
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic nickel–cobalt hydrides in H2 activation and catalytic proton reduction†
Xiaoxiao
Chu
ab,
Jihao
Jin
a,
Bangrong
Ming
a,
Maofu
Pang
a,
Xin
Yu
a,
Chen-Ho
Tung
a and
Wenguang
Wang
 *a
*a
aKey Lab for Colloid and Interface Chemistry of Education Ministry, School of Chemistry and Chemical Engineering, Shandong University, 250100, China. E-mail: wwg@sdu.edu.cn
bSchool of Chemistry and Materials Science, Ludong University, Yantai, 264025, China
First published on 30th October 2018
Abstract
The synergism of the electronic properties of nickel and cobalt enables bimetallic NiCo complexes to process H2. The nickel–cobalt hydride [(dppe)Ni(pdt)(H)CoCp*]+ ([1H]+) arising from protonation of the reduced state 1 was found to be an efficient electrocatalyst for H2 evolution with Cl2CHCOOH, and the oxidized [Ni(II)Co(III)]2+ form is capable of activating H2 to produce [1H]+. The features of stereodynamics, acid–base properties, redox chemistry and reactivity of these bimetallic NiCo complexes in processing H2 are potentially related to the active site of [NiFe]-H2ases.
Introduction
Bimetallic complexes have emerged as important components in inorganic chemistry, homogeneous catalysis and biocatalysis.1–5 Compared to monometallic catalytic processes, bimetallic catalysis can promote the rate and selectivity of a reaction catalyzed by the first metal in the bimetallic catalyst by synergism with the electronic and steric properties of the second metal, or through a concerted activation process in which both metals participate in activation of the substrate, lowering the activation barrier.6,7 Bimetallic catalysis is common in metalloenzymes that perform multi-electron redox reactions.8–10 Examples are the active sites of [FeFe]-H2ases and [NiFe]-H2ases, which catalyze the production and uptake of dihydrogen.11,12 Both of the catalysis invoke bimetallic hydride intermediates, but the [FeFe]-H2ases are thought to feature terminal-hydride intermediates while [NiFe]-H2ases are proposed to operate through bridging hydrides in catalytically significant states (Fig. 1).13–16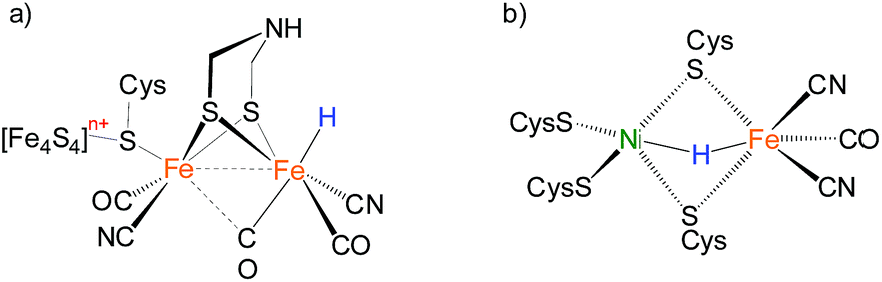 | ||
| Fig. 1 Hypothesized “hydride structure” for the active site of (a) [FeFe]-H2ases and (b) [NiFe]-H2ases. | ||
Reflecting interest in utilization of earth abundant metals for controlling energy storage and release, functional modeling of hydrogenases continues to attract considerable attention. Compared to [FeFe]-H2ase mimics, modeling the active site of [NiFe]-hydrogenases has proven more challenging17–23 and very few of the models clarify the processing of H2.24–28 Especially, the nickel–iron hydride models [Ni(II)HFe(II)]+ remain rare although they are important in modeling research.29 The established [Ni(II)HFe(II)]+ models are [(dppe)Ni(pdt)(H)Fe(PR3)x(CO)3−x]+,30 [Ni(N2S2)Fe(H)(P(OEt)3)3]+,26 [(dppe)Ni(pdt)(H)Fe(CNBArF3)2(CO)]− (ref. 27) and [(pnp)Ni(pdt)(H)Fe(CO)(dppv)]+.28 They were derived from protonation of the corresponding reduced Ni(I)Fe(I) state or were generated from the activation of H2 with an oxidized [Ni(II)Fe(II)]2+ precursor (Fig. 2a).
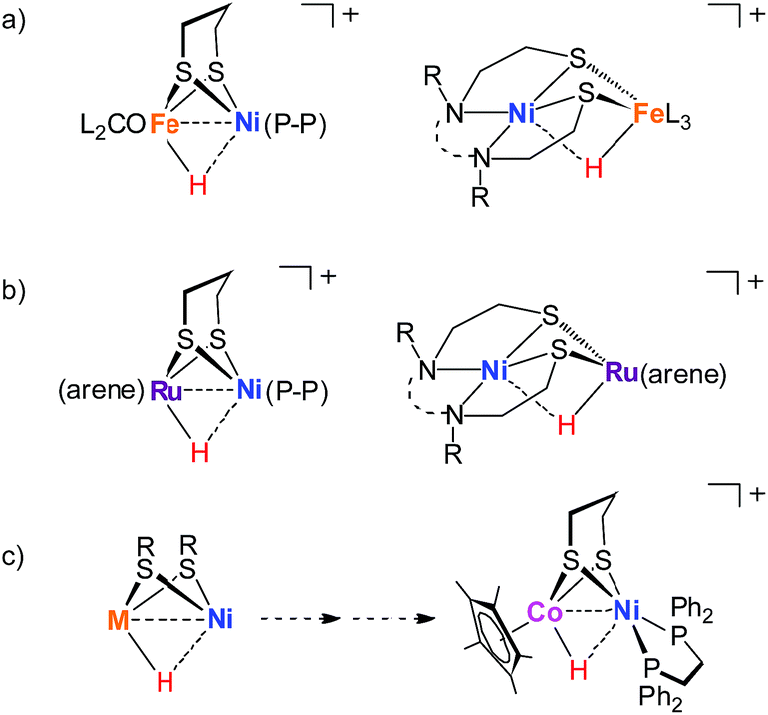 | ||
| Fig. 2 Examples of nickel-based heterobimetallic hydride complexes: (a) reported Ni(II)–H–Fe(II), (b) reported Ni(II)–H–Ru(II), and (c) Ni(II)–H–Co(III) described in this work. | ||
Biological catalysis in a bimetallic manner offers guidance for the exploration of heteronuclear redox chemistry to precede small molecule activations under mild conditions. Four-coordinate nickel(II) complexes such as Ni(N2S2)24,31 and (dppe)Ni(pdt)32,33 are optimal modules to assemble diverse bimetallic complexes, which expand the synthetic NiFe models to the nickel-based heterobimetallic NiM platform and encourage the exploration of fast bioinspired catalysts for the production or activation of H2.31,34–37 For example, [NiRu] complexes bearing the Ni(N2S2) metalloligand are active for H2 heterolysis providing [Ni(II)HRu(II)]+ hydrides with the assistance of an external base (Fig. 2b).33,38
Cobalt is earth-abundant and many cobalt complexes have been shown to be efficient catalysts for H2 production11,39 and hydrogenation reactions.40,41 However, bimetallic NiCo complexes have been reported only infrequently42 and the related heteronuclear hydride complexes have never been documented. In this work, we produced a heterobimetallic [Ni(II)Co(III)H]+ hydride [(dppe)Ni(pdt)(H)CoCp*]+ ([1H]+, pdt2− = 1,3-(CH2)3S22−, Cp*− = Me5C5−, and dppe = Ph2PC2H4PPh2) by protonating the reduced state [(dppe)Ni(pdt)CoCp*] (1) for a catalytic proton reduction (Fig. 2c). Processing of H2 with a class of Ni–Co complexes, which are potentially related to the structure and properties of the [NiFe]-H2ase, has been studied.
Results and discussion
[(dppe)Ni(pdt)(Cl)CoCp*]+ and (dppe)Ni(pdt)CoCp*
The synthesis of a heteronuclear Ni(II)Co(III) dithiolate [1Cl]+ entailed the assembly of (dppe)Ni(pdt) to [Cp*CoCl2]2. Treatment of [Cp*CoCl2]2 with two equiv. of (dppe)Ni(pdt) and KPF6 solids in CH2Cl2 results in the formation of the product [1Cl]+ in a high yield (Scheme 1). The 31P NMR spectrum of [1Cl]+ has a sharp singlet at δ 54.4 corresponding to dppe, indicating that the two phosphine groups are chemically equivalent. Crystallographic analysis of [1Cl]+ reveals that the Ni center adopts a square-planar geometry (Fig. 3) and is linked to the Cp*Co fragment through the pdt2− ligand. The Ni⋯Co distance of 2.926(2) Å far exceeds the sum of the covalent atomic radii of Ni (1.24 Å) and Co (1.26 Å, low-spin).43 | ||
| Scheme 1 Synthesis of Ni–Co bimetallic complexes. | ||
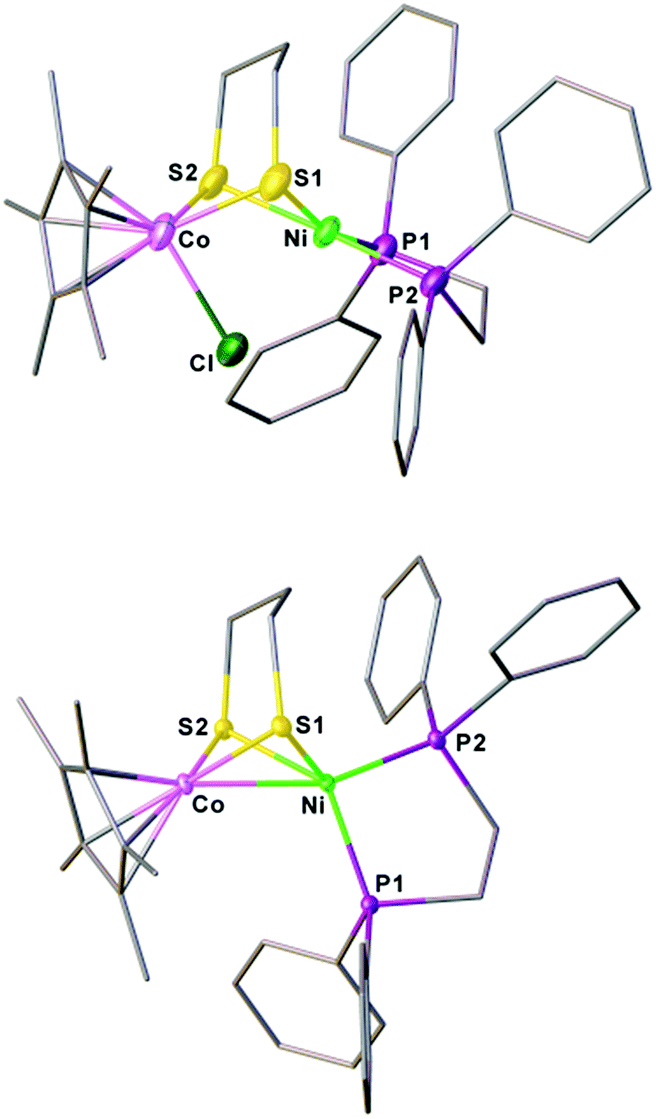 | ||
| Fig. 3 Structures of [1Cl]+ and 1 with thermal ellipsoids drawn at the 50% probability level. The counter anion PF6− in [1Cl]+ and all hydrogen atoms are omitted for clarity. Selected distances (Å): for [1Cl]+, Co–Ni, 2.926(2); Co–Cl, 2.316(2); Co–S1, 2.252(2); Co–S2, 2.259(2); Ni–S1, 2.258(2); Ni–S2; 2.247(2). For 1, Co–Ni, 2.4722(5); Co–S1, 2.1947(7); Co–S2, 2.1820(7); Ni–S1, 2.2645(7); Ni–S2; 2.2268(7). | ||
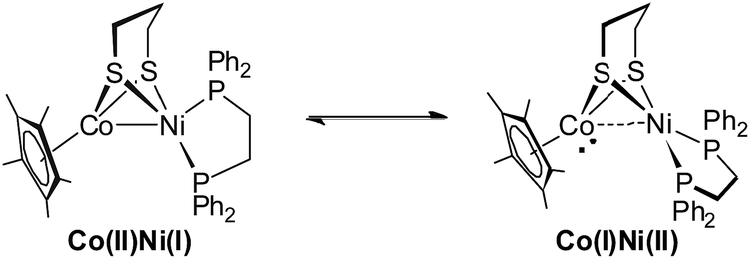
Reduction of the cationic Ni(II)Co(III) complex [1Cl]+ with two equivalents of Cp2Co affords the reduced compound 1. It dissolves well in nonpolar solvents such as toluene or benzene to form deep red solutions, which is very air sensitive and decomposes into insoluble species. Crystallographic analysis of 1 confirms a neutral complex with the formula (dppe)Ni(pdt)CoCp* (Fig. 3). The structure of 1 is more compact than that of [1Cl]+ and this is reflected by the Ni–Co distance, which is 0.454 Å shorter after the 2e− reduction. The Ni–Co distance of 2.4722(5) Å in 1 is comparable to the Ni–Fe distance of 2.4666(6) Å in (dppe)Ni(pdt)Fe(CO)3.17 Given the sum of the covalent atomic radii of Ni and Co, the Ni–Co separation suggests a metal–metal bond. The 2e− reduction causes the Cp*–Co distance to decrease from 1.699 Å to 1.686 Å. Since the Ni center adopts an approximately tetrahedral geometry, compound 1 can be appropriately considered as a Ni(I)Co(II) species in the solid state.
Interestingly, the 31P NMR spectroscopic analysis shows that compound 1 exhibits two isomers in solution at room temperature (eqn (1)). The phosphorus resonance signals appeared as two broad signals at δ 42.9 and 41.8 in a ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. 4). When the temperature was increased to 318 K, the broad resonance at δ 41.8 became resolved but the ratio of the two signals was maintained. Decreasing the temperature to 253 K, the two resonances coalesced into a single broad peak at δ 44. At this lower temperature, however, the broad signal decoalesced into two peaks at 45.7 and 43.6 ppm, which is thought to be consistent with the tetrahedral geometry of Ni that was observed in the solid state structure. Overall, the results of 31P NMR observations at temperatures ranging from 233 K to 298 K reveal a dynamic process in which the 31P sites in dppe are interchanged. The rotation of dppe at the Ni site could proceed through an intermediate or transition state with square-planar Ni.30b It is more likely that in solution at room temperature, 1 consists of two isomers with resonance states of Ni(I)Co(II) and Ni(II)Co(I). Such rotation-induced redox behavior has also been described by Rauchfuss et al.30a for (CO)3Fe(pdt)Ni(dppe). In addition, the reduced NiRu compound (cymene)Ru(pdt)Ni(dppe) features a rigid tetrahedral Ni(0) center.33a
:1 (Fig. 4). When the temperature was increased to 318 K, the broad resonance at δ 41.8 became resolved but the ratio of the two signals was maintained. Decreasing the temperature to 253 K, the two resonances coalesced into a single broad peak at δ 44. At this lower temperature, however, the broad signal decoalesced into two peaks at 45.7 and 43.6 ppm, which is thought to be consistent with the tetrahedral geometry of Ni that was observed in the solid state structure. Overall, the results of 31P NMR observations at temperatures ranging from 233 K to 298 K reveal a dynamic process in which the 31P sites in dppe are interchanged. The rotation of dppe at the Ni site could proceed through an intermediate or transition state with square-planar Ni.30b It is more likely that in solution at room temperature, 1 consists of two isomers with resonance states of Ni(I)Co(II) and Ni(II)Co(I). Such rotation-induced redox behavior has also been described by Rauchfuss et al.30a for (CO)3Fe(pdt)Ni(dppe). In addition, the reduced NiRu compound (cymene)Ru(pdt)Ni(dppe) features a rigid tetrahedral Ni(0) center.33a
 | (1) |
 | ||
| Fig. 4 31P NMR spectra of 1 recorded at various temperatures in toluene. | ||
Protonation of (dppe)Ni(pdt)CoCp*
The reduced compound 1 undergoes protonation, affording the [Ni(II)Co(III)H]+ hydride complex [1H]+. Addition of an equivalent amount of an acid such as [HPPh3]BF4 (pKMeCNa = 8.0)44 or Cl2CHCOOH (pKMeCNa = 13.2)45 to solutions of 1 in CH2Cl2 immediately leads to the color changing from brown to dark. The resulting complex [1H]+ was isolated by filtration after dilution of the reaction solution with Et2O. The 1H NMR spectrum of [1H]+ features a hydride signal at δ −9.77 as a singlet (Fig. S9†). No 31P coupling was resolved, indicating that the hydride is located at the Co(III) center rather than being bound to Ni.46 The 31P signal of the dppe group appears as a singlet at δ 67.5, about 13.1 ppm down-field shifted relative to that of [1Cl]+ (Fig. S8†). An alternative approach to synthesize [1H]+ is by displacement of the Cl− ligand of [1Cl]+ by a hydride ligand donated from NaBH4.Crystallographic analysis agrees with the NMR spectroscopic assignments of the nickel–cobalt dithiolates as a hydride. The framework of [(dppe)Ni(pdt)(X)CoCp*]+ is very similar to that of [1Cl]+ (Fig. 5). The striking difference is the Ni⋯Co separation (2.556(1) Å), which is 0.37 Å shorter than that in [1Cl]+. Protonation causes the Ni–Co distance to increase only by 0.1 Å, which is consistent with the metal–metal bond changes associated with the protonation of reduced NiFe models.30a The hydride ligand was located and refined and the Co–H bond length of 1.45(5) Å is much shorter than the Ni–H bond length (1.91(5) Å), suggesting that the hydride is strongly coordinated at the Co center instead of adopting a bridging position between the bimetallic centers.
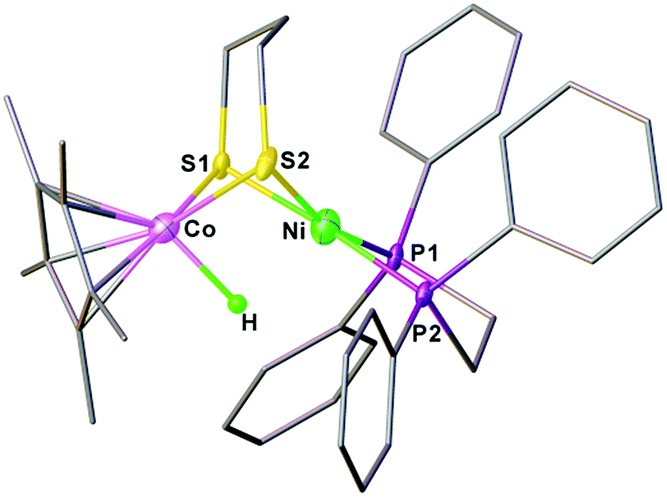 | ||
| Fig. 5 Structure of [1H]+. Selected bond distances (Å): Co–Ni, 2.556(1); Co–H, 1.45(5); Co–S1, 2.22(1); Co–S2, 2.225(1); Ni–S1, 2.222(1); Ni–S2, 2.247(1). | ||
Given the rotation-induced redox behavior of the reduced compound, we propose that the protonation reaction proceeds via the mixed-valent conformer of Ni(II)Co(I). With dppe rotation, nickel is stabilized in the oxidized Ni(II) form with concomitant cleavage of the metal–metal bond resulting in cobalt being in a strongly reduced Co(I) state.29,30b The formation of the [Ni(II)Co(III)H]+ hydride arises from protonation of the Cp*Co site rather than the metal–metal bond. This “redox isomerization–protonation” mechanism is close to that proposed for (CO)3Fe(pdt)Ni(dppe) and reflects the redox-flexibility of the (dppe)Ni(pdt) module.30
Acidity of [1H]+
The acidic character of the hydride ligand in [1H]+ was manifested by the H/D exchange reaction with D2O giving [1D]+ solely and with no liberation of HD or H2. After addition of excess D2O (20 equiv.) to the CD3CN solution (0.6 mL) of [1H]+ in a J. Young tube, the reaction process was monitored by NMR spectroscopy. 1H NMR spectroscopic analysis showed that the hydride signal disappears slowly, while the 31P resonance remains as a singlet at δ 67.4. The appearance of a Co–D signal was observed at δ −9.66 in the 2H NMR spectrum (Fig. S14†). In agreement with the calculated isotopic distribution of [1D]+, ESI-MS spectral analysis featured a characteristic peak at m/z = 758.1216 for [1D]+vs. m/z = 757.1175 for [1H]+. The exchange reaction between [1H]+ and D2O is relatively slow. A plot of the integrated hydride signal vs. time suggests that the reaction follows unimolecular kinetics and has a rate constant of 5.023 × 10−5 s−1 at 25 °C (t1/2 = 3.83 h, Fig. S16†). | (2) |
This bimetallic [Ni(II)Co(III)H]+ hydride was found to be much less acidic than the reported [Ni(II)(H)Fe(II)]+ hydrides such as (CO)3Fe(pdt)Ni(dppe) (pKMeCNa = 10.7) and (CO)2(PPh3)Fe(pdt)Ni(dppe) (pKMeCNa = 14.9),30a and the diiron bridging hydride [(μ-H)Fe(pdt)(PMe3)2(CO)4]+ (pKMeCNa = 12).47 The acidity of [1H]+ was further evaluated by the protonation of 1 with [Et3NH]BF4 (pKMeCNa = 18.8).44 An equilibrium was established between 1 and 1.0 equiv. of [Et3NH]BF4 in PhCN (eqn (2)), which provided an equilibrium mixture of [1H]+ and 1 in a ratio of 0.22:1 as determined from the 31P NMR spectrum. A pKa of 17.5 was estimated for [1H]+ on the basis of the calculated equilibrium constant Keq (0.05) (Fig. S17†). The results indicate that the acidity of [1H]+ is weak, while its conjugated base, the Ni(II)Co(I) complex, exhibits a strong basic character.
Homolytic and heterolytic bond energies for NiCo–H
Because of the centrality of metal hydrides in catalytic H2 production, we determined the homolytic and heterolytic bond energies for NiCo–H in an effort to learn more about the thermodynamic properties of the [Ni(II)(H)Co(III)]+ hydride. The cleavage of the M–H bond can involve transfer of electrons, a proton, a hydrogen atom or a hydride48–51 and is very dependent on the bond energy. Scheme 2 illuminates the relationship between acidity (pKa), homolytic bond dissociation energy (BDFE, ΔGH˙), thermodynamic hydricity (ΔGH−), and redox potentials (E1/2) for the NiCo–H cleavage.| ΔGH˙ = 1.37 pKa + 23.06 E1/2[1]0/+ + 54.9 | (3) |
| ΔGH− = 1.37 pKa + 23.06 E1/2[1]0/+ + 23.06 E1/2[1]+/2+ + 79.6 | (4) |
 | ||
| Scheme 2 Schematic pathways to the cleavage of the NiCo–H bond in [1H]+. | ||
According to the thermodynamic cycle, the redox potentials of the [1]0/+ and [1]+/2+ couples are required to calculate ΔGH˙ and ΔGH−. Cyclic voltammetry on PhCN solutions of 1 indicates oxidations at E1/2 = −0.45 V (ipa/ipc = 1) and E1/2 = −0.01 V (quasi-reversible, Fig. S19†). These couples are assigned as one-electron processes for [1]0/+ and [1]+/2+, respectively. The direct evidence is derived from oxidations conducted on a preparative scale (see below). Using the thermodynamic equations derived from eqn (3) and (4),30a,48,49 ΔGH˙ and ΔGH− are calculated to be 69 and 93 kcal mol−1, respectively.
Oxidizing 1 for H2 activation
Oxidation of 1 with one equiv. of FcBF4 provided a cationic complex [1]+, while using AgBF4 (E1/2 = 0.65 V, CH2Cl2)52 allowed for the further oxidization of [1]+ to [1]2+. The formation of a new diamagnetic species was signaled by a 31P peak at δ 59.5 (s) (Fig. S20†). As a 32e− bimetallic species, [1]2+ is unstable and cannot be isolated. When the reaction was conducted in the presence of MeCN, the dicationic complex [Ni(II)Co(III)]2+ was stabilized in the form of [(dppe)Ni(pdt)(MeCN)CoCp*]2+ ([1(NCMe)]2+).The structures of [1]+ and [1(NCMe)]2+ were characterized crystallographically. In both structures, the Ni center adopts a square-planar coordination geometry (Fig. 6). In [1(NCMe)]2+, the MeCN is bound to the Co, consistent with electrochemical assignment that the second oxidation occurs at the Cp*Co site. The 1e− oxidation increases the Ni⋯Co distance from 2.6572(10) Å for [1]+ to 3.005 Å for [1(NCMe)]2+. The principal change in the oxidation of 1 is the coordination sphere of nickel, which turns from roughly tetrahedral to a strictly square-planar geometry. According to the structures of 1 and [1]+, the first oxidation event occurs at the Ni(dppe) site. Related to 1, the Ni⋯Co distance in [1]+ is longer by 0.18 Å, meanwhile the Cp*–Co distance increases slightly from 1.686 Å to 1.693 Å.
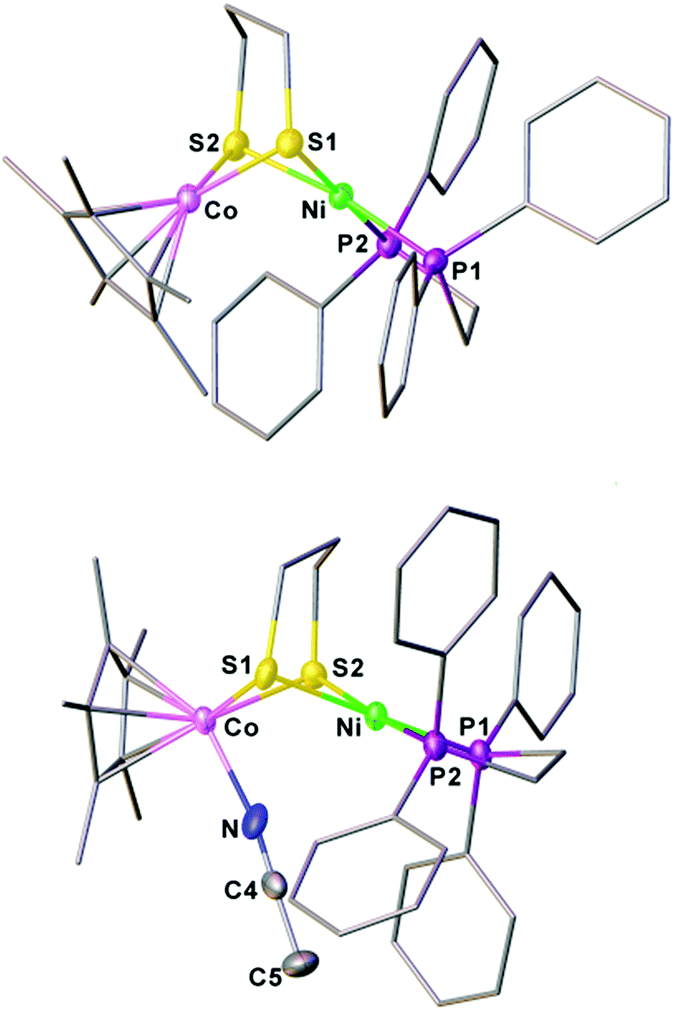 | ||
| Fig. 6 Structures of [1]+ and [1(NCMe)]2+ with thermal ellipsoids drawn at the 50% probability level. BF4− anions, hydrogen atoms and solvent are omitted for clarity. Selected bond distances (Å): for [1]+, Co–Ni, 2.657 (1); Co–S1, 2.210(1); Co–S2, 2.215(1); Ni–S1, 2.239(1); Ni–S2, 2.229(1); for [1(NCMe)]2+, Co–Ni, 3.005; Co–S1, 2.246(1); Co–S2, 2.263(1); Ni–S1, 2.237(1); Ni–S2, 2.239(1). | ||
The high affinity of the dicationic [Ni(II)Co(III)]2+ complex toward the hydride was further demonstrated by the reaction of activating H2 (Scheme 3). The MeCN solution of [1(NCMe)]2+ contained in a Schlenk flask was bubbled with H2 (10 psi) for 3 min, and then a solution of CH3ONa in MeOH was added (Fig. S24†). Over the course of 10 min at room temperature, the reaction mixture turns from red to dark brown. As confirmed by 1H NMR and 31P NMR spectra, the organometallic product is [1H]+. Interestingly, in addition to CH3ONa, alkali metal carbonates such as Na2CO3 can be employed as the base to assist [1(NCMe)]2+ to activate H2.
 | ||
| Scheme 3 Hydrogen activation by [1(NCMe)]2+. | ||
Electrocatalytic H2 production
Cyclic voltammetry indicates that [1H]+ only undergoes a reversible reduction process at −1.36 V (ipc/ipa = 0.98), which is tentatively assigned to the [Ni(II)Co(III)H]+/[Ni(II)Co(II)H]0 couple (Fig. S26†). To evaluate the catalytic activity of [1H]+ as an electrocatalyst for H2 production, Cl2CHCOOH (E0 = −0.92 V)45 was selected as the proton source. Upon the addition of Cl2CHCOOH to the CH2Cl2 solution of [1H]+, the reduction events for [1H]+/0 became irreversible, and the cathodic current intensity increased linearly with sequential increasing of the acid concentration (Fig. 7, inset). These observations are consistent with the aspects of proton reduction catalysis.53,54 Plots of ic/ipvs. [Cl2CHCOOH] are linear up to 218 equiv. of acid, indicating that the catalysis is second order with respect to the acid. The turnover frequency was estimated to be 244 s−1 (Table S1†). In the controlled experiment, proton reduction with a glassy carbon electrode was performed at a potential nearly 0.37 V more negative than that in the catalysis performed with [1H]+ (Fig. S25†).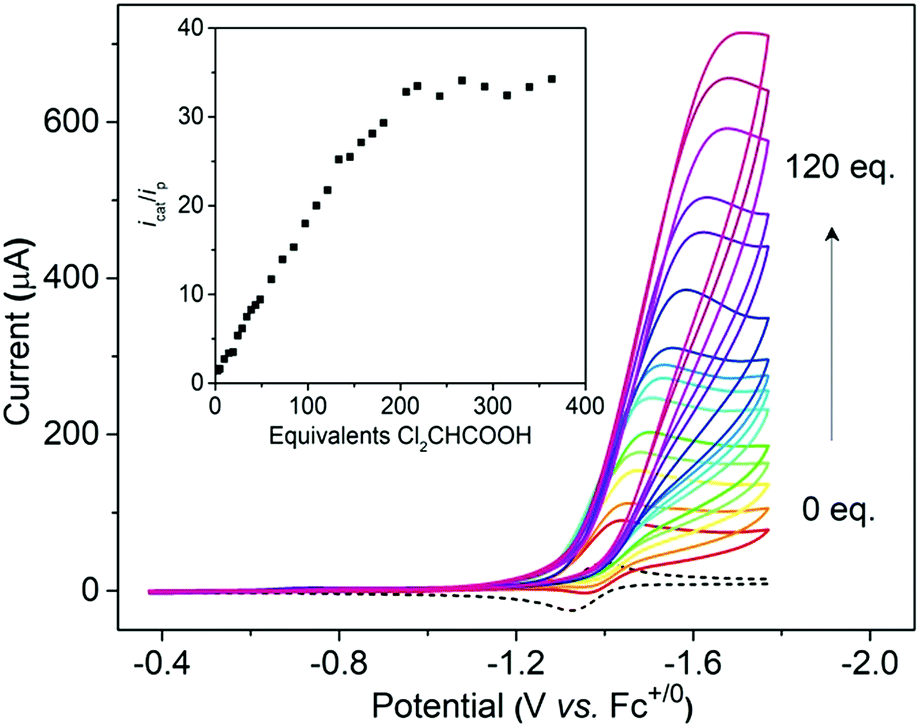 | ||
| Fig. 7 Cyclic voltammograms of [1H]+ with increments of Cl2CHCOOH (from 0 to 120 equiv.), inset: plot of catalytic current ic/ip for [1H]+vs. equivalent of Cl2CHCOOH. Conditions: 1 mM [1H]+ in MeCN, 0.1 M n-NBu4PF6, scan rate = 100 mV s−1, and V vs. Fc0/+. | ||
To gain insight into the catalytic mechanism, we examined the chemical reduction of [1H]+ with Cp*2Co (E1/2 = −1.94 V in CH2Cl2). The reaction was indicated by an immediate change of the color of the solution from brown to black. Efforts to isolate and characterize the reduced [1H]0 were unsuccessful since it is converted to 1 and loses H2 in a matter of minutes. Addition of protic acids such as Cl2CHCOOH or H(OEt2)2BArF4 into a solution of [1H]0 in THF, generated in situ by reaction of [1H]+ with Cp*2Co, resulted in the formation of [1]+ and release of H2. The yield of H2, quantified by GC analysis, is 91% ± 5 (in three experiments) and is close to the stoichiometric value. We propose that the catalytic H2 evolution is based on the reduction of the [Ni(II)Co(III)H]+ hydride, operating through an ECEC mechanism (Scheme 4).
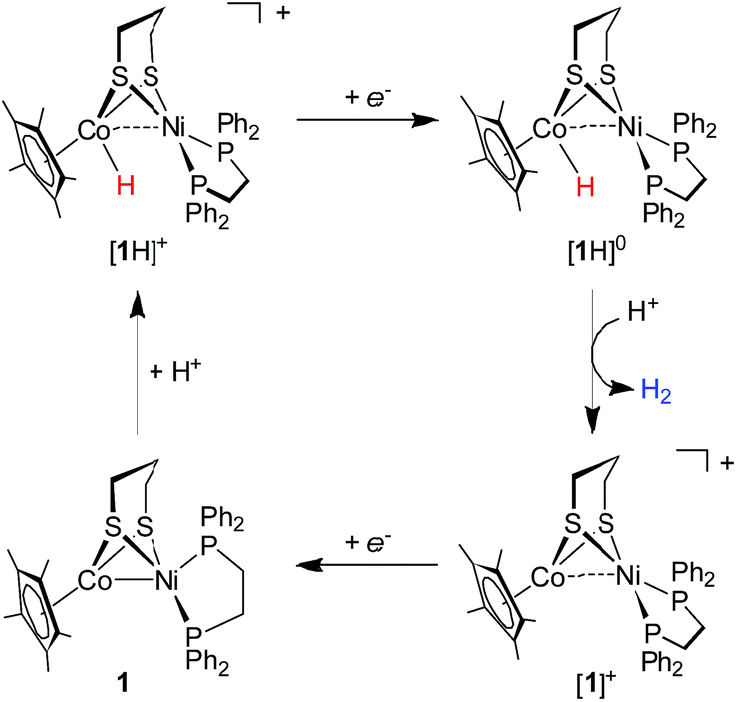 | ||
| Scheme 4 Proposed catalytic cycle for proton reduction by [1H]+. | ||
Conclusions
A class of new NiCo complexes, relevant to the active site models of [NiFe]-H2ases, have been studied and their features of stereodynamics, acid–base properties, redox chemistry and reactivity are disclosed. Incorporating the (dppe)Ni(pdt) module with a Cp*Co fragment enables the bimetallic NiCo complexes to achieve Co-centered H2 evolution and uptake. Such a bimetallic way can disperse the strong effects of redox over the two metal sites.29 Owing to the redox flexibility of Ni(dppe), distortion at the Ni center of the reduced state affects the oxidation state of the bimetallic centers, notably interconversion of Ni(I)Co(II) and Ni(II)Co(I). The protonation reaction can proceed via the mixed valence isomer Ni(II)Co(I) with enhanced basicity to afford [Ni(II)Co(III)H]+. The bimetallic hydride is catalytically active and was demonstrated to be an efficient electrocatalyst for the reduction of weak protic acid at a mild potential. Despite a stoichiometric reaction, the oxidized state [Ni(II)Co(III)]2+ is capable of activating H2 to give the hydride.Bimetallic hydride species are commonly encountered in hydrogenase modeling. Bimetallic active sites in hydrogenases provide an elegant means of softening the effect of redox reactions on the acid–base properties of the hydride.30a In addition to biocatalysis, metal hydrides play a central role in a variety of chemical transformations.55–59 Studies of metal hydride chemistry are essential to control the elementary steps for catalysis.60–62 However, the related thermodynamic properties of bimetallic systems have been inadequately examined to date.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the “Thousand Plan” Youth Program and the National Natural Science Foundation of China (21871166 and 91427303) for the financial support. We also thank Prof. Di Sun for assistance with the X-ray crystallography.Notes and references
- M. Iglesias, E. Sola and L. A. Oro, in Homo- and Heterobimetallic Complexes in Catalysis: Cooperative Catalysis, ed. P. Kalck, Springer International Publishing, Cham, Switzerland, 2016, ch. 2, pp. 31–58 Search PubMed.
- F. A. Cotton, Acc. Chem. Res., 1978, 11, 225–232 CrossRef CAS.
- L. H. Gade, Angew. Chem., Int. Ed., 2000, 39, 2658–2678 CrossRef CAS PubMed.
- D. G. Huang and R. H. Holm, J. Am. Chem. Soc., 2010, 132, 4693–4701 CrossRef CAS PubMed.
- I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef CAS.
- M. J. Page, D. B. Walker and B. A. Messerle, in Homo- and Heterobimetallic Complexes in Catalysis: Cooperative Catalysis, ed. P. Kalck, Springer International Publishing, Cham, Switzerland, 2016, ch. 4, pp. 103–138 Search PubMed.
- J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 3, 363–375 CrossRef.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- R. Pulukkody and M. Y. Darensbourg, Acc. Chem. Res., 2015, 48, 2049–2058 CrossRef CAS PubMed.
- P. V. Rao, S. Bhaduri, J. F. Jiang and R. H. Holm, Inorg. Chem., 2004, 43, 5833–5849 CrossRef CAS PubMed.
- (a) V. Artero, G. Berggren, M. Atta, G. Caserta, S. Roy, L. Pecqueur and M. Fontecave, Acc. Chem. Res., 2015, 48, 2380–2387 CrossRef CAS PubMed; (b) C. H. Hsieh, S. Ding, Ö. F. Erdem, D. J. Crouthers, T. Liu, C. C. Mccrory, W. Lubitz, C. V. Popescu, J. H. Reibenspies, M. B. Hall and M. Y. Darensbourg, Nat. Commun., 2014, 5, 3684 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef CAS PubMed.
- P. P. Liebgott, A. L. de Lacey, B. Burlat, L. Cournac, P. Richaud, M. Brugna, V. M. Fernandez, B. Guigliarelli, M. Rousset, C. Leger and S. Dementin, J. Am. Chem. Soc., 2011, 133, 986–997 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- B. L. Greene, C. H. Wu, G. E. Vansuch, M. W. W. Adams and R. B. Dyer, Biochemistry, 2016, 55, 1813–1825 CrossRef CAS PubMed.
- (a) P. A. Ash, R. Hidalgo and K. A. Vincent, ACS Catal., 2017, 7, 2471–2485 CrossRef CAS PubMed; (b) B. J. Murphy, R. Hidalgo, M. M. Roessler, R. M. Evans, P. A. Ash, W. K. Myers, K. A. Vincent and F. A. Armstrong, J. Am. Chem. Soc., 2015, 137, 8484–8489 CrossRef CAS PubMed; (c) R. Hidalgo, P. A. Ash, A. J. Healy and K. A. Vincent, Angew. Chem., Int. Ed., 2015, 54, 7110–7113 CrossRef CAS PubMed.
- W. Zhu, A. C. Marr, Q. Wang, F. Neese, D. J. E. Spencer, A. J. Blake, P. A. Cooke, C. Wilson and M. Schroder, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18280–18285 CrossRef CAS PubMed.
- (a) S. Canaguier, V. Fourmond, C. U. Perotto, J. Fize, J. Pecaut, M. Fontecave, M. J. Field and V. Artero, Chem. Commun., 2013, 49, 5004–5006 RSC; (b) Y. Oudart, V. Artero, J. Pecaut and M. Fontecave, Inorg. Chem., 2006, 45, 4334–4336 CrossRef CAS PubMed; (c) S. Canaguier, M. Field, Y. Oudart, J. Pecaut, M. Fontecave and V. Artero, Chem. Commun., 2010, 46, 5876–5878 RSC; (d) T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270, 127–150 CrossRef.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- Y. Ohki, K. Yasumura, M. Ando, S. Shimokata and K. Tatsumi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3994–3997 CrossRef CAS PubMed.
- K. Weber, O. F. Erdem, E. Bill, T. Weyhermuller and W. Lubitz, Inorg. Chem., 2014, 53, 6329–6337 CrossRef CAS PubMed.
- (a) D. Schilter, T. B. Rauchfuss and M. Stein, Inorg. Chem., 2012, 51, 8931–8941 CrossRef CAS PubMed; (b) D. Schilter, M. J. Nilges, M. Chakrabarti, P. A. Lindahl, T. B. Rauchfuss and M. Stein, Inorg. Chem., 2012, 51, 2338–2348 CrossRef CAS PubMed.
- X. Chu, X. Yu, S. Raje, R. Angamuthu, J. Ma, C.-H. Tung and W. Wang, Dalton Trans., 2017, 46, 13681–13685 RSC.
- (a) P. Ghosh, S. D. Ding, R. B. Chupik, M. Quiroz, C. H. Hsieh, N. Bhuvanesh, M. B. Hall and M. Y. Darensbourg, Chem. Sci., 2017, 8, 8291–8300 RSC; (b) S. D. Ding, P. Ghosh, M. Y. Darensbourg and M. B. Hall, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9775–E9782 CrossRef CAS PubMed; (c) S. Ding, P. Ghosh, A. M. Lunsford, N. Wang, N. Bhuvanesh, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2016, 138, 12920–12927 CrossRef CAS PubMed.
- D. Brazzolotto, M. Gennari, N. Queyriaux, T. R. Simmons, J. Pecaut, S. Demeshko, F. Meyer, M. Orio, V. Artero and C. Duboc, Nat. Chem., 2016, 8, 1054–1060 CrossRef CAS PubMed.
- S. Ogo, K. Ichikawa, T. Kishima, T. Matsumoto, H. Nakai, K. Kusaka and T. Ohhara, Science, 2013, 339, 682–684 CrossRef CAS PubMed.
- B. C. Manor and T. B. Rauchfuss, J. Am. Chem. Soc., 2013, 135, 11895–11900 CrossRef CAS PubMed.
- L. Song, X. Yang, M. Cao, X. Gao, B. Liu, L. Zhu and F. Jiang, Chem. Commun., 2017, 53, 3818–3821 RSC.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed.
- (a) B. E. Barton and T. B. Rauchfuss, J. Am. Chem. Soc., 2010, 132, 14877–14885 CrossRef CAS PubMed; (b) M. T. Huynh, D. Schilter, S. Hammes-Schiffer and T. B. Rauchfuss, J. Am. Chem. Soc., 2014, 136, 12385–12395 CrossRef CAS PubMed; (c) O. A. Ulloa, M. T. Huynh, C. P. Richers, J. A. Bertke, M. J. Nilges, S. Hammes-Schiffer and T. B. Rauchfuss, J. Am. Chem. Soc., 2016, 138, 9234–9245 CrossRef CAS PubMed.
- (a) J. A. Denny and M. Y. Darensbourg, Chem. Rev., 2015, 115, 5248–5273 CrossRef CAS PubMed; (b) M. V. Rampersad, S. P. Jeffery, J. H. Reibenspies, C. G. Ortiz, D. J. Darensbourg and M. Y. Darensbourg, Angew. Chem., 2005, 117, 1243–1246 CrossRef; (c) K. N. Green, S. P. Jeffery, J. H. Reibenspies and M. Y. Darensbourg, J. Am. Chem. Soc., 2006, 128, 6493–6498 CrossRef CAS PubMed.
- (a) M. Schmidt and G. G. Hoffmann, Eur. J. Inorg. Chem., 1979, 112, 2190–2196 CAS; (b) M. Schmidt and G. G. Hoffmann, J. Organomet. Chem., 1977, 124, 5–8 CrossRef.
- (a) G. M. Chambers, J. Mitra, T. B. Rauchfuss and M. Stein, Inorg. Chem., 2014, 53, 4243–4249 CrossRef CAS PubMed; (b) G. M. Chambers, M. T. Huynh, Y. Li, S. Hammes-Schiffer, T. B. Rauchfuss, E. Reijerse and W. Lubitz, Inorg. Chem., 2016, 55, 419–431 CrossRef CAS PubMed.
- V. Fourmond, S. Canaguier, B. Golly, M. J. Field, M. Fontecave and V. Artero, Energy Environ. Sci., 2011, 4, 2417–2427 RSC.
- D. Schilter, A. L. Fuller and D. L. Gray, Eur. J. Inorg. Chem., 2015, 28, 4638–4642 CrossRef.
- X. Chu, X. Xu, H. Su, S. Raje, R. Angamuthu, C.-H. Tung and W. Wang, Inorg. Chem. Front., 2017, 4, 706–711 RSC.
- A. M. Lunsford, K. F. Goldstein, M. A. Cohan, J. A. Denny, N. Bhuvanesh, S. D. Ding, M. B. Hall and M. Y. Darensbourg, Dalton Trans., 2017, 46, 5175–5182 RSC.
- (a) S. Ogo, R. Kabe, K. Uehara, B. Kure, T. Nishimura, S. C. Menon, R. Harada, S. Fukuzumi, Y. Higuchi, T. Ohhara, T. Tamada and R. Kuroki, Science, 2007, 316, 585–587 CrossRef CAS PubMed; (b) T. Matsumoto, B. Kure and S. Ogo, Chem. Lett., 2008, 37, 970–971 CrossRef CAS.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- W. Liu, B. Sahoo, K. Junge and M. Beller, Acc. Chem. Res., 2018, 51, 1858–1869 CrossRef CAS PubMed.
- P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687–1695 CrossRef CAS PubMed.
- H. Han, Z. Wei, M. C. Barry, J. C. Carozza, M. Alkan, A. Y. Rogachev, A. S. Filatov, A. M. Abakumov and E. V. Dikarev, Chem. Sci., 2018, 9, 4736–4745 RSC.
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- I. Kaljurand, A. Kutt, L. Soovali, T. Rodima, V. Maemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- G. A. N. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Inorg. Chem., 2006, 45, 9181–9184 CrossRef CAS PubMed.
- (a) F. Zhang, X. Xu, Y. Zhao, J. Jia, C.-H. Tung and W. Wang, Organometallics, 2017, 36, 1238–1244 CrossRef CAS; (b) J. Liu, F. Zhang, A. Zhang, Q. Tong, C.-H. Tung and W. Wang, Chem.–Asian J., 2016, 11, 2271–2277 CrossRef CAS PubMed; (c) F. Zhang, J. Jia, S. Dong, W. Wang and C.-H. Tung, Organometallics, 2016, 35, 1151–1159 CrossRef CAS.
- G. Eilers, L. Schwartz, M. Stein, G. Zampella, L. de Gioia, S. Ott and R. Lomoth, Chem.–Eur. J., 2007, 13, 7075–7084 CrossRef CAS PubMed.
- Y. Hu, A. P. Shaw, D. P. Estes and J. R. Norton, Chem. Rev., 2016, 116, 8427–8462 CrossRef CAS PubMed.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed.
- M. Bourrez, R. Steinmetz, S. Ott, F. Gloaguen and L. Hammarstrom, Nat. Chem., 2015, 7, 140–145 CrossRef CAS PubMed.
- Y. Matsubara, E. Fujita, M. D. Doherty, J. T. Muckerman and C. Creutz, J. Am. Chem. Soc., 2012, 134, 15743–15757 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. R. DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS PubMed.
- C. P. Andrieux, C. Blocman, J. M. Dumas-Bouchiat, F. M'Halla and J. M. Savéant, J. Electroanal. Chem., 1980, 113, 19–40 CrossRef CAS.
- M. Peruzzini and R. Poli, Recent Advances in Hydride Chemistry, Elsevier, Amsterdam, 2001 Search PubMed.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- H. Nakazawa and M. Itazaki, Iron Catalysis: Fundamentals and Applications, 2011, vol. 33, pp. 27–81 Search PubMed.
- (a) R. M. Bullock, Catalysis with precious metals, Wiley-VCH, Weinheim, 2010 CrossRef; (b) R. M. Bullock, Science, 2013, 342, 1054–1055 CrossRef CAS PubMed.
- S. Chakraborty, P. Bhattacharya, H. G. Dai and H. R. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed.
- C. L. Pitman, K. R. Brereton and A. J. M. Miller, J. Am. Chem. Soc., 2016, 138, 2252–2260 CrossRef CAS PubMed.
- C. M. Zall, J. C. Linehan and A. M. Appel, J. Am. Chem. Soc., 2016, 138, 9968–9977 CrossRef CAS PubMed.
- (a) R. M. Henry, R. K. Shoemaker, D. L. DuBois and M. R. DuBois, J. Am. Chem. Soc., 2006, 128, 3002–3010 CrossRef CAS PubMed; (b) T. B. Liu, D. L. DuBois and R. M. Bullock, Nat. Chem., 2013, 5, 228–233 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental results, procedures and characterization including NMR spectra and crystallographic data. CCDC 1841904–1841908. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04346a |
| This journal is © The Royal Society of Chemistry 2019 |