
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A copper-catalyzed double coupling enables a 3-step synthesis of the quassinoid core architecture†
Matthew L.
Condakes
 ,
Rachel Z.
Rosen
,
Stephen J.
Harwood
and
Thomas J.
Maimone
*
,
Rachel Z.
Rosen
,
Stephen J.
Harwood
and
Thomas J.
Maimone
*
Department of Chemistry, University of California, Berkeley, 826 Latimer Hall, Berkeley, California 94720, USA. E-mail: maimone@berkeley.edu
First published on 29th October 2018
Abstract
The quassinoids are a fascinating class of degraded triterpene natural products which possess, among other attributes, potent anti-cancer activity. Their complex polycyclic ring systems also serve as inspiration for the development of new chemical methods and strategies – especially those pertaining to C–C bond formation. Herein we disclose a novel tandem cross coupling/SN2′ reaction of vicinal epoxy vinyl triflates with simple Grignard reagents catalyzed by Cu(I). Using this transformation, the polycyclic core architecture of the quassinoids can be generated in only three linear steps from carvone epoxide, forming five carbon–carbon bonds in the process.
Introduction
Rapid generation of molecular complexity is a major driving force for 21st century organic synthesis, and natural products continue to provide an ideal testing ground for such exploration.1 In particular, the highly complex family of quassinoid natural products appears to be an area wherein novel methodologies could lead to great synthetic simplification. Derived from triterpenes, quassinoids are characterized by a tetracyclic ring system often containing a D-ring lactone (see 1–4, Fig. 1A).2 Moreover, the exciting biological profiles displayed by many quassinoids – particularly potent anti-cancer activity – provides an additional biological impetus for synthetic exploration.3 Not surprisingly, this family of natural products have historically been popular synthetic targets and have succumbed to multiple total synthesis efforts over the past several decades.4–6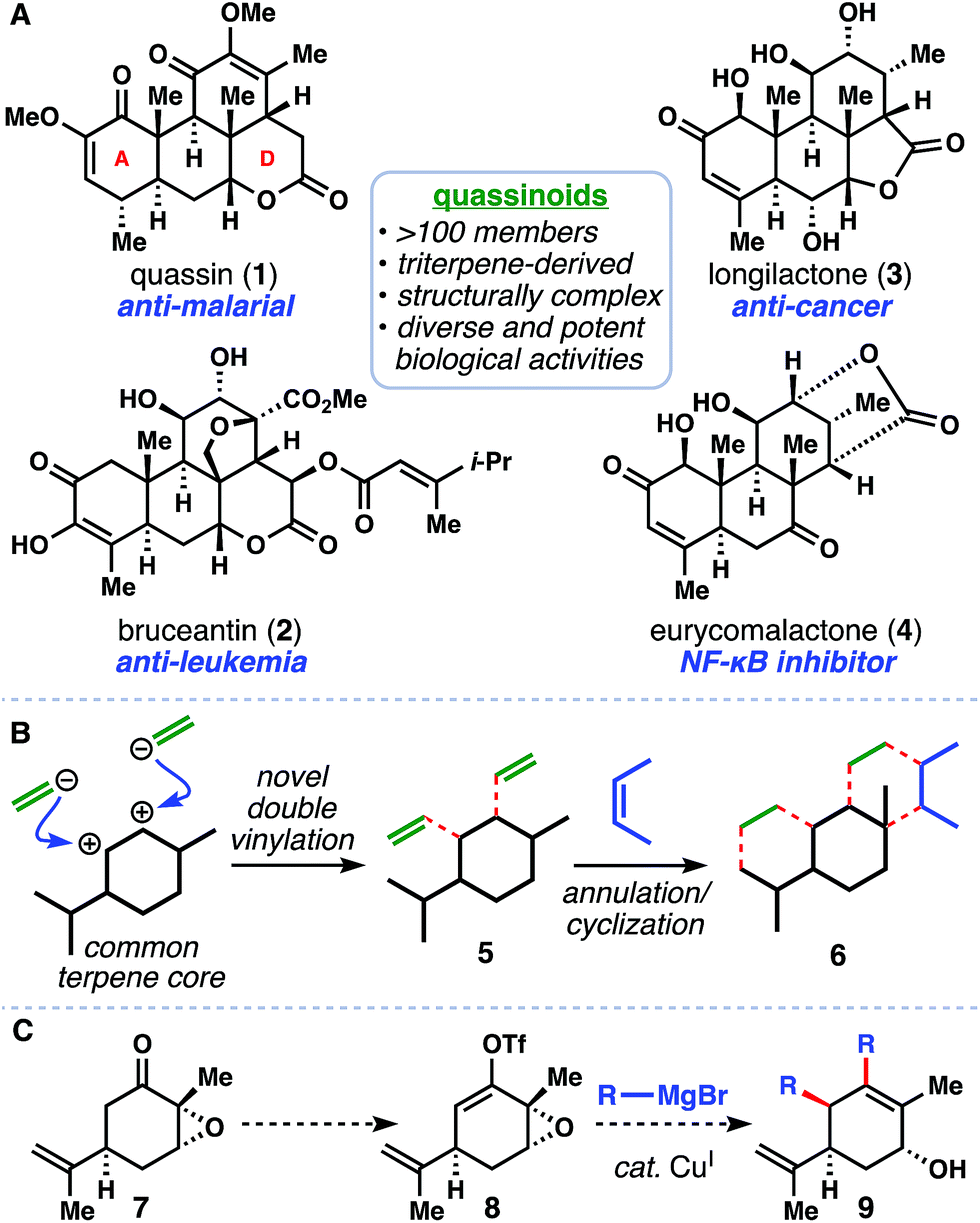 | ||
| Fig. 1 (A) Selected quassinoid natural products and their associated biological activities. (B) Synthetic strategy to access the quassinoid architecture featuring a novel double coupling reaction and successive cyclizations. (C) Desired coupling methodology. | ||
We identified a hypothetical double vinylation transform of a chiral pool monoterpene as a way of accessing 5, an intermediate we envisioned advancing to the quassinoid ring system (see 6) by way of two successive ring-forming reactions (Fig. 1B).7 Inspired by Wender's report of various organometallic reagents engaging epoxy enol ethers, enol phosphates, and enolates in SN2′ reactions,8 and McMurry's seminal work on the cross coupling of vinyl triflates with cuprates,9 we envisioned developing a Cu(I)-catalyzed process to convert carvone epoxide (7) directly into functionalized cyclohexenol 9 by way of vinyl triflate 8 and simple Grignard reagents (Fig. 1C). In principle, this reaction could formally introduce two sp2 fragments in a single step and with high diastereocontrol. From the outset, however, we were aware of the paucity of Kumada-type couplings of vinyl triflates and Grignard reagents catalyzed by copper, a preferred metal for SN2′ substitution processes.10,11 Moreover, a mechanism by which we could distinguish between the cross-coupling and allylic substitution reactivities would be ideal in realizing the selective coupling of two different nucleophiles, but potentially challenging to implement given the similarities between the two reaction modalities. Herein we realize the one-pot double coupling of epoxy ketones with Grignard reagents catalyzed by a Cu(I) complex. Using this tandem process, we then show that the quassinoid core architecture can be accessed in only three steps, forming five C–C bonds in the process.
Results and discussion
We began our investigations by examining the model coupling of epoxy ketone 10 with methyl Grignard to form double coupled product 12 (Table 1). We quickly realized that it was not necessary to isolate the vinyl triflate intermediate (see 11) for this reaction to be successful, and in fact we encountered challenges in doing so for a variety of ketones, including 10. Much to our delight, we found that multiple copper(I) salts were competent at catalyzing this transformation, albeit with variable levels of diastereoselectivity. Most copper catalysts enforced the expected anti stereochemistry for the allylic substitution;12 however, when the copper center was ligated with a bulky NHC ligand (entry 5), a slight preference for syn stereochemistry was observed. Of the copper sources examined, tetrakis(acetonitrile)copper(I) hexafluorophosphate [Cu(MeCN)4][PF6] (entry 4) seemed the most promising and was thus selected for further studies.|
|
|||||
|---|---|---|---|---|---|
| Entry | [Cu] | X (mol%) | Additivec | Yieldd (%) | d.r.e |
| a Standard reaction conditions: epoxide (0.1 mmol, 1.0 equiv.), LHMDS (0.1 mmol, 1.0 equiv.), PhNTf2 (0.1 mmol, 1.0 equiv.), −78 → 0 °C, 5 min; then add a solution of MeMgBr (0.3 mmol, 3.0 equiv.), [Cu] (X mol%), and additive, 0 °C, 1 h. b mol% with respect to epoxide starting material. c Additives included at 5.0 equiv. d Isolated yield of 12 after column chromatography. e Determined by 1H NMR of the crude reaction mixture. f Reaction performed using 1.0 mmol of epoxide. | |||||
| 1 | CuCl | 15 | — | 43% | 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | CuBr | 15 | — | 42% | 3:1 |
| 3 | Cul | 15 | — | 43% | 4:1 |
| 4 | [Cu(MeCN)4][PF6] | 15 | — | 43% | 7:1 |
| 5 | (IPr)CuCl | 15 | — | 41% | 1:2 |
| 6 | [Cu(MeCN) 4 ][PF 6 ] | 15 | HMPA | 46% |
>20:1 |
| 7 | [Cu(MeCN)4][PF6] | 15 | DMPU | 32% | 4:1 |
| 8 | [Cu(MeCN)4][PF6] | 15 | TMEDA | 23% | 4:1 |
| 9 | [Cu(MeCN)4][PF6] | 7.5 | HMPA | 23% | >20:1 |
| 10 | [Cu(MeCN)4][PF6] | 30 | HMPA | 41% | >20:1 |
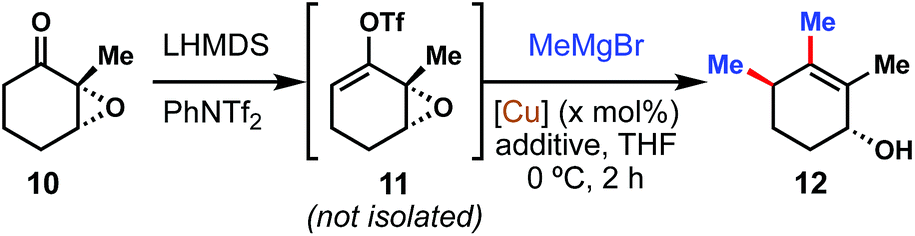
Diastereoselectivity could be further impacted by the inclusion of polar additives (entries 6–8). HMPA (5.0 equiv.) in particular was found to bias the allylic substation to the anti isomer almost exclusively. Varying the catalyst loading (entries 9, 10) did not further impact diastereoselectivity but did have a deleterious effect on yield if the amount of catalyst was decreased below 15 mol% (5 mol% with respect to MeMgBr).
We then proceeded to explore the scope of this tandem coupling process (Fig. 2A). Traditionally, copper catalysis has struggled to cross-couple aryl nucleophiles, due to their decreased reactivity relative to alkyl congeners.10,11a Much to our delight, however, products derived from aromatic Grignard reagents with diverse substituents (13–19) were afforded cleanly and in good yield.13 Other sp2 nucleophiles relevant to the quassinoid synthetic problem including dioxenyl (20) and vinyl (21) could also be coupled, generating products as single diastereomers. Single crystal X-ray diffraction studies of 18 and 21 confirmed the anti-stereochemistry. Two substrates deserve special mention (Fig. 2B). First, 22 – the product formed when using the di-Grignard reagent derived from 2,2′-dibromobiphenyl – exclusively underwent cross-coupling and SN2 opening of the epoxide, likely due to the intramolecular, and perhaps non-catalyzed, nature of the second step. Secondly, we note that 23 – the product of coupling allyl Grignard – was formed as a 1:1 mixture of syn and anti diastereomers, perhaps due to the high nucleophilicity of this reagent.14
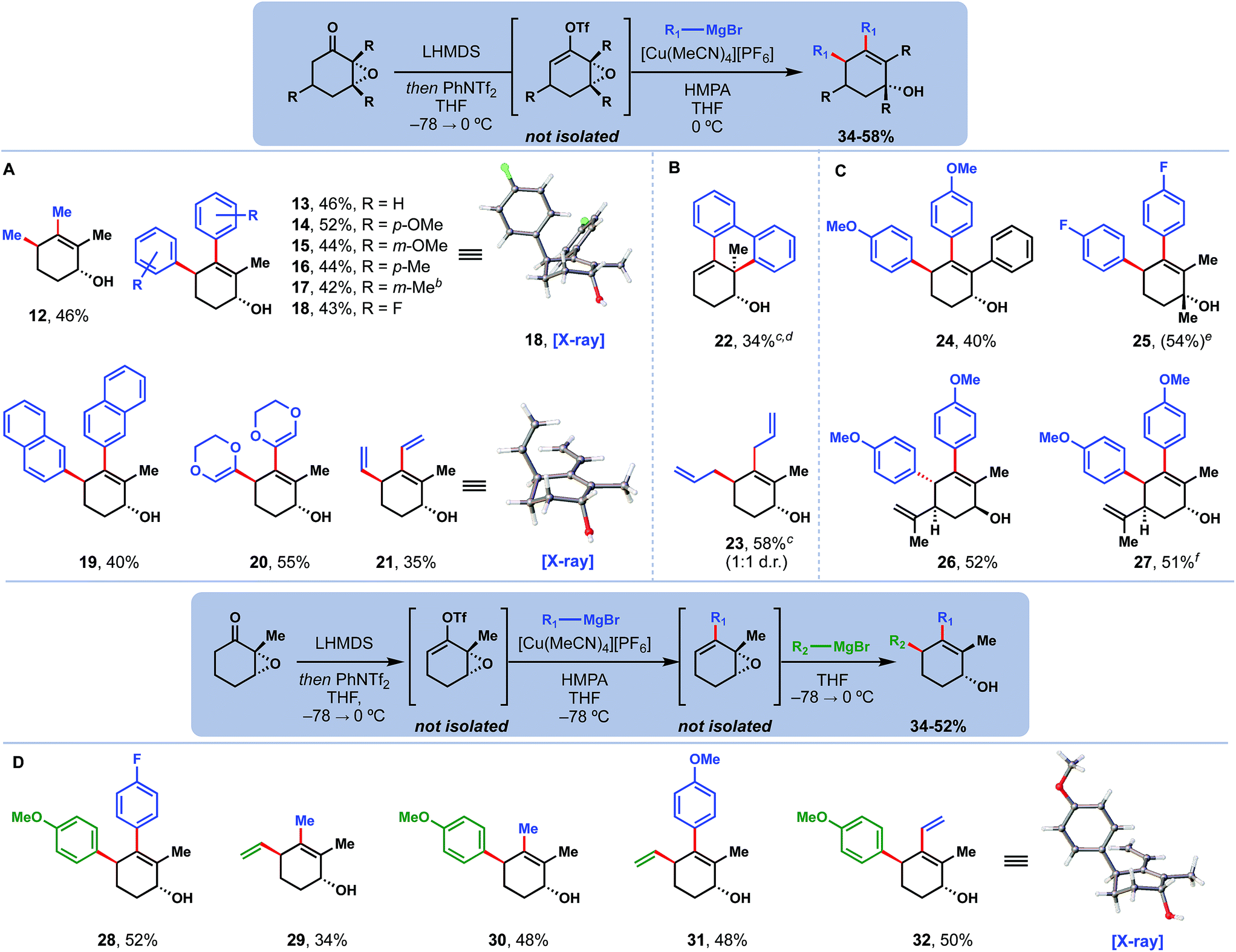 | ||
| Fig. 2 Copper-catalyzed double coupling of epoxy ketones and Grignard reagents: (A) Scope with two of the same Grignard reagents. (B) Anomalous products, see main text for further discussion. (C) Use of different epoxides. (D) Coupling of two different nucleophiles. aReactions were run with 1.0 mmol of epoxide under the optimized conditions. Reported yields are of products isolated as single isomers, except where otherwise indicated. b10:1 d.r. cCuI used in place of [Cu(MeCN)4][PF6] and no HMPA added. d2.0 equiv. of 2,2′-biphenyldimagnesium bromide used as nucleophile. eNMR yield using 1,3,5-trimethoxybenzene as an internal standard. f30 mol% loading of [Cu(MeCN)4][PF6] used. | ||
Different epoxide substrates were also well tolerated with this methodology (Fig. 2C). Replacement of the 2-methyl group with a larger phenyl group did not hinder product formation (see 24). Likewise, addition of a methyl group at the 3-position did not impact the reaction to prepare 25; however, that compound's tertiary allylic alcohol motif was highly acid sensitive and would rapidly dehydrate under even mildly acidic conditions. Products from both diastereomers of carvone epoxide (26, 27) were also obtained cleanly. Notably, this system was able to override the intrinsic steric bias of cis-carvone epoxide to still deliver anti product 27.
Finally, we were then able to expand this methodology to perform couplings with two different nucleophiles in the same pot (Fig. 2D). We determined that at low temperatures (−78 °C), the cross-coupled product could be formed selectively. This intermediate could then be treated with a different nucleophile, which would only perform the allylic substitution step when warmed to 0 °C. Through this process, differentially substituted products (28–32) were assembled in a modular manner. Importantly, nucleophile identity (aryl, vinyl, alkyl, etc.) did not change the selectivity of this transformation; rather, the controlling factors in all cases were order of nucleophile addition and reaction temperature. Thus, it proved straightforward to prepare regioisomeric products 31 and 32 by simply switching which one of the nucleophiles was added first. The connectivity and selectivity of 32 were additionally confirmed by single crystal X-ray diffraction studies.
With this methodology successfully established, we then turned our attention toward application to the quassinoid ring system (Scheme 1). We began by coupling carvone epoxide with the dioxane-based Grignard reagent to afford 33 as a single isomer in 58% yield; the anti stereochemistry of the product was confirmed by X-ray crystallographic analysis. Notably, we have been able to prepare multiple grams of this unique product using this chemistry. At this point, we reasoned that the C- and D-rings of the quassinoids could be forged through an intramolecular Diels–Alder reaction of the vinyl dioxane fragment with a dienophile side chain tethered to allylic alcohol 33.15,16 Many initial attempts to realize this transformation were foiled due to the significant lability of the allylic alcohol under thermal and acidic conditions. Eventually we determined that the depicted methyl chloroalkyl ether could first alkylate the free alcohol under basic conditions (DIPEA), creating a pair of diastereomeric acetals (ca. 1:1 d.r.) that would then undergo the Diels–Alder reaction upon further heating. Interestingly, the acetal stereochemistry exerted a strong controlling effect on the endo/exo selectivity of the cycloaddition reaction. Specifically, one acetal isomer gave exclusively the endo product (34) while the other favored exo product 35 over endo product 36 (ca. 3:1 exo:endo). Overall, this led to isolation of two major diastereomers, 34 and 35 – one endo isomer and one exo isomer (1.3:1 d.r.) – which were used in subsequent chemistry. The stereochemistry of the exo product 35 was confirmed by single crystal X-ray diffraction studies (see ESI†). Although 34 and 35 differed in three stereocenters, they were carried forward as a mixture since we anticipated epimerization or ablation of those centers before arriving at various quassinoid natural products.17
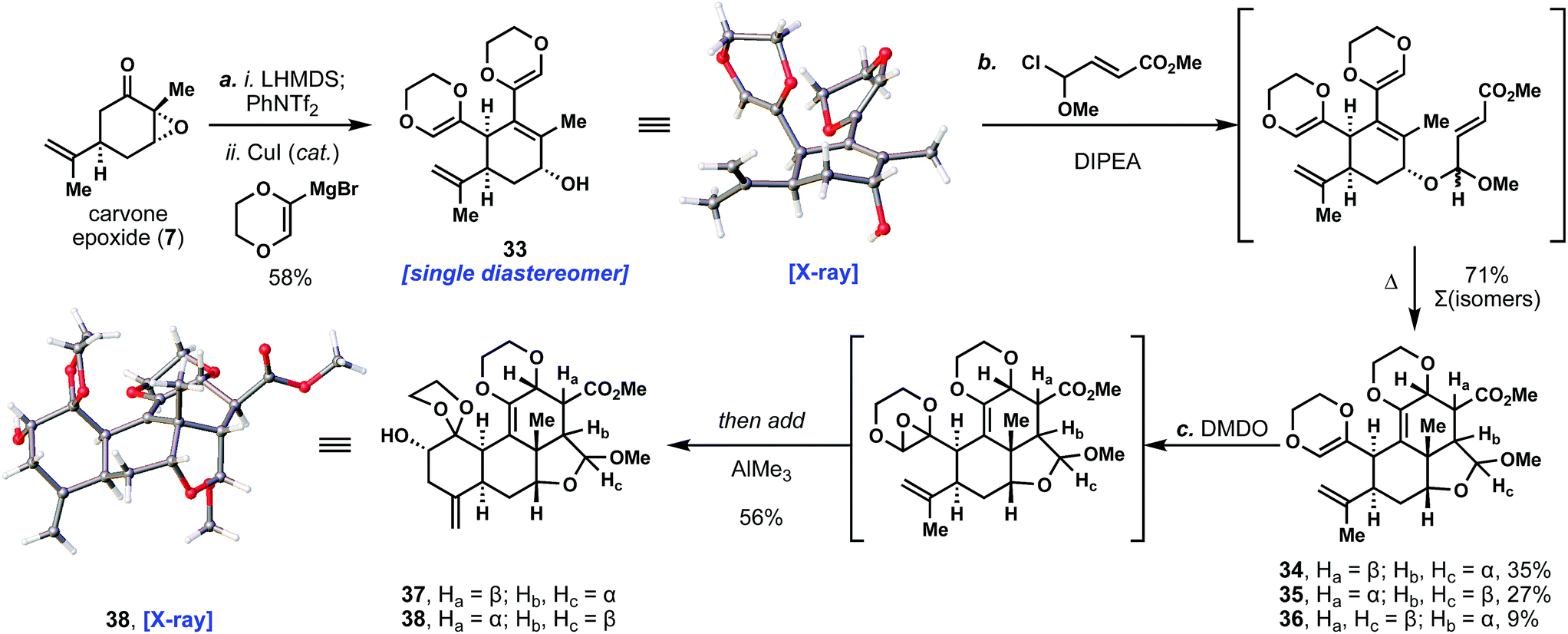 | ||
| Scheme 1 3-Step synthesis of the quassinoid core architecture. Reagents and conditions: (a) LHMDS (1.0 equiv.), PhNTf2 (1.0 equiv.), THF, −78 → 0 °C, 5 min, then add a solution of CuI (0.35 equiv.) and dioxenyl Grignard (3.5 equiv.), THF, 0 °C, 16 h, 58%; (b) DIPEA (4.0 equiv.), dienophile (2.0 equiv.), PhMe, 23 °C, 12 h, then add PhMe, HMDS, 110 °C, 3d, 71% (∑ of isomers); (c) DMDO (1.0 equiv.) then add AlMe3 (1.5 equiv.), DCM/acetone −40 °C → 0 °C, 10 min, 56%. LHMDS = lithium bis(trimethylsilyl)amide, Tf = trifluoromethanesulfonyl, DIPEA = N,N-diisopropylethylamine, HMDS = hexamethyldisilazane, DMDO = dimethyldioxirane. | ||
To complete the quassinoid core architecture, we envisioned a cascade dioxane oxidation/rearrangement/cyclization to form the A-ring. Prior work by Hanna and co-workers demonstrated that epoxidation of substituted dioxenes and rearrangement to an aldehyde occurs readily.18 In our case, we wondered whether an acid could be identified that not only performed this rearrangement, but also promoted an intramolecular ene reaction between the newly formed aldehyde and the neighboring isopropenyl group. In practice, the desired epoxide intermediate (see bracketed intermediate, Scheme 1) was cleanly generated by treatment with DMDO and then subjected in the same pot to an assortment of Lewis acids. We found that trimethylaluminum proved to be an optimal mediator for both transformations, cleanly delivering secondary alcohols 37 and 38 in 56% combined yield. Both ene products were formed with comparable efficiency and as single isomers at the newly formed alcohol stereocenter. The stereochemistry of 38 was further confirmed by single crystal X-ray crystallographic analysis. Significantly, the quassinoid core architecture was thus completed in only three steps from 7. Five carbon–carbon bonds and seven stereocenters were formed in the process, speaking to the power of tandem catalysis combined with synthetic strategy to assemble ornate, highly complex structures with high efficiency.
Conclusions
In summary, we have developed a novel copper-catalyzed difunctionalization reaction that converts readily available epoxy ketones to highly substituted, diastereomerically pure allylic alcohols in one pot. This transformation features a diverse substrate scope, with many classes of nucleophiles and epoxides being tolerated. Additionally, this chemistry can generate modular products by careful control of reaction conditions. The utility of this process was then demonstrated in a rapid synthesis of the quassinoid core architecture, a structure common to many bioactive natural products and one whose syntheses have historically required numerous chemical steps. Future studies are focused on employing this methodology to rapidly access high value intermediates and other complex natural product-like structures as well as synthesizing diverse quassinoid natural products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. J. M. acknowledges unrestricted financial support from Novartis, Bristol-Myers Squibb, Amgen, and Eli Lilly. T. J. M. is a Research Corporation Cottrell Scholar. M. L. C. acknowledges UC-Berkeley and the NSF for a Berkeley Graduate Fellowship and NSF Predoctoral Fellowship (DGE-1106400) respectively. S. J. H. thanks the UC-Berkeley College of Chemistry for a summer undergraduate research award. We are grateful to Dr Hasan Celik for NMR spectroscopic assistance. Dr Nicholas Settineri, Dr Simon Teat, and Mr Peter Waller are acknowledged for X-ray crystallographic analysis wherein support from NIH Shared Instrument Grant (S10-RR027172) is also acknowledged.Notes and references
-
(a) A. M. Armaly, Y. C. DePorre, E. J. Groso, P. S. Riehl and C. S. Schindler, Chem. Rev., 2015, 115, 9232 CrossRef CAS PubMed
; (b) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010 RSC
- Z. Guo, S. Vangapandu, R. W. Sindelar, L. A. Walker and R. D. Sindelar, Front. Med. Chem., 2009, 4, 285 Search PubMed
-
(a) F. L. Winckler, Repert. Pharm., 1835, 4, 85 Search PubMed
- For syntheses of quassin, see:
(a) G. Vidari, S. Ferriño and P. A. Grieco, J. Am. Chem. Soc., 1984, 106, 3539 CrossRef CAS
- For syntheses of bruceantin, see:
(a) M. Sasaki and T. Murae, Tetrahedron Lett., 1989, 30, 355 CrossRef CAS
- For syntheses of miscellaneous quassinoids, see:
(a) P. A. Grieco, R. Lis, S. Ferriño and J. Y. Jaw, J. Org. Chem., 1982, 47, 601 CrossRef CAS
- Z. G. Brill, M. L. Condakes, C. P. Ting and T. J. Maimone, Chem. Rev., 2017, 117, 1753 CrossRef PubMed
- P. A. Wender, J. M. Erhardt and L. Letendre, J. Am. Chem. Soc., 1981, 103, 2114 CrossRef CAS
-
(a) J. E. McMurry and W. J. Scott, Tetrahedron Lett., 1980, 21, 4313 CrossRef CAS
- For a recent review on Cu-catalyzed cross couplings, see: S. Thapa, B. Shrestha, S. K. Gurung and R. Giri, Org. Biomol. Chem., 2015, 13, 4816 RSC
- For recent copper-catalyzed Kumada and Negishi-type cross-couplings of sp2 electrophilies, see:
(a) S. Thapa, A. S. Vangala and R. Giri, Synthesis, 2016, 48, 504 CrossRef CAS
- In the absence of other factors, copper-catalyzed allylic substitutions often proceed with anti stereochemistry and SN2′ regiochemistry; see:
(a) E. J. Corey and N. W. Boaz, Tetrahedron Lett., 1984, 25, 3063 CrossRef CAS
- It is important to note that three reactions each occurring at around 80% yield would give an overall product yield of ca. 50%. Indeed, yield for the triflation step alone is 82% in our hands. Wender's and McMurry's works likewise benchmark the allylic substitution and cross coupling processes respectively at 50–80% yields each. For a discussion on the challenges and opportunities of tandem process, see: D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365 CrossRef CAS
- In light of the high reactivity of allyl-type nucleophiles, this process may involve a non copper-mediated background reaction. Similar challenges are noted in copper-catalyzed conjugate addition reactions employing allylmagnesium halides.
- For the use of vinyl dioxenes as dienes in cycloadditions, see:
(a) M. Fétizon, P. Goulaouic, I. Hanna and T. Prangé, J. Org. Chem., 1998, 53, 5672 CrossRef
- For a recent, impressive use of dioxene in total synthesis, see: A. G. Kravina and E. M. Carreira, Angew. Chem., Int. Ed., 2018, 57, 13159 CrossRef CAS PubMed
- Valenta has demonstrated the remarkable exhaustive deuteration of neoquassin under basic conditions:
 see: Z. Valenta, S. Papadopoulos and C. Podešva, Tetrahedron, 1961, 15, 100 CrossRef
see: Z. Valenta, S. Papadopoulos and C. Podešva, Tetrahedron, 1961, 15, 100 CrossRef - C. Baylon and I. Hanna, Tetrahedron Lett., 1995, 36, 6475 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data, and X-ray crystallographic data of compounds 18, 21, 32, 33, 35, and 38. CCDC 1856948, 1856946, 1856945, 1856947, 1856950, 1856949 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03835j |
| This journal is © The Royal Society of Chemistry 2019 |