroni, C. Bezuidenhout, S. Canossa, P. P. Mazzeo, L. Marchio' and P. Sozzani, Flexible porous molecular materials responsive to CO2, CH4 and Xe stimuli, J. Mater. Chem. A, 2018, 6, 14231–14239 RSC.
- M. Baroncini, S. D'Agostino, G. Bergamini, P. Ceroni, A. Comotti, P. Sozzani, I. Bassanetti, F. Grepioni, T. M. Hernandez, S. Silvi, M. Venturi and A. Credi, Photoinduced reversible switching of porosity in molecular crystals based on star-shaped azobenzene tetramers, Nat. Chem., 2015, 7, 634–640 CrossRef CAS PubMed.
- Y. He, S. Xiang and B. Chen, A Microporous Hydrogen-Bonded Organic Framework for Highly Selective C2H2/C2H4 Separation at Ambient Temperature, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS PubMed.
- W. Yang, et al., Exceptional Thermal Stability in a Supramolecular Organic Framework: Porosity and Gas Storage, J. Am. Chem. Soc., 2010, 132, 14457–14469 CrossRef CAS PubMed.
- H. Wang, et al., A Flexible Microporous Hydrogen-Bonded Organic Framework for Gas Sorption and Separation, J. Am. Chem. Soc., 2015, 137, 9963–9970 CrossRef CAS PubMed.
- W. Yan, et al., A triptycene-based porous hydrogen-bonded organic framework for guest incorporation with tailored fitting, Chem. Commun., 2017, 53, 3677–3680 RSC.
- C. A. Zentner, H. W. H. Lai, J. T. Greenfield, R. A. Wiscons, M. Zeller, C. F. Campana, O. Talu, S. A. FitzGeralde and J. L. C. Rowsel, High surface area and Z′ in a thermally stable 8-fold polycatenated hydrogen-bonded framework, Chem. Commun., 2015, 51, 11642–11645 RSC.
- P. Li, et al., A rod-packing microporous hydrogen-bonded organic framework for highly selective separation of C2H2/CO2 at room temperature, Angew. Chem., Int. Ed., 2015, 54, 574–577 CAS.
- F. Hu, et al., An Ultrastable and Easily Regenerated Hydrogen-Bonded Organic Molecular Framework with Permanent Porosity, Angew. Chem., Int. Ed., 2017, 56, 2101–2104 CrossRef CAS PubMed.
- I. Hisaki, S. Nakagawa, N. Tohnai and M. Miyata, A C3-Symmetric Macrocycle Based, Hydrogen Bonded, Multiporous Hexagonal Network as a Motif of Porous Molecular Crystals, Angew. Chem., Int. Ed., 2015, 54, 3008–3012 CrossRef CAS PubMed.
- T. Adachi and M. D. Ward, Versatile and Resilient Hydrogen-Bonded Host Frameworks, Acc. Chem. Res., 2016, 49, 2669–2679 CrossRef CAS PubMed.
- Y. Liu, C. Hu, A. Comotti and M. D. Ward, Supramolecular Archimedean cages assembled with 72 hydrogen bonds, Science, 2011, 333, 436–440 CrossRef CAS PubMed.
- A. Comotti, S. Bracco, A. Yamamoto, M. Beretta, T. Hirukawa, N. Tohnai, M. Miyata and P. Sozzani, Engineering Switchable Rotors in Molecular Crystals with Open Porosity, J. Am. Chem. Soc., 2014, 136, 618–621 CrossRef CAS PubMed.
- S. Bracco, T. Miyano, M. Negroni, I. Bassanetti, L. Marchio', P. Sozzani, N. Tohnai and A. Comotti, CO2 regulates molecular rotor dynamics in porous materials, Chem. Commun., 2017, 53, 7776–7779 RSC.
- G. Xing, I. Bassanetti, T. Ben, S. Bracco, P. Sozzani, L. Marchio' and A. Comotti, Multifunctional Organosulfonate Anions Self-Assembled with Organic Cations by Charge-Assisted Hydrogen Bonds and the Cooperation of Water, Cryst. Growth Des., 2018, 18, 2082–2092 CrossRef CAS.
- G. Xing, T. Yan, S. Das, T. Bena and S. Qiu, Synthesis of Crystalline Porous Organic Salts with High Proton Conductivity, Angew. Chem., Int. Ed., 2018, 57, 5345–5349 CrossRef CAS PubMed.
- X. Wang, M. Simard and J. Wuest, Molecular Tectonics. Three-Dimensional Organic Networks with Zeolitic Properties, J. Am. Chem. Soc., 1994, 116, 12119–12120 CrossRef CAS.
- M. J. Zaworotko, Crystal engineering of diamondoid networks, Chem. Soc. Rev., 1994, 23, 283–288 RSC.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qui and G. Zhu, Targeted synthesis of a porous aromatic framework with high stability and exceptionally high surface area, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Porous Polymer Networks: Synthesis, Porosity, and Applications in Gas Storage/Separation, Chem. Mater., 2010, 22, 5964–5972 CrossRef CAS.
- W. Lu, D. Yuan, J. Sculley, D. Zhao, R. Krishna and H.-C. Zhou, Sulfonate-Grafted Porous Polymer Networks for Preferential CO2 Adsorption at Low Pressure, J. Am. Chem. Soc., 2011, 133, 18126–18129 CrossRef CAS PubMed.
- P. Pandey, O. K. Farha, A. M. Spokoyny, C. A. Mirkin, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, A “click-based” porous organic polymer from tetrahedral building blocks, J. Mater. Chem., 2011, 21, 1700–1703 RSC.
- S. S. Han, H. Furukawa, O. M. Yaghia and W. A. Goddard, Covalent Organic Frameworks as Exceptional Hydrogen Storage Materials, J. Am. Chem. Soc., 2008, 130, 11580–11581 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, Storage of Hydrogen, Methane, and Carbon Dioxide in Highly Porous Covalent Organic Frameworks for Clean Energy Applications, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- A. Comotti, J. Perego, D. Piga, S. Bracco and P. Sozzani, Expandable porous organic frameworks with built-in amino and hydroxyl functions for CO2 and CH4 capture, Chem. Commun., 2018, 54, 9321–9324 RSC.
- R. E. Hibbs and E. Gouaux, Principles of activation and permeation in an anion-selective Cys-loop receptor, Nature, 2011, 474, 54–60 CrossRef CAS PubMed.
- A. O. Yazaydin, R. Snurr, T.-H. Park, K. Koh, J. Liu, M. D. LeVan, A. I. Benin, P. Jakubczak, M. Lanuza, D. B. Galloway, J. J. Low and R. R. Willis, Screening of Metal–Organic Frameworks for Carbon Dioxide Capture from Flue Gas Using a Combined Experimental and Modeling Approach, J. Am. Chem. Soc., 2009, 131, 18198–18199 CrossRef CAS PubMed.
- J. Lu, C. P. Krap, M. Suyetin, N. H. Alsmail, Y. Yan, S. Yang, W. Lewis, E. Bichoutskaia, C. C. Tang, A. J. Blake, R. Cao and M. Schoeder, A Robust Binary Supramolecular Organic Framework (SOF) with High CO2 Adsorption and Selectivity, J. Am. Chem. Soc., 2014, 136, 12828–12831 CrossRef CAS PubMed.
- S. Bourrelly, P. L. Llewellyn, C. Serre, F. Millange, T. Loiseau and G. Férey, Different Adsorption Behaviors of Methane and Carbon Dioxide in the Isotypic Nanoporous Metal Terephthalates MIL-53 and MIL-47, J. Am. Chem. Soc., 2005, 127, 13519–13521 CrossRef CAS PubMed.
- P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. De Weireld, J.-S. Chang, D.-Y. Hong, Y.-K. Hwang, S.-H. Jhung and G. Férey, High Uptakes of CO2 and CH4 in Mesoporous Metal–Organic Frameworks MIL-100 and MIL-101, Langmuir, 2008, 24, 7245–7250 CrossRef CAS PubMed.
- J.-S. Lee, J.-H. Kim, J.-T. Kim, J.-K. Suh, J.-M. Lee and C.-H. Lee, Adsorption Equilibria of CO2 on Zeolite 13X and Zeolite X/Activated Carbon Composite, J. Chem. Eng. Data, 2002, 47, 1237–1242 CrossRef CAS.
- B. Li, H.-M. Wen, W. Zhou, Q. Xu and B. Chen, Porous Metal–Organic Frameworks: Promising Materials for Methane Storage, Chem, 2016, 1, 557–580 CAS.
- J. A. Mason, M. Veenstra and J. R. Long, Evaluating metal–organic frameworks for natural gas storage, Chem. Sci., 2014, 5, 32–51 RSC.
- H. Li, K. Wang, Y. Sun, C. T. Lollar, J. Li and H.-C. Zhou, Recent advances in gas storage and separation using metal–organic frameworks, Mater. Today, 2018, 21, 108–121 CrossRef CAS.
- A. Pines, J. S. Gibby and J. S. Waugh, Proton-enhanced NMR of dilute spins in solids, J. Chem. Phys., 1973, 59, 569–590 CrossRef CAS.
- I. Bassanetti, A. Comotti, P. Sozzani, S. Bracco, G. Calestani, F. Mezzadri and L. Marchiò, Porous Molecular Crystals by Macrocyclic Coordination Supramolecules, J. Am. Chem. Soc., 2014, 136, 14883–14895 CrossRef CAS PubMed.
- A. Comotti, A. Fraccarollo, S. Bracco, M. Beretta, G. Distefano, M. Cossi, L. Marchese, C. Riccardi and P. Sozzani, Porous dipeptide crystals as selective CO2 adsorbents: experimental isotherms vs. grand canonical Monte Carlo simulations and MAS NMR spectroscopy, CrystEngComm, 2013, 15, 1503–1507 RSC.
- C. R. Bowers, H. W. Long, T. Pietrass, H. C. Gaede and A. Pines, A. Cross polarization from laser-polarized solid xenon to 13CO2 by low-field thermal mixing, Chem. Phys. Lett., 1993, 205, 168–170 CrossRef CAS.
- X. Kong, E. Scott, W. Ding, J. A. Mason, J. R. Long and A. A. Reimer, CO2 Dynamics in a Metal–Organic Framework with Open Metal Sites, J. Am. Chem. Soc., 2012, 134, 14341–14344 CrossRef CAS PubMed.
- N. Uehlein, C. Lovisolo, F. Siefritz and R. Kaldenhoff, The tobacco aquaporin NtAQP1 is a membrane CO2 pore with physiological functions, Nature, 2003, 425, 734–737 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1865475 and 1865476. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04376k |
| This journal is © The Royal Society of Chemistry 2019 |