
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalyzed allylic defluorinative alkylation of trifluoromethyl alkenes with reductive decarboxylation of redox-active esters†
Xi
Lu‡
 *,
Xiao-Xu
Wang‡
,
Tian-Jun
Gong
,
Jing-Jing
Pi
,
Shi-Jiang
He
and
Yao
Fu
*
*,
Xiao-Xu
Wang‡
,
Tian-Jun
Gong
,
Jing-Jing
Pi
,
Shi-Jiang
He
and
Yao
Fu
*
Hefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Urban Pollutant Conversion, Anhui Province Key Laboratory of Biomass Clean Energy, iChEM, University of Science and Technology of China, Hefei 230026, China. E-mail: luxi@mail.ustc.edu.cn; fuyao@ustc.edu.cn
First published on 7th November 2018
Abstract
Herein, we report a nickel-catalyzed allylic defluorinative alkylation of trifluoromethyl alkenes through reductive decarboxylation of redox-active esters. The present reaction enables the preparation of functionalized gem-difluoroalkenes with the formation of sterically hindered C(sp3)–C(sp3) bonds under very mild reaction conditions, while tolerating many sensitive functional groups and requiring minimal substrate protection. Therefore, this method provides an efficient and convenient approach for late-stage modification of biologically interesting molecules.
Introduction
Efficient strategies for introducing fluorine-containing fragments into organic compounds exert positive influences on biochemical sciences, because these fluorochemicals have superior bioactivity and physicochemical characteristics compared to their non-fluorinated counterparts.1 Among them, gem-difluoroalkenes are a class of structurally superior fluorine-containing compounds, and they have attracted substantial interest in agrochemistry and medicinal chemistry. For instance, the gem-difluoroethylene moiety is widely used as an ideal carbonyl group bioisostere in drug design.2 In addition, the gem-difluoroethylene moiety can be easily transformed into other fluorine-containing structures such as monofluoroalkenyl, difluoromethylenyl, and trifluoromethyl groups.3 To date, various strategies have been developed for the preparation of gem-difluoroalkenes, including the conventional ones, such as direct difluoroolefination of carbonyl or diazo groups.4 More recently, defluorinative functionalization of trifluoromethyl alkenes has been applied to the synthesis of gem-difluoroalkenes.5 For example, Hayashi reported a rhodium-catalyzed cross coupling of 1-(trifluoromethyl)alkenes with arylboroxines to access 1,1-difluoroalkenes with C(sp2)–C(sp3) bond construction.6 Molander realized an example of defluorinative alkylation of trifluoromethyl alkenes using radical precursors (potassium organotrifluoroborates, alkylbis(catecholato)silicates and trimethylsilylamines) under photocatalysis conditions.7 Despite the great successes achieved, general methods to obtain gem-difluoroalkenes with readily available reagents under mild conditions are still required.Reductive cross-coupling reactions represent a versatile tool for accurate construction of C–C bonds from cheap, abundant, and stable electrophiles as compared with methods using the corresponding organometallic reagents.8 Recently, reductive decarboxylative coupling has been exploited for generating alkyl radicals from alkanoic acids, complementing the use of alkyl halides beneficially.9 As part of our ongoing interest in alkene functionalization reactions9b,10 and fluorinated olefin synthesis,11 we set out to take advantage of allylic defluorination and reductive decarboxylation for the radical alkylation of trifluoromethyl alkenes (Fig. 1a).7,12–14 Herein, we report nickel-catalyzed defluorinative reductive cross-coupling of trifluoromethyl alkenes with redox-active esters to access functionalized gem-difluoroalkenes (Fig. 1b). This reaction enabled C(sp3)–C(sp3) bond formation through trifluoromethyl C–F bond cleavage and a decarboxylation process under mild reaction conditions. In terms of practicality and usability, this reaction shows a high degree of tolerance to many sensitive functional groups and requires minimal substrate protection. Therefore it can be a useful method for the synthesis of fluorinated compounds.
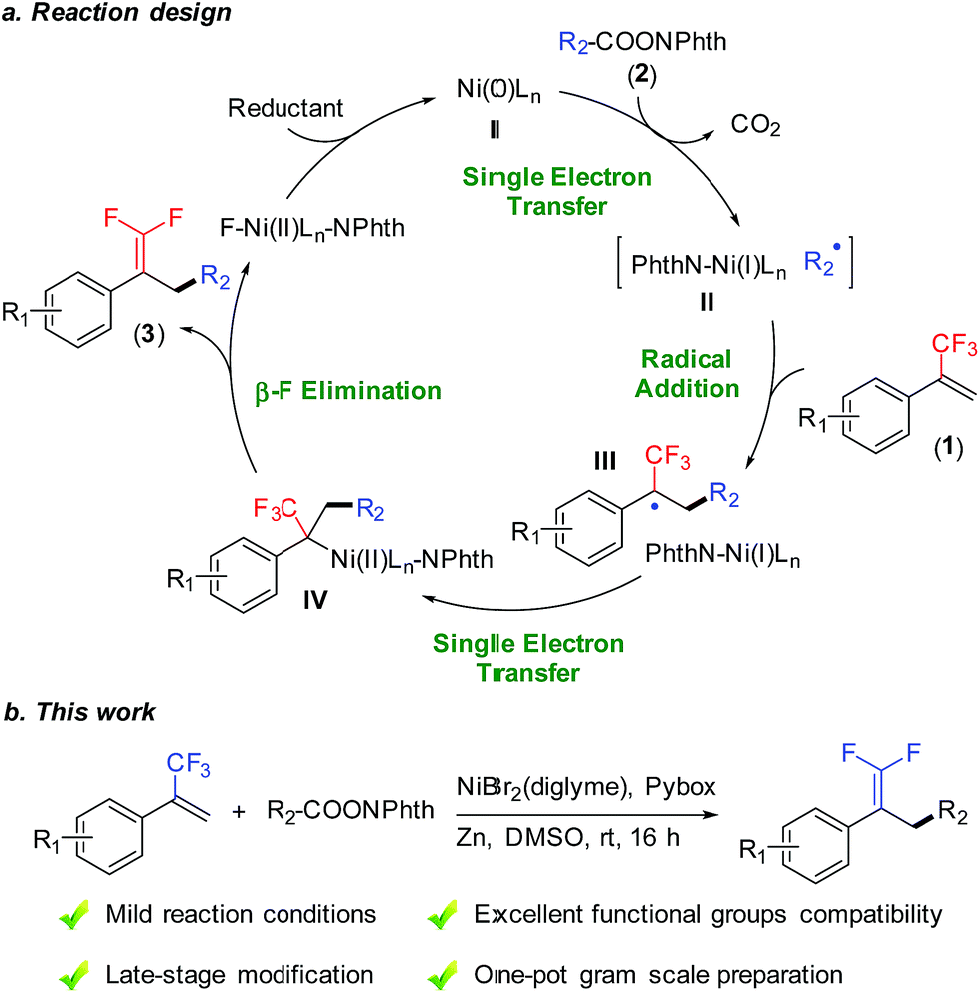 | ||
| Fig. 1 Nickel-catalyzed allylic defluorinative alkylation. NPhth = phthalimide. | ||
Results and discussion
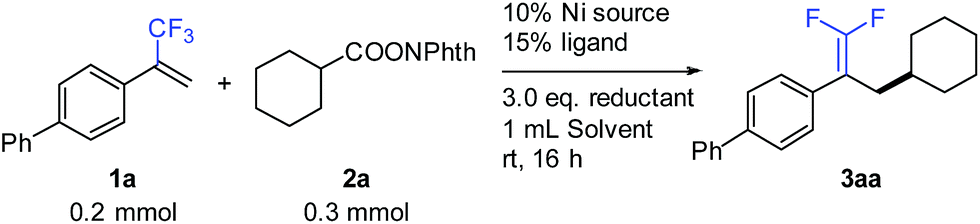
We commenced the study with the synthesis of gem-difluoroalkene 3aa through defluorinative reductive cross-coupling between 1a and 2a. Systematic screening of all the reaction parameters was carried out for optimizing the reaction performance (Table 1). A nickel-bipyridine-reductant system was tested first, affording the desired product in low yields (entries 1 and 2). Use of tridentate N-ligands (entries 3 and 4) led to dramatic improvements: Pybox (L4) increased the yield to 79% (entry 4). The selection of the nickel source was critical to this reaction: compared to NiBr2(diglyme), only NiCl2(Py)4 provided 75% yield, while other nickel catalysts were much less effective (entries 5–10). A number of solvents were also examined: ether solvents (entries 11–13), acetonitrile (entry 14), NMP (entry 15), and DMF (entry 16) were inferior. Fortunately, the use of DMSO (entry 17) provided a nearly quantitative 95% GC yield with 92% isolated yield. Because of the formation of observable by-products (see the ESI† for more details), Zn exhibited better efficiency over other reductants such as Mn, silane, or a diboron reagent (entries 18–20).|
|
|||||
|---|---|---|---|---|---|
| Entry | Nickel source | Ligand | Reductant | Solvent | Yielda/% |
| a GC yield. Triphenylmethane was used as an internal standard. b Isolated yield. rt = room temperature. Diglyme = 2-methoxyethyl ether. acac = Acetylacetone. Py = pyridine. Cy = cyclohexyl. DMAc = N,N-dimethylacetamide. DME = 1,2-dimethoxyethane. THF = tetrahydrofuran. NMP = 1-methyl-2-pyrrolidinone. DMF = N,N-dimethylformamide. DMSO = dimethyl sulfoxide. DEMS = diethoxymethylsilane. (BPin)2 = bis(pinacolato)diboron. | |||||
| 1 | NiBr2(diglyme) | L1 | Zn | DMAc | 23 |
| 2 | NiBr2(diglyme) | L2 | Zn | DMAc | 32 |
| 3 | NiBr2(diglyme) | L3 | Zn | DMAc | 47 |
| 4 | NiBr2(diglyme) | L4 | Zn | DMAc | 79 |
| 5 | NiCl2 | L4 | Zn | DMAc | 33 |
| 6 | Ni(NO3)2 | L4 | Zn | DMAc | <5 |
| 7 | Ni(acac)2 | L4 | Zn | DMAc | 26 |
| 8 | NiCl2(Py)4 | L4 | Zn | DMAc | 75 |
| 9 | NiCl2(PPh3)2 | L4 | Zn | DMAc | 23 |
| 10 | NiCl2(PCy3)2 | L4 | Zn | DMAc | 17 |
| 11 | NiBr2(diglyme) | L4 | Zn | Dioxane | <5 |
| 12 | NiBr2(diglyme) | L4 | Zn | DME | 22 |
| 13 | NiBr2(diglyme) | L4 | Zn | THF | 43 |
| 14 | NiBr2(diglyme) | L4 | Zn | MeCN | <5 |
| 15 | NiBr2(diglyme) | L4 | Zn | NMP | 54 |
| 16 | NiBr2(diglyme) | L4 | Zn | DMF | 60 |
| 17 | NiBr 2 (diglyme) | L4 | Zn | DMSO | 95 (92 ) |
| 18 | NiBr2(diglyme) | L4 | Mn | DMSO | 64 |
| 19 | NiBr2(diglyme) | L4 | DEMS/Na2CO3 | DMSO | 18 |
| 20 | NiBr2(diglyme) | L4 | (BPin)2/K3PO4 | DMSO | 22 |
|
|||||
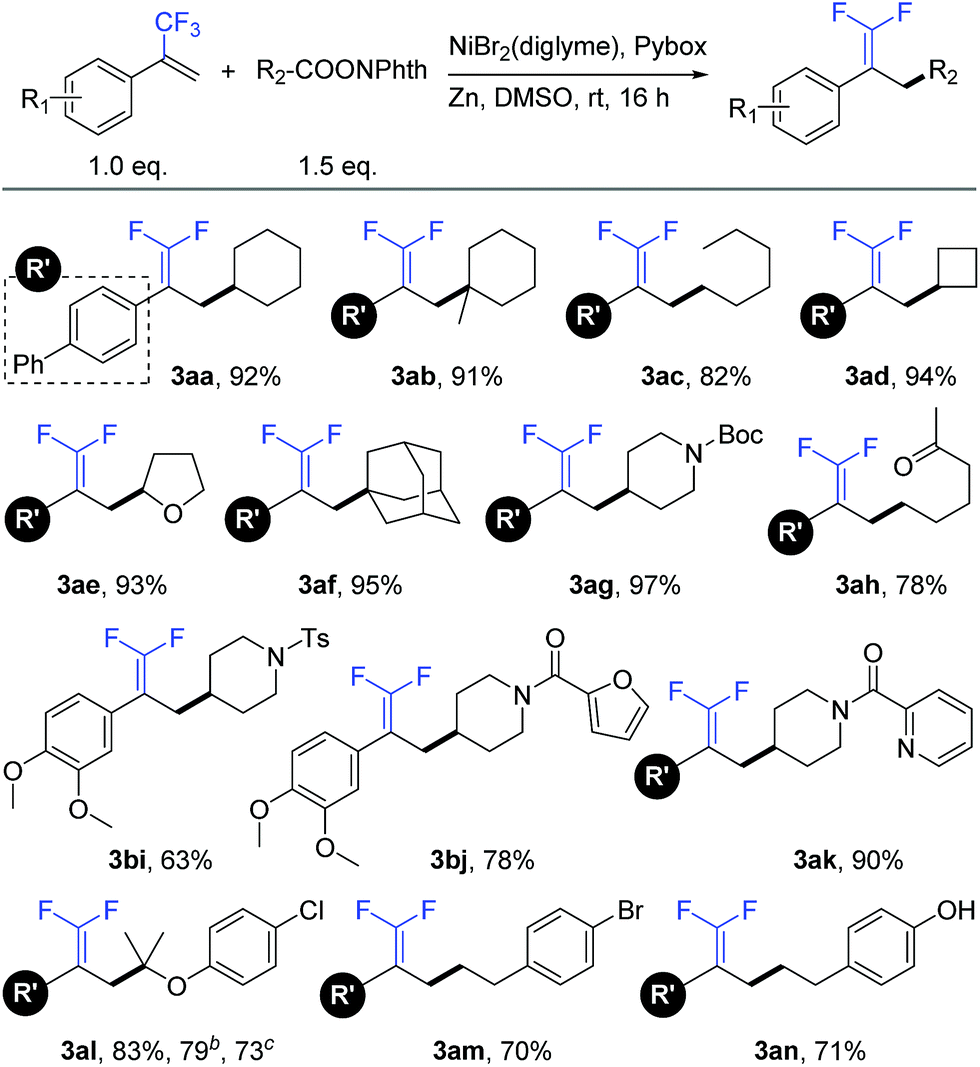
With the optimal reaction conditions in hand, we sought to examine the generality of this defluorinative reductive cross-coupling by exploring a wide range of redox-active esters (Table 2). These substrates were successfully transformed into the corresponding products, obtaining in all cases good to excellent isolated yields (63–97%). This reaction could be applied to secondary (3aa), tertiary (3ab), and primary (3ac) aliphatic redox-active esters. Both cyclic and acyclic esters were good substrates in this transformation. With respect to cyclic esters, modification of the ring size posed no problem. For example, cyclobutyl (3ad), tetrahydrofuran (3ae), and adamantyl (3af) substrates provided the respective products with nearly quantitative yields. Under mild reaction conditions, this reaction exhibited good compatibility with many synthetically useful functional groups such as carbamate (3ag), ketone (3ah), and sulfonamide (3bi). In addition, heterocycles including furan (3bj) and pyridine (3ak) were well tolerated. The compatibility to aryl chloride (3al) and aryl bromide (3am) provided opportunities for convenient transformations at the retained carbon–halogen bonds through the use of other cross-coupling reactions. Moreover, this reaction could even be conducted in the presence of an acidic phenolic hydroxyl group (3an).15 It is worth mentioning that a number of products (e.g., 3ae, 3al, 3ao, 3ap, 3aq and 3ar) were difficult and even chemically infeasible to prepare through the use of a cross-coupling with the corresponding alkyl halides.16 A one-pot synthesis at the gram scale further demonstrated the simplicity and usability of this new method. A representative substrate (3al) was conveniently prepared without any prior preparation and isolation of the corresponding redox-active esters (see the ESI† for more details).
a Isolated yield for 0.2 mmol scale reaction. Reaction conditions are the same as those for Table 1, entry 17.
b Isolated yield for 0.2 mmol scale one-pot reaction.
c Isolated yield for 5.0 mmol scale one-pot reaction. Ratio of desired product/addition by-product >50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 unless otherwise noted. Boc = tert-butoxycarbonyl. Ts = tosyl. :1 unless otherwise noted. Boc = tert-butoxycarbonyl. Ts = tosyl.
|
|---|
|
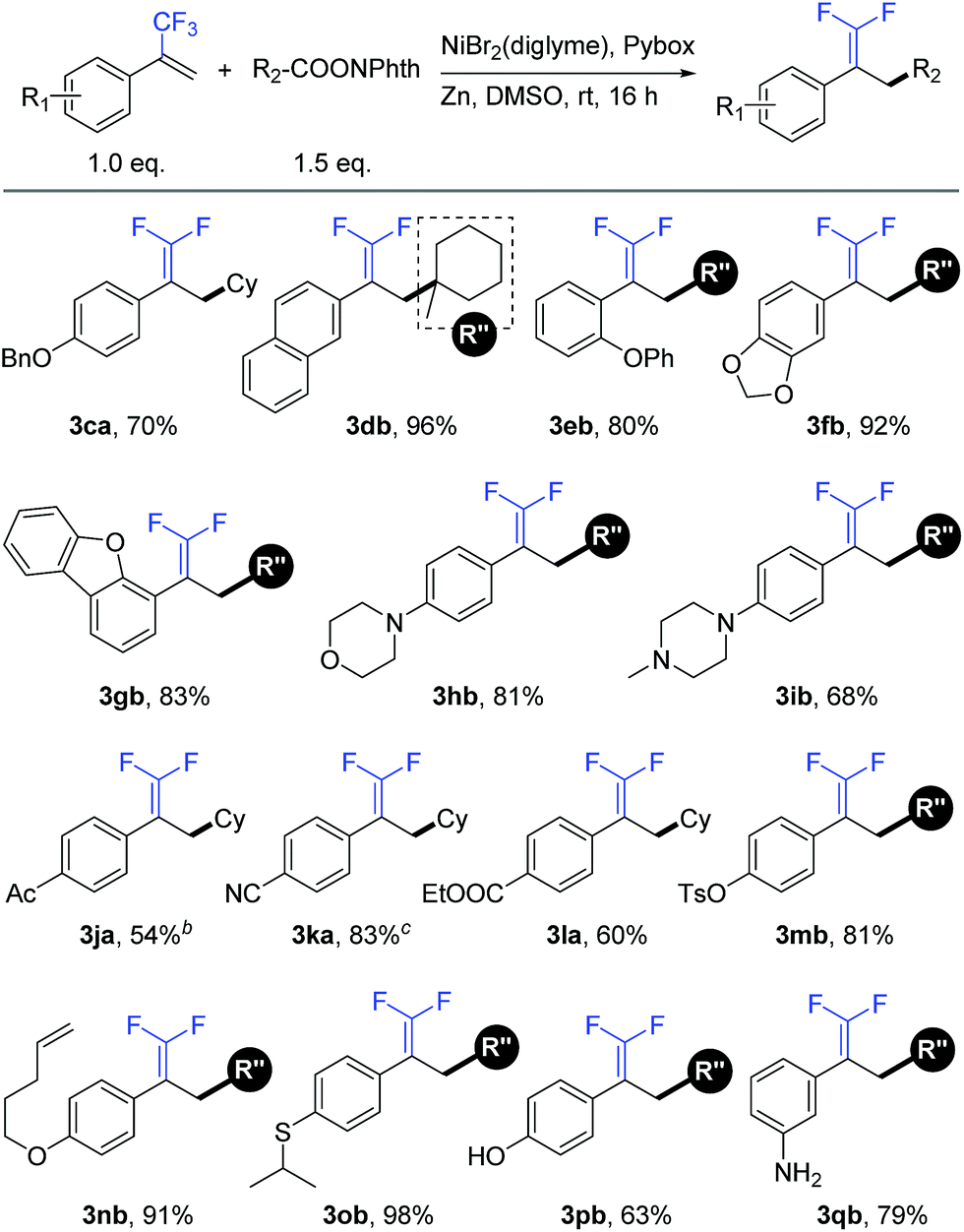
We further examined the applicability of this reaction by evaluating the substrate scope of trifluoromethyl alkenes. As shown in Table 3, trifluoromethyl alkenes with different functional groups could be converted to the desired products successfully. For instance, benzyl ether (3ca), naphthalene (3db), and diphenyl ether (3eb) were well tolerated. Heterocycles such as dioxolane (3fb), dibenzofuran (3gb), morpholine (3hb), and piperazine (3ib) could be used in this transformation. Moreover, some base-sensitive groups such as acetyl (3ja), cyano (3ka), and ethoxycarbonyl (3la) also survived during the defluorinative reductive cross-coupling process. The tolerance of aryl tosylate (3mb) and intramolecular terminal alkene (3nb) afforded further functionalization possibilities. Finally, more active groups that have been difficult substrates in transition-metal-catalyzed cross-coupling reactions, such as sulfoether (3ob), unprotected phenolic hydroxyl (3pb), and primary amine (3qb), were compatible with this defluorinative reductive cross-coupling.17
|
a Isolated yield for 0.2 mmol scale reaction. Reaction conditions are the same as those for Table 1, entry 17. Ratio of desired product/addition by-product >50:1 unless otherwise noted.
b Ratio of desired product/addition by-product = 14:1.
c Ratio of desired product/addition by-product = 35:1. Bn = benzyl. Ac = acetyl.
|
|---|
|
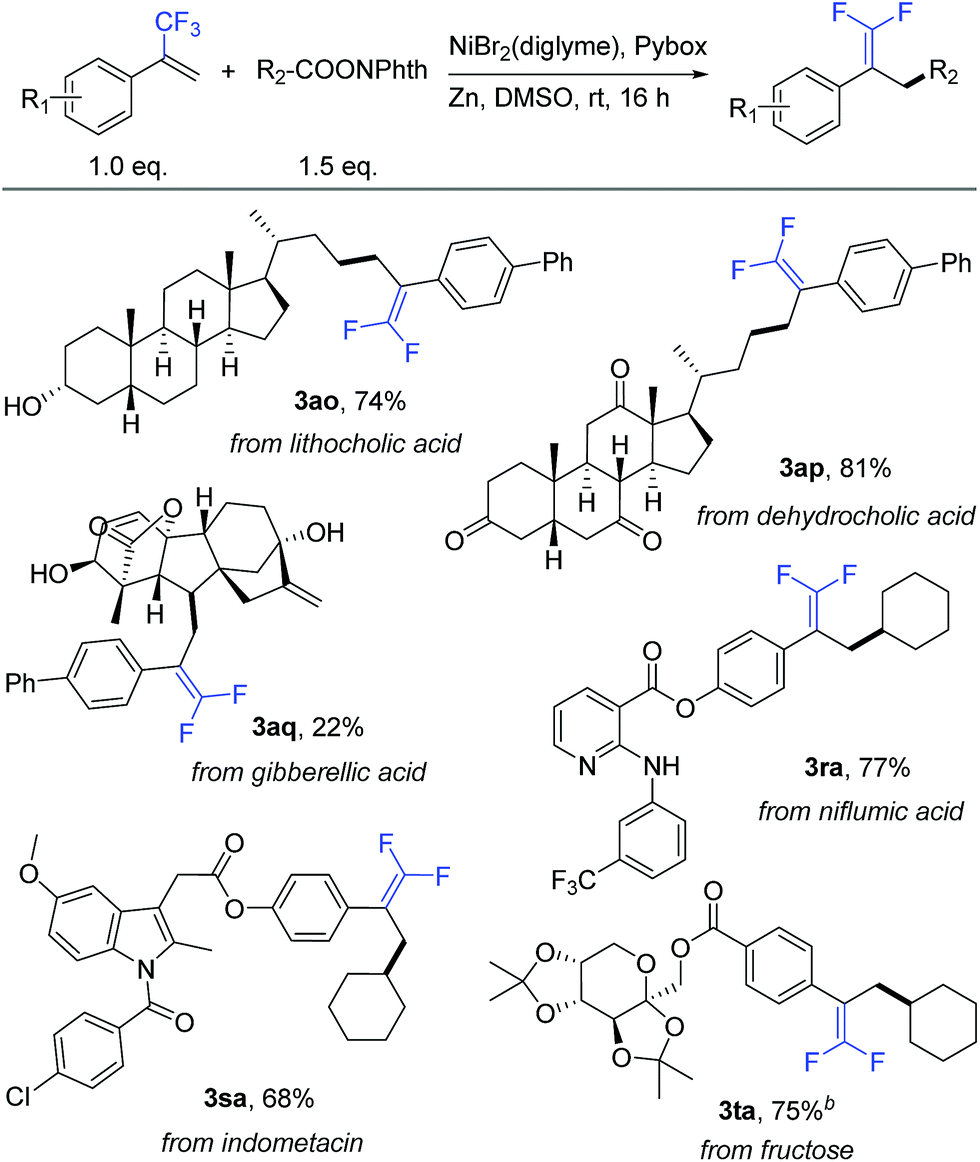
To further demonstrate the high compatibility of this reaction with diverse functional groups, we exploited its application as an easy-to-use tool for the modification of natural products and drug molecules (Table 4). As an illustration, lithocholic acid derivative 2o smoothly reacted with 1a to afford the desired product 3ao with 74% isolated yield. Another example is of dehydrocholic acid ester 2p containing three base-sensitive ketone groups, which performed well during this modification process. In the modification of more complex gibberellic acid ester 2q, the desired product 3aq was obtained in 22% yield despite the presence of an ester group, internal and terminal alkenes, and unprotected secondary and tertiary alcohol groups in the reactant. Modification of a niflumic acid derivative 1r produced the corresponding product 3ra while tolerating the ester group, pyridine ring, and secondary amine. Indometacin derivative 1s could react with 2a to provide product 3sa in 68% yield, without affecting either the indole ring or aryl chloride. Finally, fructose derivative 1t was also a good substrate and afforded product 3ta with a satisfactory 75% isolated yield. Therefore, this defluorinative reductive cross-coupling presents an attractive opportunity for late-stage protecting-group-free modification of biologically interesting molecules.
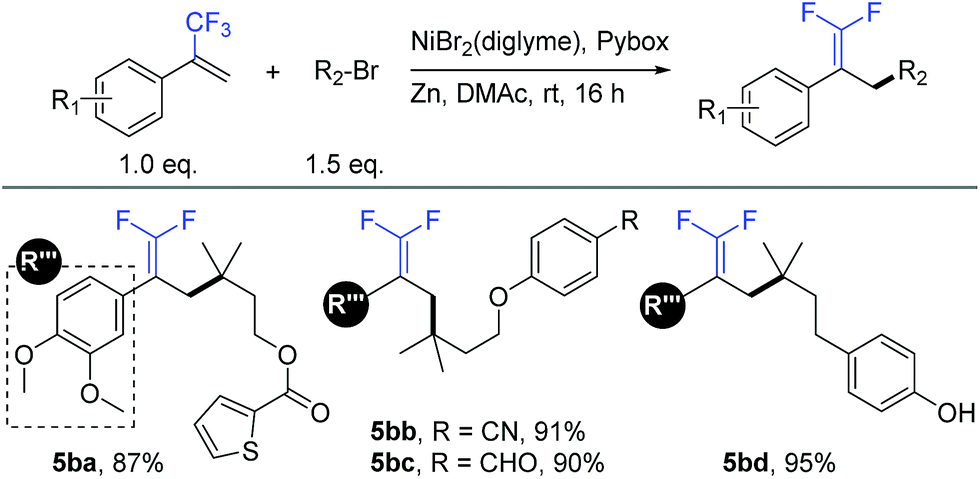
Similar to our previous studies,9b,10a,11a we herein show that this allylic defluorinative alkylation reaction could be applied to alkyl halides (Table 5), which perhaps less surprising is also practical. Several sensitive functional groups were examined, such as thiophene (5ba), cyano (5bb), aldehyde (5bc), and phenolic hydroxyl (5bd), and good to excellent yields were obtained in all cases.
|
a Isolated yield for 0.2 mmol scale reaction. Reaction conditions: trifluoromethyl alkenes (1.0 eq.), alkyl halides (1.5 eq.), NiBr2(diglyme) (10%), Pybox (15%), Zn (3.0 eq.), DMAc (0.2 M), rt, 16 h. Ratio of desired product/addition by-product >50:1 unless otherwise noted.
|
|---|
|
In competition experiments, tertiary alkyl electrophiles exhibited better reactivity than both primary and secondary ones. For instance, we obtained 5be and 5bf as the sole products (Scheme 1, eqn (1)), in which carbon–carbon bonds were formed at the tertiary alkyl bromide sites, while the primary and secondary alkyl sulfonates survived. Interesting results were obtained for the substrates (5ag and 5ah) containing tertiary and primary or secondary alkyl bromides (Scheme 1, eqn (2)). Cyclization products (as the sole product for 5ag and the main product for 5ah) were generated firstly through allylic defluorinative alkylation of the tertiary alkyl bromide and then intramolecular cyclization at the primary or secondary sites.18 Finally, using a trifluoromethyl alkene containing an acrylamide (1u) provided a mixture of mono-alkylation (3uba, defluorinative alkylation) and di-alkylation (3ubb, defluorinative alkylation and Giese addition) products (Scheme 1, eqn (3)).19
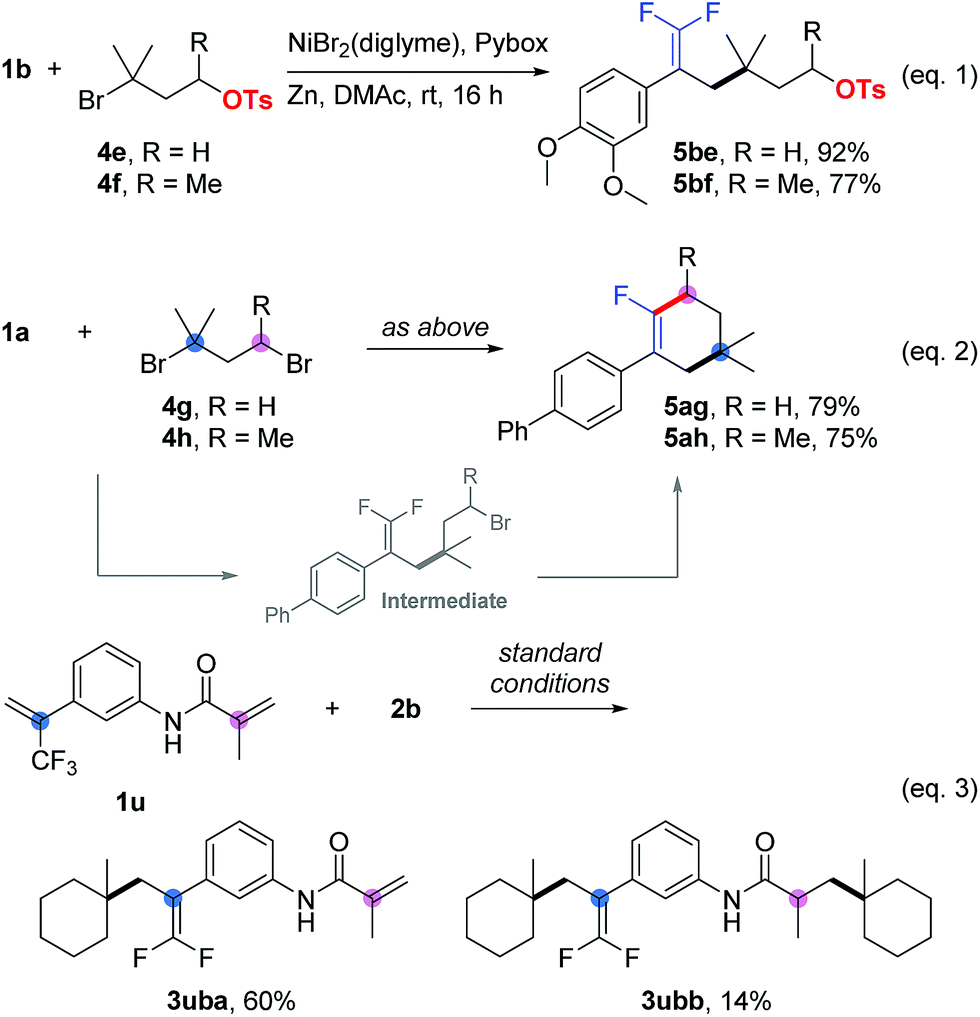 | ||
| Scheme 1 Competition experiments. Isolated yield for 0.2 mmol scale reaction. Reaction conditions for eqn (1) and eqn (2) are the same as those for Table 5. Reaction conditions for eqn (3) are the same as those for Table 1, entry 17. Ratio of desired product/addition by-product >50:1 unless otherwise noted. | ||
To examine the reaction mechanism, the nonmetallic reducing agent TDAE was used to replace Zn(0), which provided an appreciable amount of product and revealed that the activation of redox-active esters might proceed through a single-electron-transfer (SET) process rather than in situ formation of organozinc reagents (Scheme 2, eqn (4)).20 An optically pure redox-active ester (1r) was used to study the stereochemistry, which led to a racemic product (3ar) in 85% isolated yield (Scheme 2, eqn (5)). Collectively, the above results supported a radical-type reaction mechanism for this defluorinative reductive cross-coupling.21
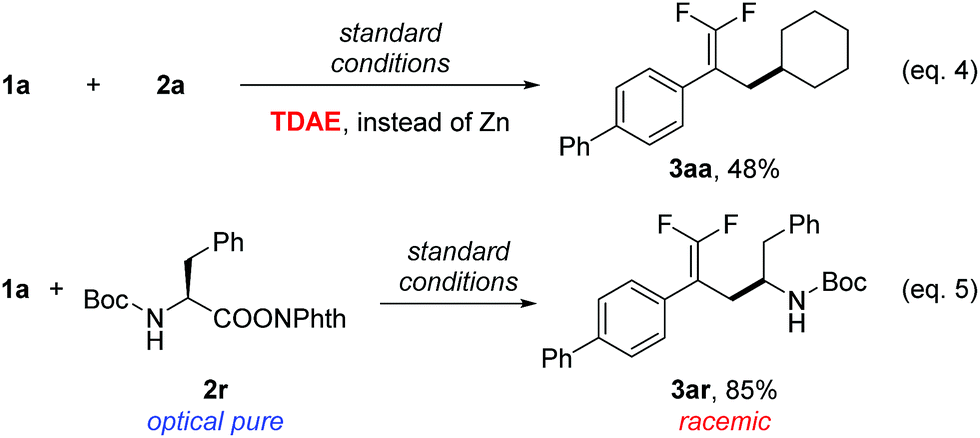 | ||
| Scheme 2 Mechanistic probes. Isolated yield for 0.2 mmol scale reaction. Reaction conditions are the same as those for Table 1, entry 17. Ratio of desired product/addition by-product >50:1 unless otherwise noted. TDAE = 1,1,2,2-tetrakis(dimethylamino)ethylene. | ||
Conclusions
We developed a nickel-catalyzed defluorinative reductive cross-coupling of trifluoromethyl alkenes with redox-active esters. This reaction enables convenient and efficient preparation of gem-difluoroalkenes through C(sp3)–F bond cleavage and C(sp3)–C(sp3) bond formation. Under mild reaction conditions, many sensitive functional groups were tolerated, therefore providing a robust approach for late-stage protecting-group-free modification of natural products or drug molecules. A one-pot synthesis at the gram scale further demonstrated the usability and applicability of this new method. Preliminary mechanistic studies suggested a nickel-catalyzed radical-type process. Our next challenge is the extension of the reaction to an asymmetric version.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We are grateful for the support from the National Natural Science Foundation of China (21572212, 21502184, 21732006, and 21702200), Strategic Priority Research Program of CAS (XDB20000000, XDA21060101), Major Program of Development Foundation of Hefei Center for Physical Science and Technology (2017FXZY001), Ministry of Science and Technology of China (2017YFA0303502).Notes and references
- (a) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, John Wiley & Sons, 2013 CrossRef; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (c) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed; (d) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, John Wiley & Sons, 2009 CrossRef; (e) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (f) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (g) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- (a) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (b) H. Yanai and T. Taguchi, Eur. J. Org. Chem., 2011, 2011, 5939 CrossRef CAS; (c) G. Magueur, B. Crousse, M. Ourévitch, D. Bonnet-Delpon and J.-P. Bégué, J. Fluorine Chem., 2006, 127, 637 CrossRef CAS; (d) C. Leriche, X. He, C.-w. T. Chang and H.-w. Liu, J. Am. Chem. Soc., 2003, 125, 6348 CrossRef CAS PubMed.
- (a) X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375 CrossRef CAS; (b) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS PubMed.
- (a) J. Zheng, J. Cai, J.-H. Lin, Y. Guo and J.-C. Xiao, Chem. Commun., 2013, 49, 7513 RSC; (b) M. Hu, Z. He, B. Gao, L. Li, C. Ni and J. Hu, J. Am. Chem. Soc., 2013, 135, 17302 CrossRef CAS PubMed; (c) Z. Zhang, W. Yu, C. Wu, C. Wang, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2016, 55, 273 CrossRef CAS PubMed; (d) M. Hu, C. Ni, L. Li, Y. Han and J. Hu, J. Am. Chem. Soc., 2015, 137, 14496 CrossRef CAS PubMed; (e) Y. Zhao, W. Huang, L. Zhu and J. Hu, Org. Lett., 2010, 12, 1444 CrossRef CAS PubMed; (f) S. Krishnamoorthy, J. Kothandaraman, J. Saldana and G. K. S. Prakash, Eur. J. Org. Chem., 2016, 2016, 4965 CrossRef; (g) K. Aikawa, W. Toya, Y. Nakamura and K. Mikami, Org. Lett., 2015, 17, 4996 CrossRef CAS PubMed.
- (a) T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., DOI:10.1002/anie.201805292; (b) F. Jaroschik, Chem.–Eur. J., 2018, 24, 14572 CrossRef CAS PubMed; (c) J.-P. Bégué, D. Bonnet-Delpon and M. H. Rock, J. Chem. Soc., Perkin Trans. 1, 1996, 1409 RSC; (d) R. Corberán, N. W. Mszar and A. H. Hoveyda, Angew. Chem., Int. Ed., 2011, 50, 7079 CrossRef PubMed; (e) W. Dai, Y. Lin, Y. Wan and S. Cao, Org. Chem. Front., 2018, 5, 55 RSC; (f) P. Gao, C. Yuan, Y. Zhao and Z. Shi, Chem, 2018, 4, 2201 CrossRef CAS; (g) R. Kojima, S. Akiyama and H. Ito, Angew. Chem., Int. Ed., 2018, 57, 7196 CrossRef CAS PubMed; (h) Y. Liu, Y. Zhou, Y. Zhao and J. Qu, Org. Lett., 2017, 19, 946 CrossRef CAS PubMed; (i) K. Fuchibe, H. Hatta, K. Oh, R. Oki and J. Ichikawa, Angew. Chem., Int. Ed., 2017, 56, 5890 CrossRef CAS PubMed; (j) X. Wu, F. Xie, I. D. Gridnev and W. Zhang, Org. Lett., 2018, 20, 1638 CrossRef CAS PubMed; (k) K. Fuchibe, M. Takahashi and J. Ichikawa, Angew. Chem., Int. Ed., 2012, 51, 12059 CrossRef CAS PubMed; (l) T. Xiao, L. Li and L. Zhou, J. Org. Chem., 2016, 81, 7908 CrossRef CAS PubMed; (m) T. Ichitsuka, T. Fujita and J. Ichikawa, ACS Catal., 2015, 5, 5947 CrossRef CAS; (n) M. Wang, X. Pu, Y. Zhao, P. Wang, Z. Li, C. Zhu and Z. Shi, J. Am. Chem. Soc., 2018, 140, 9061 CrossRef CAS PubMed.
- Y. Huang and T. Hayashi, J. Am. Chem. Soc., 2016, 138, 12340 CrossRef CAS PubMed.
- S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073 CrossRef CAS PubMed.
- (a) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed; (b) X. Wang, Y. Dai and H. Gong, Top. Curr. Chem., 2016, 374, 43 CrossRef PubMed; (c) D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793 CrossRef CAS PubMed; (d) C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. Jacobi von Wangelin, Chem.–Eur. J., 2014, 20, 6828 CrossRef CAS PubMed; (e) E. Richmond and J. Moran, Synthesis, 2018, 50, 499 CrossRef CAS; (f) J.-H. Liu, C.-T. Yang, X.-Y. Lu, Z.-Q. Zhang, L. Xu, M. Cui, X. Lu, B. Xiao, Y. Fu and L. Liu, Chem.–Eur. J., 2014, 20, 15334 CrossRef CAS PubMed; (g) Z. Duan, W. Li and A. Lei, Org. Lett., 2016, 18, 4012 CrossRef CAS PubMed; (h) L. K. G. Ackerman, M. M. Lovell and D. J. Weix, Nature, 2015, 524, 454 CrossRef CAS PubMed; (i) C. Xu, W.-H. Guo, X. He, Y.-L. Guo, X.-Y. Zhang and X. Zhang, Nat. Commun., 2018, 9, 1170 CrossRef PubMed; (j) V. Bacauanu, S. Cardinal, M. Yamauchi, M. Kondo, D. F. Fernández, R. Remy and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 12543 CrossRef CAS PubMed; (k) K. M. Arendt and A. G. Doyle, Angew. Chem., Int. Ed., 2015, 54, 9876 CrossRef CAS PubMed; (l) B. P. Woods, M. Orlandi, C.-Y. Huang, M. S. Sigman and A. G. Doyle, J. Am. Chem. Soc., 2017, 139, 5688 CrossRef CAS PubMed; (m) M. O. Konev, L. E. Hanna and E. R. Jarvo, Angew. Chem., Int. Ed., 2016, 55, 6730 CrossRef CAS PubMed; (n) X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562 CrossRef CAS PubMed; (o) F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84 CrossRef PubMed; (p) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835 CrossRef PubMed; (q) X.-B. Yan, C.-L. Li, W.-J. Jin, P. Guo and X.-Z. Shu, Chem. Sci., 2018, 9, 4529 RSC; (r) J. Sheng, H.-Q. Ni, H.-R. Zhang, K.-F. Zhang, Y.-N. Wang and X.-S. Wang, Angew. Chem., Int. Ed., 2018, 57, 7634 CrossRef CAS PubMed; (s) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084 CrossRef CAS PubMed; (t) F. Chen, K. Chen, Y. Zhang, Y. He, Y.-M. Wang and S. Zhu, J. Am. Chem. Soc., 2017, 139, 13929 CrossRef CAS PubMed; (u) X. Zhao, H.-Y. Tu, L. Guo, S. Zhu, F.-L. Qing and L. Chu, Nat. Commun., 2018, 9, 3488 CrossRef PubMed.
- (a) N. Suzuki, J. L. Hofstra, K. E. Poremba and S. E. Reisman, Org. Lett., 2017, 19, 2150 CrossRef CAS PubMed; (b) X. Lu, B. Xiao, L. Liu and Y. Fu, Chem.–Eur. J., 2016, 22, 11161 CrossRef CAS PubMed; (c) L. Huang, A. M. Olivares and D. J. Weix, Angew. Chem., Int. Ed., 2017, 56, 11901 CrossRef CAS PubMed; (d) S. Ni, A. F. Garrido-Castro, R. R. Merchant, J. N. de Gruyter, D. C. Schmitt, J. J. Mousseau, G. M. Gallego, S. Yang, M. R. Collins, J. X. Qiao, K.-S. Yeung, D. R. Langley, M. A. Poss, P. M. Scola, T. Qin and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 14560 Search PubMed; (e) H. Li, C. P. Breen, H. Seo, T. F. Jamison, Y.-Q. Fang and M. M. Bio, Org. Lett., 2018, 20, 1338 CrossRef CAS PubMed.
- (a) X. Lu, B. Xiao, Z. Zhang, T. Gong, W. Su, J. Yi, Y. Fu and L. Liu, Nat. Commun., 2016, 7, 11129 CrossRef PubMed; (b) L. Li, T. Gong, X. Lu, B. Xiao and Y. Fu, Nat. Commun., 2017, 8, 345 CrossRef PubMed; (c) G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, J. Am. Chem. Soc., 2017, 139, 18307 CrossRef CAS PubMed; (d) W. Su, T.-J. Gong, X. Lu, M.-Y. Xu, C.-G. Yu, Z.-Y. Xu, H.-Z. Yu, B. Xiao and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 12957 CrossRef CAS PubMed.
- (a) X. Lu, Y. Wang, B. Zhang, J.-J. Pi, X.-X. Wang, T.-J. Gong, B. Xiao and Y. Fu, J. Am. Chem. Soc., 2017, 139, 12632 CrossRef CAS PubMed; (b) J. Xu, Y. Fu, D.-F. Luo, Y.-Y. Jiang, B. Xiao, Z.-J. Liu, T.-J. Gong and L. Liu, J. Am. Chem. Soc., 2011, 133, 15300 CrossRef CAS PubMed; (c) J. Xu, E.-A. Ahmed, B. Xiao, Q.-Q. Lu, Y.-L. Wang, C.-G. Yu and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 8231 CrossRef CAS PubMed.
- (a) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS PubMed; (b) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, eaam7355 CrossRef PubMed; (c) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283 CrossRef CAS PubMed; (d) X. G. Liu, C. J. Zhou, E. Lin, X. L. Han, S. S. Zhang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 13096 CrossRef CAS PubMed; (e) W. Zhao, R. P. Wurz, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 12153 CrossRef CAS PubMed; (f) W.-M. Cheng, R. Shang and Y. Fu, ACS Catal., 2017, 7, 907 CrossRef CAS.
- (a) C. M. McMahon and E. J. Alexanian, Angew. Chem., Int. Ed., 2014, 53, 5974 CrossRef CAS PubMed; (b) K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016 CrossRef CAS PubMed; (c) H. Kim and C. Lee, Org. Lett., 2011, 13, 2050 CrossRef CAS PubMed.
- (a) L. Gao, G. Wang, J. Cao, D. Yuan, C. Xu, X. Guo and S. Li, Chem. Commun., 2018, 54, 11534 RSC; (b) T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. E. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2016, 56, 260 CrossRef PubMed; (c) C. R. Jamison and L. E. Overman, Acc. Chem. Res., 2016, 49, 1578 CrossRef CAS PubMed; (d) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401 CrossRef CAS; (e) L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886 CrossRef CAS PubMed; (f) C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270 CrossRef CAS PubMed; (g) L. Yu, M.-L. Tang, C.-M. Si, Z. Meng, Y. Liang, J. Han and X. Sun, Org. Lett., 2018, 20, 4579 CrossRef CAS PubMed.
- (a) F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS; (b) Y. Fu, L. Liu, R.-Q. Li, R. Liu and Q.-X. Guo, J. Am. Chem. Soc., 2004, 126, 814 CrossRef CAS PubMed.
- Y. Lan, F. Yang and C. Wang, ACS Catal., 2018, 8, 9245 CrossRef CAS.
- I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193 CrossRef CAS PubMed.
- (a) H. Chen, T. Xiao, L. Li, D. Anand, Y. He and L. Zhou, Adv. Synth. Catal., 2017, 359, 3642 CrossRef CAS; (b) L. Li, T. Xiao, H. Chen and L. Zhou, Chem.–Eur. J., 2017, 23, 2249 CrossRef CAS PubMed.
- The reduction potentials of α-trifluoromethyl benzylic radicals are unknown; however, they lie below the known potential of their corresponding alkyl congeners, indicating that α-trifluoromethyl benzylic radicals might be reduced by either a Ni catalyst or Zn metal. (a) D. D. M. Wayner, D. J. McPhee and D. Griller, J. Am. Chem. Soc., 1988, 110, 132 CrossRef CAS; (b) X.-M. Zhang, J. Org. Chem., 1998, 63, 3590 CrossRef CAS . We thank a referee for suggesting an alternative mechanism involved in the reduction of benzylic radical (III) by Zn.
- D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920 CrossRef CAS PubMed.
- J. Wang, T. Qin, T.-G. Chen, L. Wimmer, J. T. Edwards, J. Cornella, B. Vokits, S. A. Shaw and P. S. Baran, Angew. Chem., Int. Ed., 2016, 55, 9676 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental protocols and spectral data. See DOI: 10.1039/c8sc04335c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |