
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of metal coordination and light irradiation on hierarchical self-assembly processes†
Kalathil K.
Kartha
a,
Naveen Kumar
Allampally
b,
Antiope T.
Politi
c,
Deepak D.
Prabhu
d,
Hayato
Ouchi
 d,
Rodrigo Q.
Albuquerque
*c,
Shiki
Yagai
*de and
Gustavo
Fernández
*a
d,
Rodrigo Q.
Albuquerque
*c,
Shiki
Yagai
*de and
Gustavo
Fernández
*a
aOrganisch-Chemisches Institut, Universität Münster, Corrensstraße 40, 48151 Münster, Germany. E-mail: fernandg@uni-muenster.de
bInstitut für Organische Chemie, Universität Würzburg am Hubland, 97074 Würzburg, Germany
cSchool of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, Liverpool L3 3AF, UK. E-mail: ralbuque@uni-muenster.de
dDepartment of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, 1-33-Yayoi-cho, Inage-Ku, Chiba 263-8522, Japan. E-mail: yagai@faculty.chiba-u.jp
eInstitute for Global Prominent Research (IGPR), Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan
First published on 24th October 2018
Abstract
Smart light-responsive supramolecular materials have been extensively investigated in the past decade, but so far the impact of metal coordination on hierarchical supramolecular structures of light-responsive building blocks has remained nearly unexplored. Herein, we unravel the hierarchical self-assembly of a small π-conjugated azo-containing pyridyl ligand that is able to respond to UV-light and metal complexation. The ligand self-assembles in an antiparallel fashion into long twisted fibers, which are then disassembled upon photoisomerization of the azobenzene groups, resulting in shorter rigid rods with a different packing motif. Complexation of Pd(II) ions enhances the cooperativity of the aggregation and induces a molecular rearrangement into slipped stacks with subsequent formation of long thin fibers. These are then transformed into thinner, shorter rods upon light irradiation. The observed different light-responsiveness, besides clearing up the influence of metal coordination and light irradiation in self-assembly processes, paves the way towards the design of novel supramolecular photochromic systems.
Introduction
Self-assembly of functional small molecules has become a promising approach to create smart materials1 that can respond to changes in various external variables such as temperature, concentration, pH and solvent or stimuli such as light,2 sound,3 mechanical forces4 or cations/anions.5 Light-responsive supramolecular assemblies are of particular interest in this regard because of the sensitivity of the molecular shape, size and polarity upon photoisomerization, which enables the modulation of the structural and functional properties associated with the self-assembly pathway.2 The incorporation of photochromic moieties such as azobenzenes,6 dithienylethenes7 and spiropyrans8 offers great potential to create light-responsive supramolecular assemblies. In particular, azobenzenes have been extensively exploited as photoswitchable units in light-responsive host–guest systems,9 liquid crystals,10 vesicles,11 gels,12 biomaterials13 and self-assembled structures of π-conjugated molecules.14–16 For the construction of one-dimensional (1D) photoresponsive supramolecular assemblies, the major molecular design is the use of photochromic molecules as single-molecular building blocks.14,16 A particularly attractive approach is the oligomerization of photochromic moieties via non-covalent interactions, i.e. hydrogen bonds, to form supramolecular building blocks, as this provides access to hierarchically organized systems with unique photoresponsive behaviour not achievable by single-molecule based systems.15b,c In this regard, we envisaged that the introduction of a metal to link photochromic units would have a strong impact on the photoresponsive behaviour and hierarchy levels of the assemblies due to the modification in the molecular conformation and packing modes. For example, we have reported that coordination of a Pt(II)/Pd(II) dihalogen metal center to pyridyl-based π-conjugated moieties can induce new non-covalent interaction sites different from the ligand thereby enabling new self-assembly pathways.17 To date, photoresponsive metallosupramolecular systems have been limited to polymeric materials,18 metallacycles19a and metal–organic frameworks.19b,d Despite these advances, understanding the influence of metal coordination20 and light-irradiation on hierarchical self-assembly processes remains elusive.19cHerein, we unravel the impact of metal complexation and photoisomerization on hierarchical self-assembly processes via detailed mechanistic studies by means of combining experimental and theoretical techniques. Our molecular design features a light-responsive azobenzene unit, an amide group for hydrogen bonding, a metal ion-responsive pyridyl ligand and dodecyloxy side chains (L1, Scheme 1). The target ligand L1 and its corresponding Pd(II) complex C1 (Scheme S1†) have been synthesized by modified literature procedures,21 as described in the (ESI†). Additionally, a related ligand L2 bearing ethoxy groups as well as its corresponding Pd(II) complex C2 have been prepared to facilitate crystal growth (Scheme S1†).
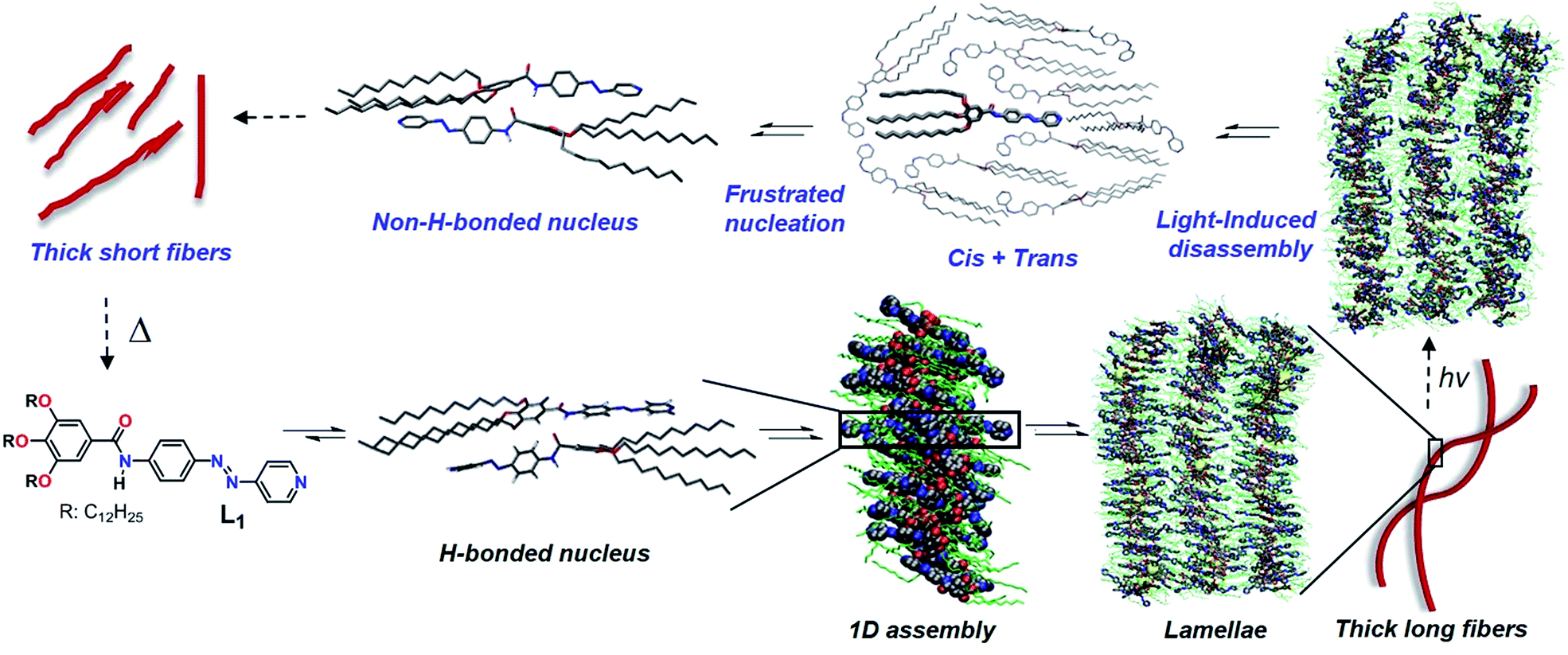 | ||
| Scheme 1 Proposed hierarchical self-assembly and photoresponsive behavior of L1. Structures of monomers and small aggregates were optimized using DFT and dispersion-corrected PM6 calculations, respectively, while structures with large aggregates (270 monomers) were obtained from classical molecular dynamics. | ||
Results and discussion
Hierarchical self-assembly of L1
We examined the self-assembly of ligand L1 in a poor solvent (methylcyclohexane; MCH) by cooling a hot solution (c = 5 × 10−4 M) from 363 K to 283 K with 1 K min−1 ramp. The freshly prepared solution showed negligible absorption spectral changes in various good solvents (i.e. dichloromethane; DCM), suggesting the molecularly dissolved state (Fig. S1†) at 298 K. However, a gradual decrease of the absorption maximum at 363 nm and a concomitant increase of a shoulder at ca. 420 nm were observed upon keeping the MCH solution at 283 K over time (Fig. 1a). This, along with the appearance of an isosbestic point at 407 nm, strongly suggests the formation of self-assembled species. A plot of absorbance at 360 nm (A360) vs. time reveals that around 80% of the aggregation process is complete within 5 h (Fig. 1a, inset). Likewise, a lower concentrated solution (2.5 × 10−4 M) was also monitored over time at 283 K, but in this case negligible changes were observed, indicating that the critical aggregation concentration is close to 5 × 10−4 M at 283 K. Variable Temperature (VT)–NMR studies of L1 in MCH-d14 (c = 5 × 10−4 M) from 363 to 283 K showed downfield shifts for the amide protons (He) and aromatic protons Hd (ca. 1 and 0.15 ppm, respectively, see Fig. 1b), indicating the proximity of an electron-rich environment (i.e. N or O atoms from a neighboring molecule) and H-bonding.22 Comparison of the FT-IR spectra of L1 (c = 5 × 10−4 M) in the monomer (CHCl3) and aggregated state (MCH) allowed us to assign these hydrogen bonds to N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C interactions between the amide groups (Fig. S2†). On the other hand, the phenyl protons Hf underwent a marked upfield shift upon cooling, strongly suggesting the involvement of the trialkoxyphenyl unit in π-stacking. Interestingly, the pyridine protons (Ha,b) remain almost unaltered during the cooling process, ruling out the possibility of a face-to-face parallel stacking.22a A noteworthy observation is the sharpening of the proton signals upon decreasing temperature, which can be ascribed to the formation of discrete aggregate species, such as dimers, during the cooling process.23 This is in agreement with the results obtained from variable-temperature dynamic light scattering (DLS) (Fig. S3; for details see ESI†). Attempts to further monitor this process by diffusion-ordered spectroscopy (DOSY) NMR proved unsuccessful due to unreliable data (Fig. S4†).
C interactions between the amide groups (Fig. S2†). On the other hand, the phenyl protons Hf underwent a marked upfield shift upon cooling, strongly suggesting the involvement of the trialkoxyphenyl unit in π-stacking. Interestingly, the pyridine protons (Ha,b) remain almost unaltered during the cooling process, ruling out the possibility of a face-to-face parallel stacking.22a A noteworthy observation is the sharpening of the proton signals upon decreasing temperature, which can be ascribed to the formation of discrete aggregate species, such as dimers, during the cooling process.23 This is in agreement with the results obtained from variable-temperature dynamic light scattering (DLS) (Fig. S3; for details see ESI†). Attempts to further monitor this process by diffusion-ordered spectroscopy (DOSY) NMR proved unsuccessful due to unreliable data (Fig. S4†).
 | ||
| Fig. 1 (a) Temperature-dependent absorption changes of L1 (5 × 10−4 M, MCH) from 363 K (red line) to 283 K (blue line) followed by time-dependent studies at 283 K for a period of 400 min. Inset: plot of A360vs. time and photographs showing the reversible sol-aggregate formation. (b) Partial VT–1H NMR spectra of L1 (5 × 10−4 M, MCH-d14) between 363 and 283 K followed by time-dependent studies at 283 K over a period of 60 min (initial lag is excluded). (c–e) AFM images of self-assembled L1 in MCH upon keeping the solution at 283 K for (c) 15 min, (d) 30 min and (e) 60 min on HOPG (initial lag is excluded) with cross-section analysis along the yellow lines. | ||
Monitoring this discrete aggregate formation by VT-UV-Vis experiments under identical conditions showed only a slight hyperchromic effect without a clear isosbestic point upon cooling from 363 K to 283 K (Fig. 1a and S5†). Though hyper/hypochromic effects can be ascribed to a weak intermolecular interaction of the π-systems on the basis of Kasha's exciton theory,24 the absence of a clear isosbestic point suggests the existence of more than one type of discrete aggregate species, most likely dimers, in equilibrium with the monomer. Time-dependent 1H NMR studies were then performed immediately after the solution used for VT–NMR studies reached 283 K (Fig. 1b). The observed strong broadening and subsequent disappearance of the signals indicate a further oligomerization of the dimers over time, which is in perfect agreement with the depletion of the monomer band and a concomitant emergence of an aggregate band in time dependent UV-Vis experiments (see Fig. 1a). Time-dependent DLS studies in MCH (c = 5 × 10−4 M) at 283 K (Fig. S3†) revealed a consistent increase in decay time during the initial 15 min, which supports the formation of oligomers. Over time, a more pronounced aggregation process is evident from the decrease in relative counts over 60 min. Comparison of the 2D COSY and ROESY spectra of an aggregate solution of L1 (11.7 mM, MCH-d14, 315 K) strongly suggests an H-bonded antiparallel molecular arrangement (see ESI for details, Fig. S6†) which is in good agreement with the findings observed by previous VT–UV and NMR studies. A related antiparallel arrangement of aromatic groups via H-bonds has been previously observed in the solid state for oligomeric zipper complexes bearing aromatic amides.25
The structure of this H-bonded packing optimized via the dispersion-corrected PM6 method is shown in Scheme 1. This conformation is appropriate to grow a stable 1D assembly of L1 (Scheme 1), as revealed by classical Molecular Dynamics (MD) simulations carried out at 300 K and 1 atm (for more details on the MD simulations, see the ESI†). These simulations also reveal that the interactions between the alkyl chains are very important to stabilize the antiparallel stacks, together with hydrogen bonding. In the MD snapshot shown in Scheme 1, the structure of the simulated supramolecular material is somewhat heterogeneous although in average the monomers tend to stack in an antiparallel fashion driven to a large extent by H-bonding. Also, interdigitation of the alkyl chains facilitates the growth into 2D lamellae (see Scheme 1) and finally long, twisted fibers by shielding the polar pyridine rings from exposing themselves to the surrounding nonpolar medium.
The hierarchical self-assembly of L1 has also been studied using atomic force microscopy (AFM) imaging by spin-coating MCH solutions of L1 (c = 5 × 10−4 M, 283 K) onto highly oriented pyrolytic graphite (HOPG) at different time intervals. After keeping the solution for 15 minutes at 283 K (excluding the initial lag), short 1D supramolecular fibers with 2–3 nm in height, 10–15 nm in width (Fig. 1c and S7a†) and a strong tendency to form lamellae were observed, which further grew into 2D lamellar structures with 2–3 nm in height and 50–100 nm in width over a period of 30 minutes (Fig. 1d and S7b†). Subsequent rolling and bundling of the tapes (Fig. S7c†) ultimately results in the formation of thick, long, twisted fibers after an overall equilibration time of 45–60 minutes (Fig. 1e and S7d†). The width of the fibers ranges from 50 to 100 nm, whereas the length goes up to several microns. The formation of these thicker fibers led to a yellow gel above 50 mM, as confirmed by AFM (Fig. S8†).
Photoisomerization of L1
Irradiation at 370 nm of the long fibers of L1 with the azo groups in the trans conformation (trans-L1, c = 5 × 10−4 M) at 283 K led to the disassembly of the fibers, which is attributed to the trans-to-cis isomerization, as demonstrated by the depletion of the π–π* transition (363 nm) along with the increase of the n–π* transition at 450 nm (Fig. 2a, purple line). Some insight into this UV-induced disassembly process could be obtained from MD simulations by inducing an instantaneous trans–cis isomerization of L1 within a fiber: a substantial reduction in the number of amide groups involved in short hydrogen-bonding contacts (dO⋯H < 2 Å) was observed, as depicted as yellow spheres in the MD snapshots, and the monomer–monomer distances increased (Scheme 1, top right). These structural changes might explain the initial driving force for the disassembly of the long fibers after light irradiation (vide infra). According to UV-Vis results, approximately 80% of the cis isomer is formed upon UV irradiation, which reverts progressively to the more stable trans isomer over the period of 120 min upon keeping the solution in the dark (blue line in Fig. 2 and S9a†). Monitoring the A360vs. time at 283 K upon the entire back isomerization process allowed us to distinguish an aggregation process that closely resembles the one shown by non-irradiated L1 (Fig. 1a and S9a†). Thus, we conclude that the cis isomer is a dormant species, whereas aggregation only occurs upon activation to the active trans monomer via back isomerization (Fig. 2a, inset).16,26 These processes (photoisomerization and self-assembly) compete partially at higher concentration (1 mM), where the cis form is likewise dormant to aggregation (Fig. S10†).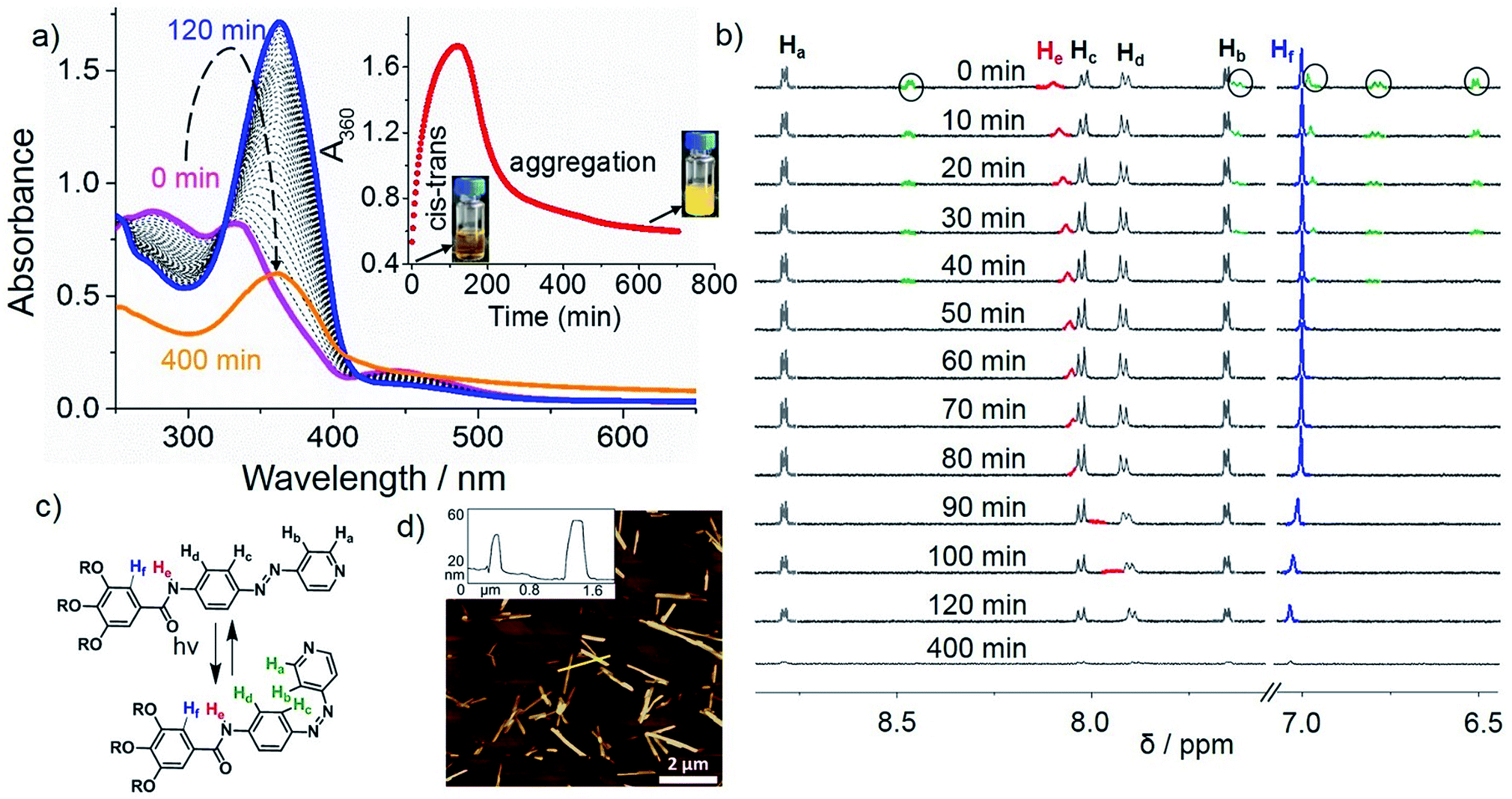 | ||
| Fig. 2 (a) Time-dependent absorption changes (0–120 min: purple to blue; 120–400 min blue to orange) of L1 (5 × 10−4 M) in MCH after irradiation for 30 min with a 370 nm LED lamp at 283 K; inset: plot of A360vs. time at 283 K, and photographs showing the irradiated clear L1cis + trans monomer (left) to turbid L1trans aggregates (right). (b) Time-dependent 1H-NMR changes at 283 K of L1 (5 × 10−4 M) after irradiation for 30 min with a 370 nm LED lamp in MCH-d14. Black encircled green peaks correspond to cis isomer. (c) Scheme showing reversible trans–cis isomerization. (d) AFM image of L1 (5 × 10−4 M) on HOPG upon UV-irradiation for 30 min in MCH and ageing at 283 K for 400 min with corresponding cross-section analysis along the yellow line. | ||
Closer insight into the influence of cis-to-trans isomerization on the hierarchical self-assembly of L1 was provided by time-dependent 1H-NMR experiments (5 × 10−4 M, MCH-d14) at 283 K. Due to the impossibility of irradiating the sample inside the NMR spectrometer and the longer equilibration time needed compared to UV-Vis, the first recorded 1H NMR spectrum (denoted as ‘0 min’ in Fig. 2b) was obtained around 15 min after the sample was irradiated for 30 min at 298 K. By this procedure, we established that 40% of the cis isomer was present for the first NMR measurement even though back isomerization took place at 298 K prior to the NMR measurement (Fig. S9b–d†). Over a period of around 40 min, complete disappearance of the signals of the cis isomer (marked in green with black circles) was concomitant with a slight upfield shift of the N–H proton (He), whereas all remaining protons showed insignificant changes. Interestingly, a more significant broadening of the He signal and further upfield shift was also observed even after complete cis–trans conversion (Fig. 2c) around 60 min. This trend was not observed for the trans isomer without irradiation (Fig. 1b), and indicates the lack of hydrogen bonding but rather the proximity of a π-surface. We hypothesize that the high excess of dormant cis monomers (80% according to UV-Vis) formed immediately after photoirradiation at 283 K could sequester the trans-monomers and prevent them to form antiparallel hydrogen-bonded stacks, leading to a non-H-bonded arrangement different from the non-irradiated pathway (Scheme 1, “cis + trans” structure).
The above photoisomerization-regulated stepwise aggregation process of L1 (5 × 10−4 M, MCH) was examined by AFM. The aggregates formed by L1 in the absence of light at 283 K (Fig. 1e and S11a†) were irradiated with 370 nm UV-light for 30 min, kept at 283 K and finally spin-coated onto HOPG at different time intervals. AFM analysis of the sample kept at 283 K for 140 min upon irradiation (approximately the maximum value observed in the plot A360vs. time, see inset of Fig. 2a) reveals the formation of ill-defined amorphous aggregates (Fig. S11b†) that are most likely the result of a kinetically-driven off-pathway aggregation event. Interestingly, further ageing this solution at 283 K for additional 260 min (total time 400 min after irradiation) showed the transformation of the amorphous short assemblies into photo-reconstructed rod-like structures (Fig. 2d and S11c, d†) that are considerably shorter than those formed without irradiation (Fig. 1e). These results can be explained in terms of a frustrated nucleation event of the active trans isomers caused by the presence of sterically hindered, dormant cis-monomers, which is supported by a non-H-bonded antiparallel dimerization predicted by PM6 calculations and MD simulations (Scheme 1). The formation of these less organized pre-nuclei dramatically affects the addition of further active trans monomers during the subsequent elongation process, leading to a less compact non-H-bonded arrangement that ultimately results in the shortening of the ensembles. These results can be corroborated by the reversible light-induced gel–sol transition of L1 at 50 mM (Fig. S11f†).
Hierarchical self-assembly of Pd(L1)2Cl2
We envisioned that the complexation of metal ions such as Pd(II) by L1 to yield Pd(L1)2Cl2 (C1, Scheme 2) would cause a significant impact on the photoresponsive and self-assembly behavior. This metal ion was selected not only based on the expertise of our group in Pd(II)-based assemblies19 but also due to their preorganized coordination geometry and aggregation propensity. Unlike trans-L1, trans-C1 readily self-assembles at room temperature in nonpolar solvents such as MCH (see solvent-dependent UV-Vis studies in Fig. S12†). Fig. 3a shows the spectral changes upon cooling a monomer solution of C1 in MCH from 363 K to 283 K with 1 K min−1 ramp. On cooling, the metal-to-ligand charge transfer (MLCT) transition at 395 nm broadens progressively and decreases in intensity whereas a shoulder at 460 nm becomes apparent (Fig. 3a). This spectral change is suggestive of aggregation. Monitoring the absorbance at 395 nm vs. temperature (T) at four different concentrations yielded non-sigmoidal plots, which could be accurately fitted to the nucleation–elongation cooperative model (Fig. 3a, inset and Fig. S13, S14†).27 According to this model, the formation of a small aggregate (nucleus) is needed to activate the supramolecular growth into fiber-like structures (for an overview of the thermodynamic parameters, see Fig. S13, S14 and Table S1†).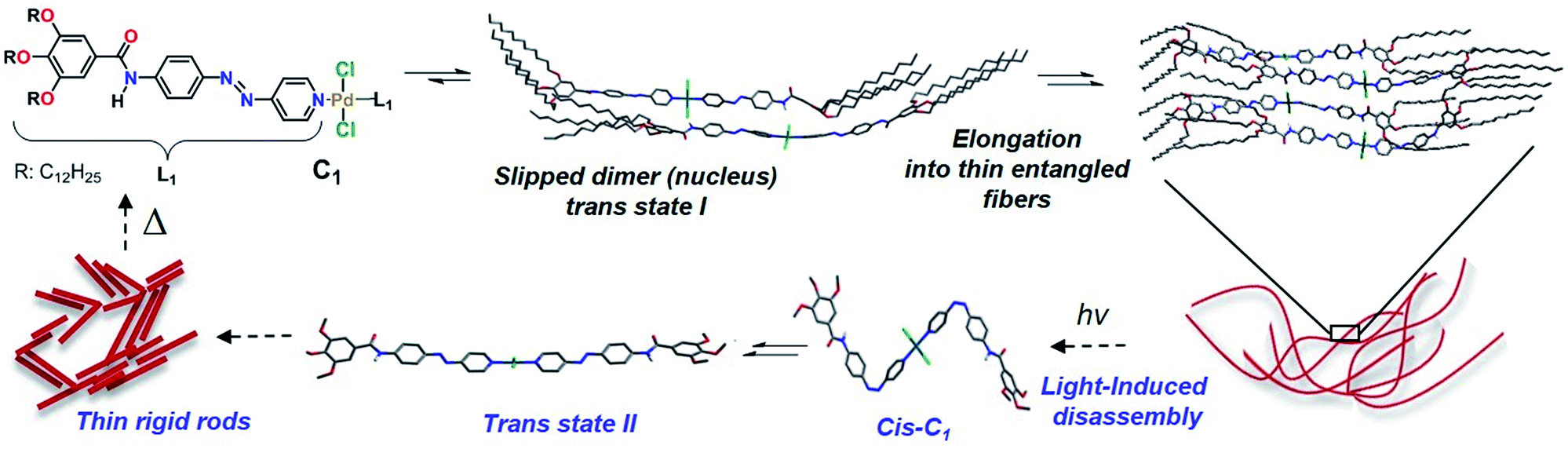 | ||
| Scheme 2 Proposed hierarchical self-assembly and photoresponsive behavior of C1. Structures of monomers and small aggregates were optimized using DFT (PBE0/6-31G*/LANL2DZ) and dispersion-corrected PM6, respectively. | ||
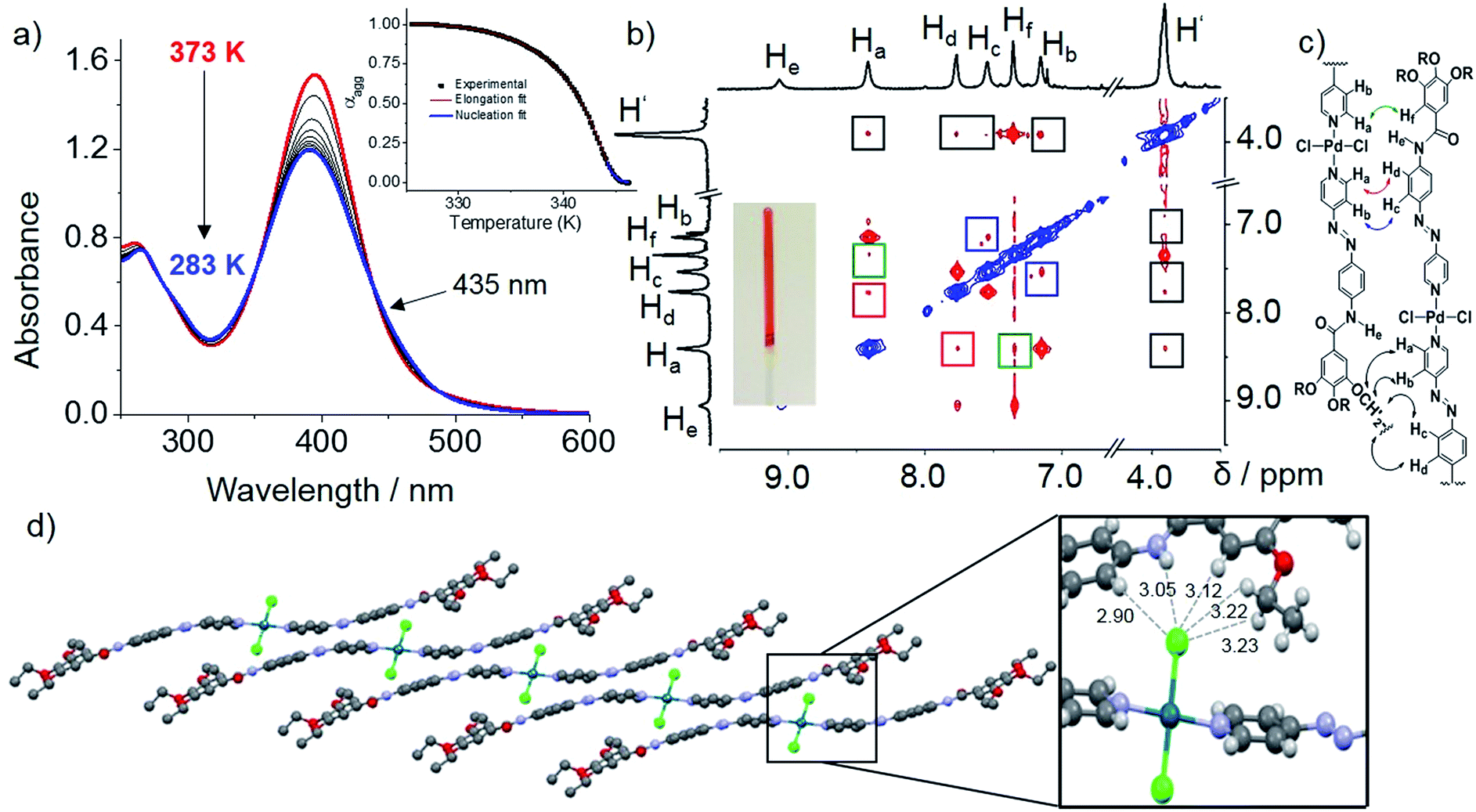 | ||
| Fig. 3 (a) Temperature-dependent UV-Vis spectra of C1 at 5 × 10−5 M; inset: plot of αaggvs. T for C1 at 5 × 10−5 M monitored at 395 nm. (b) COSY and ROESY of C1 at 7.5 mM in MCH-d14 at 358 K and photograph of gel (inset). (c) Plausible molecular arrangement. (d) Packing of C2 (b-axis) driven by N–H⋯Cl and C–H⋯Cl interactions (inset). | ||
VT–DOSY and DLS measurements of C1 further support the formation aggregates in solution (Fig. S15 and S16†). ROESY NMR of an aggregate solution (7.5 mM, 358 K) in MCH-d14 revealed the presence of five new cross-peaks (highlighted in coloured squares in Fig. 3b) that are absent in COSY studies. For instance, correlation signals between protons He and Hf as well as Hd and Hf can be distinguished (Fig. 3b). As these protons are within the vicinity of 5 Å, they should result from an intramolecular coupling. On the other hand, cross-peaks between Ha and Hf (green), Hb and Hc (blue) and Ha and Hd (red) can only be due to intermolecular contacts, as the respective protons are very far from one another (>5 Å). Furthermore, additional intermolecular interactions between –O–CH2– protons of the alkyl chains (H′) and aromatic protons Ha–d were also identified (black). This coupling pattern is in agreement with the formation of slipped stacks stabilized by π–π interactions between the aromatic rings of the ligands as well as N–H⋯Cl–Pd hydrogen bonding interactions (Fig. 3c), as recently proposed for related π-conjugated Pd(II) complexes.28 The fingerprints associated with these interactions could be also identified by FT-IR measurements in MCH (Fig. S17†).28
This stacking arrangement in solution is in perfect agreement with the molecular packing extracted from X-ray analysis of single crystals grown from DCM/acetonitrile of a nearly identical Pd-azo derivative (C2) with short ethyl groups. The crystal structure analysis showed a marked molecular curvature of the azo-based pyridyl-ligands on both sides of the metal center (Fig. S18 and S19†). The packing in the crystal structure is mainly driven by three types of cooperative weak interactions: C–H⋯Cl, N–H⋯Cl interactions and π–π stacking. In analogy with the packing deduced by ROESY studies, the monomer units are arranged in a slipped fashion driven by a combination of one N–H⋯Cl and four C–H⋯Cl intermolecular interactions. Each Cl is interacting with two aromatic protons (Hd and Hf), an NH group of the amide moiety and two polarized methylene groups belonging to the ethoxy chains of a neighboring unit (Fig. 3d, inset). A further growth of the system into layered structures is facilitated by lateral interactions of the formed 1D stacks via 4 C–H⋯Cl and 4 C–H⋯O interactions. Further, π–π interactions are stabilizing the packing along the a-axis (Fig. S20†). These overall results highlight the key impact of NH⋯Cl interactions on slipped stacking stabilization.
In contrast to the free ligand L1, C1 forms considerably thinner and shorter well-defined fibers (5–10 nm in width and 60–150 nm in length) in MCH (Fig. S21†). This difference in morphology clearly reflects different molecular packing with distinct intermolecular interactions (π–π, C–H⋯Cl and N–H⋯Cl, vide supra) compared to the free ligand L1. As the NMR signals of C1 are nearly unidentifiable in pure MCH-d14 due to strong aggregation, a solvent mixture with 10% CDCl3 was chosen for further studies (Fig. S22†). Prior to the NMR experiments, we confirmed by VT–UV-Vis studies under identical conditions that the addition of 10% CHCl3 does not influence the aggregation behavior of C1 (Fig. S23†). AFM analysis showed the formation of a network of thin entangled fibers further supporting an identical aggregation behavior in pure MCH and 10% CHCl3–MCH at 5 × 10−5 M (Fig. 4b and S24†). The powder X-ray diffraction pattern of a thin film of C1 showed the formation of a hexagonal columnar structure with the lattice parameter of a = 3.5 nm, which is larger than that of L1 (a = 2.45) in a tetragonal columnar structure (Fig. S25†). Probably, the well-defined fibers of C1 visualized by AFM (Fig. 4b and S24†) are the elementary structure composed of one-dimensionally stacked C1.
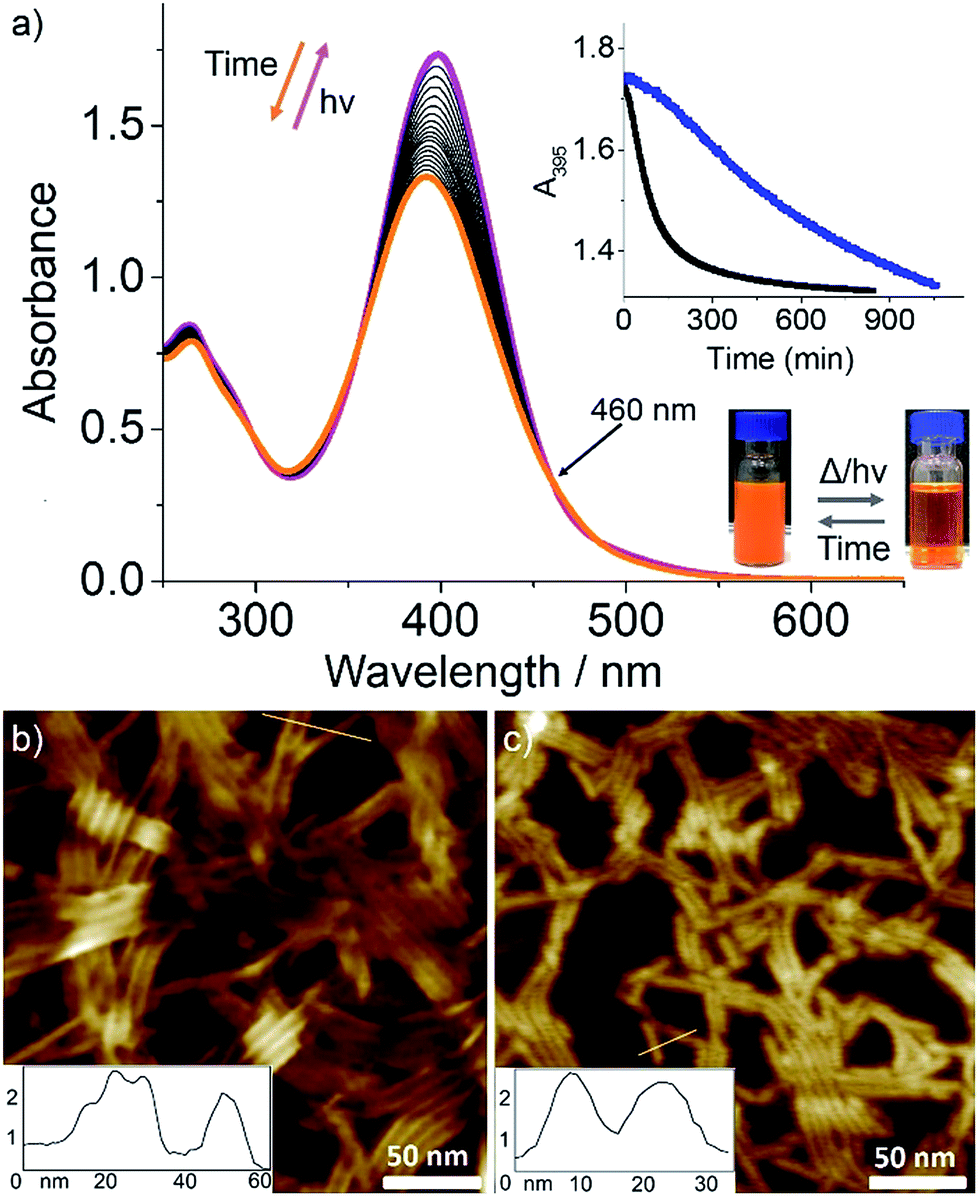 | ||
Fig. 4 (a) Time-dependent UV-Vis spectra (5 × 10−5 M MCH/CHCl3 (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10)) of C1 upon light irradiation and subsequent quenching to 283 K; inset: plot of A395vs. time before (blue) and after irradiation (black) at 283 K and photographs showing opacity changes upon irradiation for 15 min. AFM images of C1 aggregates formed under equivalent conditions as for UV-Vis: (b) before and (c) after UV irradiation for 30 min at 283 K with corresponding cross-section analysis along the yellow line. :10)) of C1 upon light irradiation and subsequent quenching to 283 K; inset: plot of A395vs. time before (blue) and after irradiation (black) at 283 K and photographs showing opacity changes upon irradiation for 15 min. AFM images of C1 aggregates formed under equivalent conditions as for UV-Vis: (b) before and (c) after UV irradiation for 30 min at 283 K with corresponding cross-section analysis along the yellow line. | ||
Photoisomerization of C1
Irradiation at 370 nm of the entangled thin fibers of C1 (MCH:CHCl3 (9:1), c = 5 × 10−5 M, see Fig. 4b and S24† for AFM images) at 283 K for 30 min caused a small red shift in the absorption maximum and significant hyperchromism (Fig. 4a). Because the UV-Vis spectral changes cannot be explained by usual trans-to-cis isomerization of azobenzene, we assume this to be due to a photo-induced disassembly via formation of cis-C1 followed by a rapid back-isomerization to trans-C1, which is most likely kinetically trapped. After finishing the UV irradiation for 30 min, we monitored the spectral changes over a period of 800 min while keeping the solution at 283 K (Fig. 4a). A blue shift in the absorption maximum from 397 nm to 391 nm with a small absorption change at around 490 nm was observed with multiple isosbestic points (Fig. 4a). Without any lag, A395 started decreasing and reached a plateau after several hours (Fig. 4a, inset), indicating the formation of photo-reconstructed aggregates. Comparison of this spectrum with that corresponding to the aggregation process of C1 without irradiation (see Fig. S23 and S3a†) reveals a shift of the isosbestic point from 435 nm to 460 nm.
In order to compare the above photochemically achieved kinetic state with that obtained by quick temperature drop (quenching), we rapidly cooled a hot MCH/CHCl3 solution of C1 (5 × 10−5 M) to 283 K and monitored the UV-Vis spectral changes for 1000 min (Fig. S26†). These studies showed significant differences compared to the UV-irradiated sample, i.e., a blue shift of the absorption maximum from 398 nm to 394 nm with only one isosbestic point at around 470 nm. A plot of A395vs. time showed a slow decay compared to the irradiated sample (Fig. 4a, inset). These results indicate that the self-assembly of C1 after the photo-induced disassembly proceeds through a different nucleation–elongation mechanism compared to that from the thermally obtained monomeric C1.
We next attempted to identify the possible cis-C1 formation by 1H-NMR measurements. The observed rapid transformation of the initially slightly opaque solution of trans-C1 in 1:9 CDCl3–MCH-d14 at 5 × 10−4 M into a clear solution upon irradiation for 30 min implies a disassembly of trans-C1 aggregates (Fig. 4, inset). However, to our surprise, no resonances corresponding to cis-C1 were identified. Dissociation of the N–H⋯Cl hydrogen bonds was evident from 1H-NMR where the amide signal shifts from 9.41 to 9.16 ppm upon irradiation (see Fig. S27†). Although nearly complete dissociation of trans-C1 (c = 9 mM) aggregates was observed upon irradiation in CDCl3 (Fig. S28†), no signals corresponding to cis-C1 were identified. To justify the UV-induced disassembly of C1 aggregates, we performed 1H-NMR experiments of a structurally related OPE-based Pd(II) complex 1 lacking the photo-responsive unit (Fig. S29†), which was previously observed to self-assemble via N–H⋯Cl hydrogen bonds.28 In contrast to C1, irradiation of 1 for 30 min showed negligible NMR shifts (Fig. S29†). Accordingly, the disassembly of trans-C1 aggregates upon irradiation is ascribable to the formation of bulky cis-C1.
A plausible explanation for the absence of cis-C1 during the NMR measurements can be related to the change in the excited state dynamics of L1 upon Pd(II) coordination,18c,29 which has been inspected by Density Functional Theory (DFT) and Time-Dependent DFT (TD-DFT) calculations. Initially, irradiation of the trans-state I (Scheme 2), in which the carbonyls of each complex are antiparallel to each other, populates a molecular orbital which is antibonding with respect to both azo nitrogens (inset in Fig. S30†). The NN double bond is then broken and rotation around this bond generates the cis-C1 isomer. The rather distorted geometry of the latter (Scheme 2 and Fig. S31†) leads to the dissociation of the aggregates. The lowest excited state of cis-C1 is almost resonant with that of trans-C1-II, where carbonyl groups are now oriented parallel to each other (Fig. S32†), suggesting a fast cis-C1 → trans-C1-II conversion. The relative orientation of carbonyl groups inside a fiber, namely parallel vs. antiparallel, can strongly influence the energy of excited states, whose differences can be as high as 0.7 eV (Fig. S33†). As anticipated from the above calculations, the photo-reconstructed C1 aggregates show appreciable morphological changes compared to the nanostructures before UV irradiation. The AFM images show that the initially formed thin flexible fibers with several μm in length (Fig. 4b and S24, S26†) transform into short rods with maximum length of 20–100 nm (Fig. 4c and S34†). Supramolecular systems in which all carbonyl groups are pointing in the same direction, like fibers of trans-C1-II, can form giant dipole moments or macrodipoles, influencing the interaction between nearby fibers as well as the final morphology of the material.30 Because the local accumulation of macrodipoles in dense regions containing supramolecular fibers is not thermodynamically favorable,31 further growth of fibers of trans-C1-II becomes frustrated, explaining why they are shorter. On the other hand, local accumulation of macrodipoles does not occur in fibers of trans-C1-I because the carbonyls are oriented antiparallel to each other, stabilizing the fibers and allowing them to grow much further (notice that the crystal structure shown in Fig. 3d has also antiparallel amide groups).
Conclusions
We have reported a new small molecule-based supramolecular system (azo-based pyridyl ligand L1) that undergoes significant changes in its hierarchical self-assembly and aggregate morphology upon response to UV-light irradiation and metal coordination. Combined experimental and theoretical studies allowed us to propose a mechanism for its hierarchical self-assembly behavior. L1 self-assembles in the absence of UV-light and metal ions into long twisted fibers driven by hydrogen bonding and π-stacking via the formation of an antiparallel dimer species. UV irradiation and subsequent photoisomerization of L1 leads to a new non-H-bonded packing mode that ultimately results in the formation of shorter rigid rods. Complexation of PdCl2 drastically changes the mode of aggregation from antiparallel to slipped stacks driven by N–H⋯Cl interactions with subsequent increase in the degree of cooperativity of the supramolecular growth. Finally, these long thin fibers transform into thinner, shorter rods upon UV irradiation via a reorganization into a different trans conformation of the Pd(II) complex. The stability of short vs. long fibers with Pd(II) was rationalized in terms of the accumulation of macrodipoles, which depend on the relative orientation of carbonyl groups inside the fibers, namely parallel or antiparallel. Our results have allowed us to unravel for the first time the impact of metal coordination and light irradiation on hierarchical self-assembly processes. In our opinion, the present study represents a starting point towards the development of a new class of stimuli-responsive self-assembled materials combining the properties of metal ions and light.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Dr Mitsuaki Yamauchi (Kwansei Gakuin University) and Dr Xu Lin (Southwest Forestry University) for experimental assistance. We thank the European Commission (ERC StG SUPRACOP 715923) and the Humboldt Foundation (Sofja-Kovalevskaja) for funding. This work was partially supported by KAKENHI grant (26102010 for S. Y.) and the Grant-in-Aid for Scientific Research on Innovative Areas “π-Figuration” (26102001) of the Japanese Ministry of Education, Culture, Sports, Science, and Technology (MEXT). We thank Ana-Maria Krause for X-Ray analysis.Notes and references
- (a) G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CrossRef CAS PubMed; (b) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed; (c) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed; (d) J. D. Tovar, Acc. Chem. Res., 2013, 46, 1527–1537 CrossRef CAS PubMed; (e) C. Kulkarni, S. Balasubramanian and S. J. George, ChemPhysChem, 2012, 14, 661–673 CrossRef PubMed; (f) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed; (g) C. Rest, R. Kandanelli and G. Fernandez, Chem. Soc. Rev., 2015, 44, 2543–2572 RSC; (h) A. Sandeep, V. K. Praveen, K. K. Kartha, V. Karunakaran and A. Ajayaghosh, Chem. Sci., 2016, 7, 4460–4467 RSC; (i) D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC; (j) M. Hifsudheen, R. K. Mishra, B. Vedhanarayanan, V. K. Praveen and A. Ajayaghosh, Angew. Chem., Int. Ed., 2017, 56, 12634–12638 CrossRef CAS PubMed; (k) A. Sorrenti, J. LeiraIglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC; (l) E. E. Greciano, B. Matarranz and L. Sánchez, Angew. Chem., Int. Ed., 2018, 57, 4697–4701 CrossRef CAS PubMed.
- (a) S. Yagai, T. Karatsu and A. Kitamura, Chem.–Eur. J., 2005, 11, 4054–4063 CrossRef CAS PubMed; (b) S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520–1529 RSC; (c) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed; (d) M. Kathan and S. Hecht, Chem. Soc. Rev., 2017, 46, 5536–5550 RSC; (e) R. Yamakado, M. Hara, S. Nagano, T. Seki and H. Maeda, Chem.–Eur. J., 2017, 23, 9244–9248 CrossRef CAS PubMed; (f) R. Yamakado, M. Hara, S. Nagano, T. Seki and H. Maeda, Chem. Lett., 2018, 47, 404–407 CrossRef CAS.
- (a) G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684–2697 RSC; (b) A. Tsuda, Y. Nagamine, R. Watanabe, Y. Nagatani, N. Ishii and T. Aida, Nat. Chem., 2010, 2, 977–983 CrossRef CAS PubMed.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- (a) Y. Geng, X.-J. Wang, B. Chen, H. Xue, Y.-P. Zhao, S. Lee, C.-H. Tung and L.-Z. Wu, Chem.–Eur. J., 2009, 15, 5124–5129 CrossRef CAS PubMed; (b) Z. Y. Xiao, X. Zhao, X. K. Jiang and Z. T. Li, Chem. Mater., 2011, 23, 1505–1511 CrossRef CAS; (c) S. Sato, T. Murase and M. Fujita, Supramolecular Chemistry: From Molecules to Nanomaterials, John Wiley & Sons, Ltd, Hoboken, 2012 Search PubMed; (d) S. Lee, E. H. Brandon, Y. Liu, R. D. James, W. B. David, L. T. Steven and A. H. Flood, Chem.–Eur. J., 2015, 22, 560–569 CrossRef PubMed; (e) B. Qiao, B. E. Hirsch, S. Lee, M. Pink, C.-H. Chen, B. W. Laursen and A. H. Flood, J. Am. Chem. Soc., 2017, 139, 6226–6233 CrossRef CAS PubMed.
- (a) M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed; (b) R. Klajn, K. J. M. Bishop and B. A. Grzybowski, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10305–10309 CrossRef CAS PubMed; (c) J. W. Fredy, A. Mendez-Ardoy, S. Kwangmettatam, D. Bochicchio, B. Matt, M. C. A. Stuart, J. Huskens, N. Katsonis, G. M. Pavan and T. Kudernac, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11850–11855 CrossRef CAS PubMed.
- (a) H. Tian and S. Yang, Chem. Soc. Rev., 2004, 33, 85–97 RSC; (b) J. J. D. de Jong, L. N. Lucas, R. M. Kellogg, J. H. van Esch and B. L. Feringa, Science, 2004, 304, 278–281 CrossRef CAS PubMed; (c) S. Yagai, K. Iwai, T. Karatsu and A. Kitamura, Angew. Chem., Int. Ed., 2012, 51, 9679–9683 CrossRef CAS PubMed; (d) S. Yagai, K. Ishiwatari, X. Lin, T. Karatsu, A. Kitamura and S. Uemura, Chem.–Eur. J., 2013, 19, 6971–6975 CrossRef CAS PubMed; (e) S. Yagai, K. Iwai, M. Yamauchi, T. Karatsu, A. Kitamura, S. Uemura, M. Morimoto, H. Wang and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 2602–2606 CrossRef CAS PubMed; (f) J. T. van Herpt, J. Areephong, M. C. A. Stuart, W. R. Browne and B. L. Feringa, Chem.–Eur. J., 2014, 20, 1737–1742 CrossRef CAS PubMed.
- P. K. Kundu, D. Samanta, R. Leizrowice, B. Margulis, H. Zhao, M. Börner, T. Udayabhaskararao, D. Manna and R. Klajn, Nat. Chem., 2015, 7, 646–652 CrossRef CAS PubMed.
- (a) A. Harada, Y. Takashima and M. Nakahata, Acc. Chem. Res., 2014, 47, 2128–2140 CrossRef CAS PubMed; (b) S. Engel, N. Möller and B. J. Ravoo, Chem.–Eur. J., 2017, 24, 4741–4748 CrossRef PubMed.
- (a) K. Ichimura, Chem. Rev., 2000, 100, 1847–1874 CrossRef CAS PubMed; (b) J. A. Sarah, F. Lancia, S. Iamsaard, B. Matt, T. Kudernac, P. F. Stephen and N. Katsonis, Angew. Chem., Int. Ed., 2017, 56, 3261–3265 CrossRef PubMed.
- (a) A. Samanta and B. J. Ravoo, Chem.–Eur. J., 2014, 20, 4966–4973 CrossRef CAS PubMed; (b) J. Moratz, L. Stricker, S. Engel and B. J. Ravoo, Macromol. Rapid Commun., 2017, 39, 1700256 CrossRef PubMed.
- R. Rajaganesh, A. Gopal, T. M. Das and A. Ajayaghosh, Org. Lett., 2012, 14, 748–751 CrossRef CAS PubMed.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- A. Gopal, M. Hifsudheen, S. Furumi, M. Takeuchi and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 10505–10509 CrossRef CAS PubMed.
- (a) M. Yamauchi, T. Ohba, T. Karatsu and S. Yagai, Nat. Commun., 2015, 6, 8936 CrossRef CAS PubMed; (b) B. Adhikari, Y. Yamada, M. Yamauchi, K. Wakita, X. Lin, K. Aratsu, T. Ohba, T. Karatsu, M. J. Hollamby, N. Shimizu, H. Takagi, R. Haruki, S.-i. Adachi and S. Yagai, Nat. Commun., 2017, 8, 15254 CrossRef PubMed; (c) B. Adhikari, X. Lin, M. Yamauchi, H. Ouchi, K. Aratsu and S. Yagai, Chem. Commun., 2017, 53, 9663–9683 RSC.
- M. Endo, T. Fukui, S. H. Jung, S. Yagai, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2016, 138, 14347–14353 CrossRef CAS PubMed.
- (a) M. J. Mayoral, C. Rest, V. Stepanenko, J. Schellheimer, R. Q. Albuquerque and G. Fernández, J. Am. Chem. Soc., 2013, 135, 2148–2151 CrossRef CAS PubMed; (b) C. Rest, M. J. Mayoral, K. Fucke, J. Schellheimer, V. Stepanenko and G. Fernández, Angew. Chem., Int. Ed., 2014, 53, 700–705 CrossRef CAS PubMed; (c) N. K. Allampally, M. J. Mayoral, S. Chansai, M. C. Lagunas, C. Hardacre, V. Stepanenko, R. Q. Albuquerque and G. Fernández, Chem.–Eur. J., 2016, 22, 7810–7816 CrossRef CAS PubMed; (d) L. Herkert, A. Sampedro and G. Fernández, CrystEngComm, 2016, 18, 8813–8822 RSC.
- (a) M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed; (b) J. R. Kumpfer and S. J. Rowan, J. Am. Chem. Soc., 2011, 133, 12866–12874 CrossRef CAS PubMed; (c) E. Borré, S. Bellemin-Laponnaz and M. Mauro, Chem.–Eur. J., 2016, 22, 18718–18721 CrossRef PubMed; (d) M. E. Garah, E. Borré, A. Ciesielski, A. Dianat, R. Gutierrez, G. Cuniberti, S. Bellemin-Laponnaz, M. Mauro and P. Samorì, Small, 2017, 13, 1701790 CrossRef PubMed.
- (a) X. Yan, J. F. Xu, T. R. Cook, F. Huang, Q. Z. Yang, C. H. Tung and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8717–8722 CrossRef CAS PubMed; (b) R. D. Mukhopadhyay, V. K. Praveen and A. Ajayaghosh, Mater. Horiz., 2014, 1, 572–576 RSC; (c) E. Borre, J. F. Stumbe, S. Bellemin-Laponnaz and M. Mauro, Chem. Commun., 2017, 53, 8344–8347 RSC; (d) R. D. Mukhopadhyay, G. Das and A. Ajayaghosh, Nat. Commun., 2018, 9, 1987 CrossRef PubMed.
- (a) M. J. Mayoral and G. Fernández, Chem. Sci., 2012, 3, 1395–1398 RSC; (b) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed; (c) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed; (d) K. Li, G. S. Ming Tong, Q. Wan, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Chem. Sci., 2016, 7, 1653–1673 RSC.
- (a) J. Garcia-Amoros, S. Nonell and D. Velasco, Chem. Commun., 2012, 48, 3421–3423 RSC; (b) A. Florian, M. J. Mayoral, V. Stepanenko and G. Fernández, Chem.–Eur. J., 2012, 18, 14957–14961 CrossRef CAS PubMed.
- (a) X. Lu, Z. Guo, C. Sun, H. Tian and W. Zhu, J. Phys. Chem. B, 2011, 115, 10871–10876 CrossRef CAS PubMed; (b) A. Rödle, B. Ritschel, C. Mück-Lichtenfeld, V. Stepanenko and G. Fernández, Chem.–Eur. J., 2016, 22, 15772–15777 CrossRef PubMed.
- J. Gershberg, F. Fennel, T. H. Rehm, S. Lochbrunner and F. Würthner, Chem. Sci., 2016, 7, 1729–1737 RSC.
- M. Kasha, Radiat. Res., 1963, 20, 55–70 CrossRef CAS PubMed.
- H. Adams, P. L. Bernad Jr, D. S. Eggleston, R. C. Haltiwanger, K. D. M. Harris, G. A. Hembury, C. A. Hunter, D. J. Livingstone, B. M. Kariuki and J. F. McCabe, Chem. Commun., 2001, 1500–1501 RSC.
- (a) S. Yagai, T. Nakajima, T. Karatsu, K.-i. Saitow and A. Kitamura, J. Am. Chem. Soc., 2004, 126, 11500–11508 CrossRef CAS PubMed; (b) S. Yagai, T. Nakajima, K. Kishikawa, S. Kohmoto, T. Karatsu and A. Kitamura, J. Am. Chem. Soc., 2005, 127, 11134–11139 CrossRef CAS PubMed.
- P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS PubMed.
- A. Langenstroer, Y. Dorca, K. K. Kartha, M. J. Mayoral, V. Stepanenko, G. Fernández and L. Sánchez, Macromol. Rapid Commun., 2018, 1800191 CrossRef PubMed.
- (a) T. Yutaka, I. Mori, M. Kurihara, N. Tamai and H. Nishihara, Inorg. Chem., 2003, 42, 6306–6313 CrossRef CAS PubMed; (b) G. Auböck and M. Chergui, Nat. Chem., 2015, 7, 629–633 CrossRef PubMed.
- M. P. Oliveira, H.-W. Schmidt and R. Q. Albuquerque, Chem.–Eur. J., 2017, 24, 2609–2617 CrossRef PubMed.
- C. S. Zehe, J. A. Hill, N. P. Funnell, K. Kreger, K. P. van der Zwan, A. L. Goodwin, H.-W. Schmidt and J. Senker, Angew. Chem., Int. Ed., 2017, 56, 4432–4437 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization, description of experimental techniques, and additional images. CCDC 1864670. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03875a |
| This journal is © The Royal Society of Chemistry 2019 |