
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA synthetic approach to ‘click’ neoglycoprotein analogues of EPO employing one-pot native chemical ligation and CuAAC chemistry†
D. J.
Lee
a,
A. J.
Cameron
 abc,
T. H.
Wright
b,
P. W. R.
Harris
abc and
M. A.
Brimble
*abc
abc,
T. H.
Wright
b,
P. W. R.
Harris
abc and
M. A.
Brimble
*abc
aSchool of Chemical Sciences, The University of Auckland, 23 Symonds St, Auckland 1142, New Zealand. E-mail: m.brimble@auckland.ac.nz; Fax: +64 9 3737422; Tel: +64 9 3737599
bSchool of Biological Sciences, The University of Auckland, 3 Symonds St, Auckland 1142, New Zealand
cMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
First published on 29th October 2018
Abstract
The clinical significance of batch-wise variability on the pharmacokinetics and potency of commercial erythropoietin (EPO), prepared recombinantly as a heterogeneous mixture of glycoforms, necessitates the development of synthetic strategies to afford homogenous EPO formulations. Herein we present a previously unexplored and divergent route towards ‘click’ neoglycoprotein analogues of EPO, employing one-pot native chemical ligation (NCL) of alkynylated peptides and copper-catalysed azide–alkyne cycloaddition (CuAAC) with azido monosaccharides. By design, our synthetic platform permits glycosylation at virtually any stage, providing flexibility for the synthesis of various glycoforms for biological analysis. Insights obtained from attempted folding of our ‘click’ neoglycoprotein EPO analogue, bearing four different neutral sugar moieties, highlight the important role played by the charged oligosaccharides present in native EPO glycoproteins.
Introduction
Erythropoietin (EPO) is the principal hormone involved in the regulation of mammalian red blood cell differentiation.1 EPO is used clinically to induce the production of red blood cells and alleviate anaemia in conditions such as chronic kidney disease2 and cancer.3 The des-Arg166 form of EPO (1), 165 amino acids in length, is the form of the protein found to be circulating in human plasma.4 In nature the EPO protein is modified extensively following translation by the covalent attachment of complex oligosaccharides at conserved residues in the polypeptide sequence.5 EPO has three N-linked complex oligosaccharides attached to asparagine side chains at positions 24, 38 and 83, and a short O-linked oligosaccharide at serine 1265. The precise identity of the oligosaccharides attached at these sites is highly variable.6 Two disulfide bonds (Cys7–Cys161 and Cys29–Cys33) stabilise the folded, helical structure.7,8 Notably, the folded protein with two disulfide bonds is active at the EPO receptor in vitro in the absence of glycosylation.9–11 Glycosylation is, however, important for in vivo activity. Glycosylation extends the circulation time of EPO in the blood by improving both the physiochemical properties and biological stability;12 the N-linked terminally sialylated groups, in particular, reduce the rate of metabolic clearance in hepatic cells by preventing binding to galactose-binding receptors.13The current commercial forms of EPO are all produced using recombinant DNA technology that results in production of a heterogeneous mixture of glycoforms.14 Such heterogeneity can result in differing pharmacokinetic properties and potency on a batch-wise basis and thus hampers therapeutic optimization. The production of homogeneous EPO glycoforms is therefore an important goal of the synthetic community that has rendered EPO a testing ground for new chemical and enzymatic methodologies. Impressive methodological advances from Danishefsky's EPO program, notably, use of metal-free desulfurization for Cys → Ala conversion15 and convergent aspartylation,16 a method for joining peptides and oligosaccharides via the late-stage amidation of unprotected aspartate residues, culminated in the 2013 synthesis of the first homogeneous ‘consensus’ glycoform of EPO.17 This purely synthetic glycoform contained N-glycans of realistic biological complexity, including the key terminal sialic acid moieties, and was fully active in stimulating red blood cell production in vivo.
Although undoubtedly a masterpiece in total synthesis, more facile synthetic methods are required for commercial application of homogeneous EPO variants. In 2003, Kochendoerfer and co-workers18 reported the synthesis of a polymer-modified EPO, containing the entire 166 amino acid EPO polypeptide chain and two hydrophilic, charged polymer groups attached at two out of the four EPO glycosylation sites. Notably, the synthetic construct was fully active and exhibited significantly improved duration of action in vivo relative to recombinant EPO. These results demonstrated that the complex glycan moieties on EPO are not strictly necessary for optimal activity in vivo and can be replaced by more synthetically accessible non-carbohydrate groups. The Kajihara group19 has reported an alternative, hybrid approach involving glycopeptide synthesis and ligation with a recombinant EPO-thioester via expressed protein ligation (EPL) methodology. A homogeneous EPO analogue with two biantennary sialyloligosaccharide glycans (at residues 24 and 30) was produced and characterised. Although active at the EPO receptor in vitro, the in vivo activity of the assembled analogue was disappointing. In 2016 the Kajihara group20 reported synthesis of a series of five EPO analogues (Gln78 to Ala) with in vivo activity, bearing varying patterns of sialyloligosaccharides at the three key sites but devoid of the short O-linked oligosaccharide at serine126. These syntheses incorporated the key sialyloligosaccharides into SPPS as a Boc-Asn-(sialyloligosaccharide diphenacyl ester)-OH prepared from egg yolk on gram scale. Despite offering an attractive synthetic route from a scalability perspective, the inflexibility in glycan installation highlights the necessity for more facile/rapid methods to systematically screen potential glycan modifications for in vivo activity.
As an alternate strategy towards the expedient synthesis of EPO analogues, avoiding extensive preparation of glycosylated amino acid build blocks requiring subsequent hydroxyl protection/deprotection, we sought to leverage our experience in the preparation of neoglycopeptides21–27 by the powerful combination of native chemical ligation and ‘click’ chemistry (Fig. 1a). It was envisaged that incorporation of propargylated amino acids within the EPO polypeptide sequence (Fig. 1c) would allow site-specific glycosylation of different carbohydrate moieties via copper-catalysed azide–alkyne cycloaddition (CuAAC) reactions using saccharides bearing an azide group. The CuAAC reaction has been extensively applied to the preparation of glycoconjugates,28–31 as it allows rapid and high-yielding ligations of unprotected peptides and carbohydrates in aqueous media but is also compatible with a wide range of solvents and protecting groups. Notably, there are no reports to date of EPO polypeptides modified by the CuAAC reaction. As the 1,2,3-triazole ring is known to be resistant to enzymatic hydrolysis,32,33 conjugation of the peptide and carbohydrate moieties by CuAAC would render the neoglycosylated EPO less susceptible to enzymatic degradation, potentially prolonging biological activity in vivo. Furthermore, the ability to readily ‘click’ different oligosaccharides at different positions on the peptide backbone in a divergent manner should enable the synthesis of homogeneous neoglycosylated EPO analogues to provide further insight into the role of glycans on the bioactivity of EPO. Herein, we report our development of a versatile chemical toolkit for the divergent chemical synthesis of ‘click’ neoglycoprotein analogues of EPO.
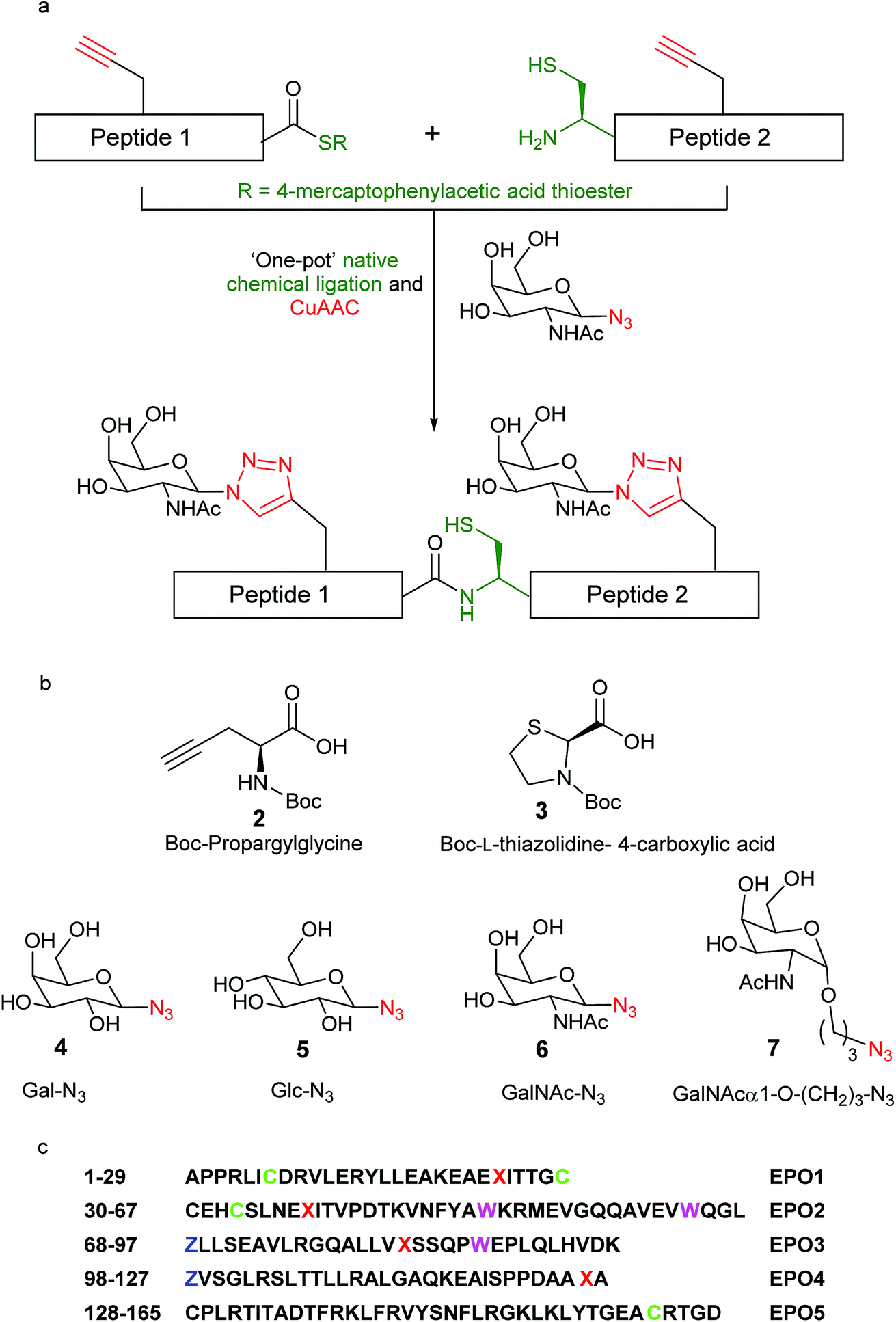 | ||
| Fig. 1 (a) One-pot synthesis of ‘click’ neoglycopeptides employing CuAAC ‘click’ chemistry and native chemical ligation (NCL); (b) specific amino acid (2 and 3) and azido-monosaccharide (4–7) building blocks employed in this work; (c) polypeptide sequence of des-Arg166 human erythropoietin (1), divided into 5 peptide fragments for convergent synthesis and highlighting the location of relevant residues: X = click neoglycosylation sites = propargylglycine = Pra; C= Cys(Acm), Acm = acetamidomethyl; Z = Thz = L-thiazolidine-4-carboxo; W = Trp(CHO). | ||
Results and discussion
General synthetic approach
We set out to synthesise ‘click’ neoglyco-variants of the biologically relevant des-Arg166 form of EPO. The initially formulated synthetic strategy towards our target neoglyco-EPO construct is shown in Scheme 1. The planned synthesis involved dividing the 165-amino acid polypeptide into five fragments, following an efficient convergent total synthesis of [lysine24,38,83] human EPO devised in the Kent laboratory.11 The two key fragments, ‘click’ EPO1-2 and ‘click’ EPO3-5, were envisioned to be combined in a final native chemical ligation (NCL) step to give the target neoglyco-EPO analogue 1, followed by protecting group removal and protein folding. The final fragments ‘click’ EPO1-2 (2) and ‘click’ EPO3-5 (3) are synthesised from a combination of mutually compatible CuAAC, NCL and kinetically controlled ligation (KCL) reactions. The use of KCL for assembly of the N-terminal half of the EPO polypeptide, from the union of Ala1–Cys(Acm)29-aryl thioester and Cys30–Leu67-alkyl thioester precursor segments, enables the convergent synthesis.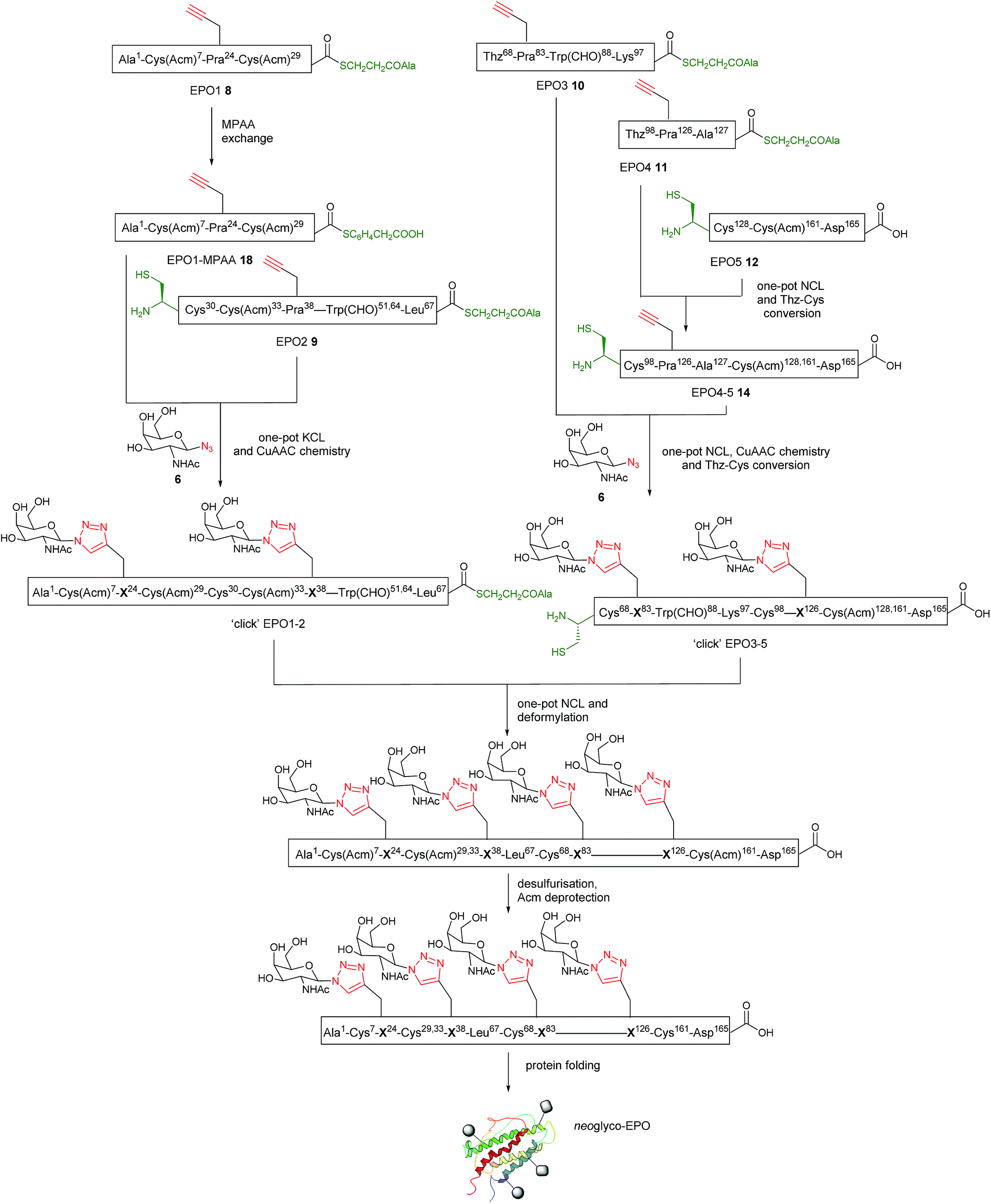 | ||
| Scheme 1 Initial synthetic strategy towards a ‘click’ neoglyco-EPO analogue. For graphical simplicity, GalNAc-N3 (6) is shown as the sugar component; other glycans can be substituted in a straightforward manner. | ||
Four propargylglycine (Pra, 2, Fig. 1b) residues are incorporated at the sites of glycosylation in native EPO (residues 24, 38, 83, and 126, Fig. 1c), allowing CuAAC reactions for glyco-conjugation to be performed at virtually any stage of the synthesis. Scheme 1 only shows one such possibility wherein two CuAAC reactions are performed at the final large fragment stage to install four glycans in total. Based on our previous results22 we expected the CuAAC reactions to be amenable to combination with NCL reactions in a one-pot fashion, thus avoiding low-yielding RP-HPLC separations in between synthetic steps. Given our ultimate goal was to develop a synthetic platform, rather than a biologically active EPO glycomimetic, we chose not to incorporate the key sialic acid moieties, instead employing four sugar azides from our previous work.21–24 Therefore, Gal-N3 (4), Glc-N3 (5), GalNAc-N3 (6) and its hydrocarbon-extended analogue GalNAcα1-O-(CH2)3N3 (7), were chosen as proof-of-principle coupling partners for our divergent synthetic strategy (Fig. 1). One key goal of our initial investigations, reported here, was to demonstrate the divergent installation of different sugars en route to a final EPO sequence. The successful synthesis of a neoglyco-EPO polypeptide using these representative sugars should then allow us to ultimately employ this strategy for the synthesis of neoglyco-EPO libraries bearing more complex oligosaccharides with potential to exhibit potent and sustained biological activity.
EPO1–EPO5 fragment preparation
The full syntheses of the requisite starting peptides 8–12 are reported in the ESI.† Briefly, EPO1 (8), EPO2 (9), EPO3 (10) and EPO4 (11) segments were synthesised using the in situ neutralization protocol for Boc (tert-butoxycarbonyl) stepwise SPPS on Boc-L-Ala-PAM resin. Boc-L-Pra-OH was incorporated into positions 24, 38, 83 and 126, where glycosylation is normally observed in native EPO. Synthesis of EPO5 (12) employed automated Fmoc SPPS methods on Fmoc-L-Asp(OtBu)-HMPP (HMPP = hydroxymethylphenoxypropionic acid) functionalised resin using a CEM Liberty microwave peptide synthesiser. The requisite N-terminal Cys residues of EPO3 (10) and EPO4 (11) peptides were side chain protected by their introduction as Boc-L-thiazolidine-4-carboxylic acid (Boc-Thz, 3, Fig. 1b), thereby allowing the free thiol to be unmasked by facile Thz to Cys conversion for sequential ligation steps.22,34 An Fmoc SPPS protocol was used for the synthesis of EPO5 (10) as it does not contain the base-sensitive thioester group. Appropriate characterisation by MS and HPLC is provided in Fig. S1–S5† for the five peptide segments.Initial synthetic approach to ‘click’ EPO3-5 (15)
With the five fragments in hand, the first native chemical ligation (NCL) was performed between EPO4 (11) and EPO5 (12) segments. Both peptides (3 mM) were dissolved in 6 M guanidinium chloride (GnHCl)/0.2 M Na2HPO4 (ligation buffer) buffered solution containing 4-mercaptophenylacetic acid (MPAA, 200 mM) and tris(2-carboxyethyl)phosphine hydrochloride TCEP·HCl, 50 mM) at pH 6.8. The reaction proceeded nearly quantitatively in 2 h to afford Thz98-EPO4-5 ligated peptide 13 (Fig. S6†). Methoxylamine hydrochloride (150 mM) was then added and the pH adjusted to 4.0 to effect conversion of Thz98 to Cys98. After 3 h, the Cys98-EPO4-5 ligated peptide EPO4-5 (14) was purified by RP-HPLC in a yield of 40% (Fig. S6†).The one-pot NCL and CuAAC sequence was first investigated using EPO3 (10) and EPO4-5 (14), with GalNac-α1-O-(CH2)3N3 (7) as the azido sugar component (Fig. 2a). For NCL, thiol–thioester exchange with 200 mM MPAA is commonplace.35–37 However, in order to minimise inhibition of the CuAAC reaction, MPAA addition is kept to a minimum (20 mM) to afford compatibility for our one-pot system.22 Briefly, NCL between EPO3 (10) and EPO4-5 (14) was carried out in ligation buffer (6 M GnHCl/0.2 M Na2HPO4 at pH 6.8) containing MPAA (20 mM) and TCEP·HCl (40 mM) and monitored by LC-MS. The ligated Thz68-EPO3-5 peptide was obtained after 7 h, then CuSO4 (20 mM), TCEP (10 mM), and GalNac-α1-O-(CH2)3N3 (7) (9 mM) were added to the reaction mixture. To effect glycosylation at both sites, the reaction mixture required heating to 50 °C; after 5 h, the ‘double-clicked’ Cys68-EPO3-5 glycopeptide product (‘click’ EPO3-5) 15 was obtained. It is noteworthy that during this reaction, Thz68 was quantitatively converted to Cys68, supporting the generality of our previous findings.22 The ‘click’ EPO3-5 glycopeptide 15 was purified by RP-HPLC, resulting in a 9% yield. The HPLC and MS data for the reaction is shown in Fig. 2b–f.
 | ||
| Fig. 2 (a) Initial synthetic approach towards ‘click’ EPO3-5 neoglycopeptide 15, employing one-pot NCL and CuAAC chemistry. Reagents and conditions: NCL: MPAA (20 mM), TCEP·HCl (40 mM), 6 M GnHCl/0.2 M Na2HPO4, pH 6.8, 7 h; CuAAC: CuSO4 (20 mM), TCEP·HCl (10 mM), GalNAcα1-O-(CH2)3N37 in the same pot, 50 °C; (b–f) analytical LC-MS traces for the one-pot NCL and click reaction between EPO4-5 (14), EPO3 (10) peptides and GalNAcα1-O-(CH2)3N37; (b) t = 1 h; (c) t = 7 h. The ligated Thz-EPO3-5 ligation peptide was observed in near quantitative yield; (d) t= 5 h after start of click reaction. The bisglycosylated ‘click’-EPO3-5 neoglycopeptide product (15) was obtained in high purity; (e) purified ‘click’-EPO3-5 (15); (f) mass spectrum of ‘click’-EPO3-5 (15) (observed: [M + 6H]6+ = 1903.22 Da, calculated: 1903.52 Da). Analytical monitoring was carried out using a Phenomenex Gemini C18 column (110 Å, 50 mm × 2.0 mm; 5 μm) using a gradient of 5–65% buffer B over 30 min (buffer A= 0.1% TFA in H2O; buffer B= 0.1% TFA in acetonitrile) at 210 nm. | ||
Alternative synthetic approach to ‘click’ EPO3-5 (16)
One powerful advantage conferred by the combination of NCL and CuAAC chemistry presented here is the ability to perform the reaction sequence in any order, enabling the CuAAC reactions for glyco-conjugation to be performed at virtually any stage of the synthesis and raising the possibility of employing different sugar azides in consecutive steps, thus expanding the diversity of neoglycopeptide structures accessible in a divergent manner. To explore this possibility, we examined an alternative route to the ‘click’ EPO3-5 neoglycopeptide fragment involving two consecutive NCL-CuAAC steps with two different sugar azides used as the coupling partners in the CuAAC reactions (Fig. 3a). Rather than employing one-pot NCL and CuAAC reactions, the revised route towards the EPO3-5 fragment made use of separate NCL reactions followed by one-pot CuAAC and Thz to Cys conversion. This allowed for a greater a concentration of MPAA (100 mM) to be used during the previously sluggish ligation of EPO3 and EPO4-5 (7 h, Fig. 2), providing enhanced reaction kinetics (2 h, see later, Fig. 3c). | ||
| Fig. 3 (a) Alternative synthetic approach towards ‘click’ EPO3-5 neoglycopeptide 16, bearing two different ‘click’ neoglycans. Reagents and conditions: NCL between EPO4 (11) and EPO5 (12): 6 M GnHCl/0.2 M Na2HPO4, MPAA (200 mM), TCEP·HCl (50 mM) pH 6.8, 2.5 h; SPE using C4 semi-prep column and lyophilization, then CuAAC with sugar azide 7: 6 M GnHCl/0.2 M Na2HPO4, GalNAcα1-O-(CH2)3–N3 (7) (5 mM), CuSO4 (40 mM) and TCEP·HCl (20 mM), 40 °C, pH 7, 2 h. NCL between ‘click’ EPO4-5 (17) and EPO3 (10): 6 M GnHCl/0.2 M Na2HPO4, MPAA (100 mM), TCEP·HCl (40 mM), pH 6.8, 2 h; SPE using C4 semi-prep column and lyophilization, then CuAAC with sugar azide 5: 6 M GnHCl/0.2 M Na2HPO4, Glc-N35 (5 mM), CuSO4 (40 mM) and TCEP·HCl (20 mM), 50 °C, pH 7, 4 h; (b) analytical LC monitoring of the NCL-CuAAC sequence between EPO4 (11), EPO5 (12) and sugar azide (7). Mass spectrum of pure ‘click’ EPO4-5 (17) (observed: [M + 5H]5+ = 1555.78 Da, calculated: 1555.99 Da). The analytical monitoring was carried out using an analytical column (Phenomenex Jupiter C4, 300 Å, 50 mm × 2.0 mm; 5 μm) using a gradient of 5–65% buffer B over 30 min (buffer A= 0.1% TFA in H2O; buffer B= 0.1% TFA in acetonitrile) at 210 nm; (c) analytical LC monitoring of the NCL-CuAAC sequence between EPO3 (10), ‘click’ EPO4-5 (17) and sugar azide 5. Mass spectrum of pure ‘click’ EPO3-5 neoglycopeptide product (16) (observed: [M + 8H]8+ = 1415.46 Da, calculated: 1415.50 Da). The analytical monitoring was carried out using an analytical column (Phenomenex Jupiter C4, 300 Å, 50 mm × 2.0 mm; 5 μm) using a gradient of 5–65% buffer B over 30 min (buffer A= 0.1% TFA in H2O; buffer B= 0.1% TFA in acetonitrile) at 210 nm. | ||
Using this revised strategy, the differentially ‘clicked’ neoglycopeptide ligation product ‘click’-EPO3-5 (16) was prepared (Fig. 3a). Native chemical ligation between EPO4 (11) and EPO5 (12) was performed similarly to before (Fig. S6†) in ligation buffer (6 M GnHCl/0.2 M Na2HPO4 at pH 6.8) containing MPAA (200 mM) and TCEP·HCl (50 mM) and monitored by LC-MS until completion was observed at 2.5 h (Fig. 3b). The ligation product was recovered by solid phase extraction (SPE, see ESI for details†) and lyophilised to afford the crude ligation product, which was subjected to CuAAC at Pra126. The coupling partner chosen for proof of principle in this reaction was the sugar azide GalNAcα1-O-(CH2)3–N3 (7) (Fig. 3b), because it mimics the natural O-linked glycoside at position 126 of EPO. The click reaction was complete in 2 h at 40 °C and the reaction mixture was purified by reverse phase HPLC to afford pure Cys98-‘click’-EPO4-5 peptide 17 bearing a single triazole-linked glycan in 37% yield over two steps (Fig. 3b). As expected, Thz98 was also quantitatively converted to Cys98 during this reaction.
With ‘click’-EPO4-5 (17) in hand, ligation between 17 and EPO3 (10) peptides was carried out. The peptides were dissolved in ligation buffer (pH 6.8) containing MPAA (100 mM) and TCEP (40 mM). After 2 h the reaction was complete and the reaction mixture recovered by HPLC and lyophilised (Fig. 3c). The crude ligation product was then subjected to CuAAC reaction with sugar azide Glc-N3 (5) to install a second alternative glycan, different from those present in the EPO3-5 polypeptide 15. As with the synthesis of the analogous bis-glycosylated peptide ‘click’ EPO3-5 (15), an elevated temperature (50 °C) was required for the ‘click’ reaction to proceed at Pra83; after 4 h, both the click reaction and Thz68 to Cys68 conversion had proceeded to completion (Fig. 3c). The reaction mixture was purified by RP HPLC to afford pure ‘click’-EPO3-5 neoglycopeptide 16 containing two different sugars in 21% yield over two steps (Fig. 3c).
Attempted assembly of EPO1-2 fragment
Having thus proved the feasibility of our strategy for installing either homo- or heterogeneous neoglycans onto EPO polypeptides (EPO3-5 (15) and EPO3-5 (16) respectively), our attention turned to synthesis of the requisite N-terminal EPO1-2 fragment. To convert the EPO1-alkyl thioester 8 into EPO1-aryl thioester 18, MPAA, was employed for thioester exchange (see ESI† for details). The MPAA-thioester peptide 18 was purified by reverse phase HPLC (Fig. S7†). With the purified EPO1-MPAA thioester 18 in hand, the critical KCL step was attempted with EPO2 (9). The two peptides (3 mM) were dissolved in ligation buffer and reacted at pH 6.1. Disappointingly, the reaction failed to effect greater than 20% conversion. After further trials with diluted reaction mixtures, including additions of organic solvents, the best result obtained was 50% conversion (Fig. S8†). This observation was significantly different to those obtained previously by the Kent group,11 whereby KCL of EPO1 and EPO2 segments proceeded to completion rapidly. The employed peptides differ only by the substitution of propargylglycine at Arg24 in EPO1 and at Arg38 in EPO2. We therefore postulate that the reduced charge and increased hydrophobicity of the product peptide due to this substitution could contribute to the incomplete KCL of our peptide segments.Revised synthetic approach to ‘click’ EPO1-2 (21)
We envisaged that ‘clicking’ the unprotected sugar azide (6) onto either EPO1-thioester 8 or EPO2-thioester 9, or both, before carrying out the KCL would increase the hydrophilic character of the peptide(s) and help solubilise the ligation product, perhaps leading to improved yields from the ligation. To compare the stability of EPO1 (8) and EPO2 (9) towards the CuAAC conditions, the peptides were separately subjected to the CuAAC reaction conditions in the absence of any sugar azides (Fig. S9†). While EPO2 (9) exhibited significant Acm deprotection at the Cys(Acm)33 residue after 30 min, EPO1 (8) (bearing Cys(Acm) at two positions) displayed no detectable Acm deprotection even after 6 h. Histidine, well known to coordinate to copper ions, has been shown to significantly accelerate the CuAAC reaction by stabilising the Cu(I) complex through strong chelation, providing a basis for peptide self-catalysis.38–41 Given that the Cys(Acm)33 residue of EPO2 (9) is adjacent to a histidine residue (His32), the interesting observation of its deprotection, unique to the EPO2 (9) fragment, is likely attributable to the chelating effects of the adjacent His32 residue. Presumably, the localisation of the stabilised Cu(I) complex facilitates Acm deprotection of the adjacent cysteine. While troublesome for the synthesis at hand, this unexpected reactivity potentially lends itself to a one-pot NCL, CuAAC and Acm deprotection system.These observations allowed us to modify our synthetic route accordingly; in the revised scheme (Scheme 2), a sugar azide is first ‘clicked’ onto EPO1 peptide (8). The ‘click’ glycosylated EPO1-alkyl thioester peptide 19 would then be converted into ‘click’ EPO1-MPAA thioester 20 in a ‘one-pot’ reaction and, following purification, undergo KCL with EPO2 (9), facilitated by the installation of the hydrophilic glycan moiety. EPO1 (8) (3 mM) was therefore dissolved in buffer (6 M GnHCl/0.2 M Na2HPO4), with CuSO4 (20 mM), TCEP (10 mM) added and the pH adjusted to 6.3 (Fig. S10†). Finally, GalNAc-N3 (6) (5 mM) was added and the reaction monitored by RP-HPLC. Pleasingly, the CuAAC reaction was complete in 3 h and MPAA (100 mM) was added to the product mixture. The thioester exchange proceeded readily and the resultant click EPO1-MPAA-thioester product 20 was purified by RP-HPLC in 19% yield. It is noteworthy that this thioester exchange gave an improved conversion in relation to our previous attempt on EPO1 (8, see Fig. S7B and S10D†). This can likely be attributed to improved solubility of the intermediate ‘click’ product (19), bearing a hydrophilic neoglycan, again highlighting the benefits of our flexible synthetic strategy. Full analytical monitoring of these manipulations can be found in Fig. S10.†
 | ||
| Scheme 2 Revised route to ‘click’ EPO1-2 (21). The initial route (Scheme 1) failed due to the inefficiency of KCL between EPO1-MPAA and EPO2 peptides. The revised strategy involved a one-pot CuAAC and MPAA thioester exchange reaction on EPO1 peptide 8, followed by one-pot KCL and click chemistry with the EPO2 peptide 9 using a different sugar. This route allows for diversity in sugars to be attached to the resulting ‘click’ EPO1-2 neoglycopeptide fragment 21. | ||
With the ‘click’ EPO1-MPAA-thioester (glycopeptide 20) in hand, KCL with EPO2 (9) was reexamined (Fig. 4). Under optimised conditions, ‘click’ EPO1-MPAA-thioester 20 (3 mM) and EPO2 (9) (3 mM) were dissolved in ligation buffer (6 M GnHCl/0.2 M Na2HPO4) and the pH adjusted to 6.3. To our delight, KCL proceeded much more efficiently (completion observed after 6 h) supporting our hypothesis that the original KCL employing EPO1-MPAA (18) was hampered by the low solubility of the ligated product. Sodium 2-mercaptoethanesulfonate (MESNa) (100 mM) was then added to remove branched thioester by-products at Cys30 (see ESI† for details). Under these conditions, thioester exchange with MESNa, was not observed at Leu67. At this point, we extended the synthetic complexity by adding CuSO4 (20 mM), TCEP·HCl (10 mM), and Gal-N34 (5 mM) to effect an overall ‘one-pot’ KCL and CuAAC reaction. After a further 5 h at room temperature, the CuAAC reaction was complete, affording the desired ‘click’ EPO1-2 neoglycopeptide product 21 in 8% yield after purification by RP-HPLC (Fig. 4b and c). Notably, relative to the initial attempts at the CuAAC reaction of EPO2 (9) (detailed above), the undesired Acm deprotection was significantly reduced. Presumably, this is due to alteration of the backbone conformation or steric effects, resulting from peptide elongation by NCL. Furthermore, this revised and divergent strategy enabled versatility in the installation of sugar-azides at successive steps for the EPO1 and EPO2 fragments. As such, two different sugar moieties were successfully installed to the final ‘click’ EPO1-2 fragment 21 by CuAAC. Notably, this result testifies to the flexibility in synthetic planning and potential unexpected benefits resultant from the combination of NCL and CuAAC chemistries. Furthermore, in the absence of MPAA during KCL, branching thioester adducts are often observed and may interfere with the subsequent CuAAC reaction. Here we demonstrate complete compatibility between the sodium MESNa treated KCL reaction, in which branched thioesters by-products are removed (by thioester exchange), and CuAAC chemistry in one-pot.
 | ||
| Fig. 4 (a) ‘One-pot’ synthetic approach to ‘click’ EPO1-2 neoglycopeptide 21, bearing two different ‘click’ neoglycans. Reagents and conditions: 6 M GnHCl/0.2 M Na2HPO4, pH 6.3, 6 h, then MESNa (100 mM); Gal-N34 (5 mM), CuSO4 (20 mM) and TCEP·HCl (10 mM), added to the same pot, r.t., 5 h; (b) analytical LC monitoring of the one-pot KCL-CuAAC sequence between ‘click’ EPO1-MPAA (20), EPO2 (9) and sugar azide 4; (c) mass spectrum of ‘click’ EPO1-2 neoglycopeptide 21 (observed: [M + 4H]2+ = 2127.63 Da, calculated: 2128.40 Da). The analytical monitoring was carried out using a Phenomenex Jupiter C4 column (300 Å, 50 mm × 2.0 mm; 5 μm) using a gradient of 5–65% buffer B over 30 min (buffer A= 0.1% TFA in H2O; buffer B= 0.1% TFA in acetonitrile) at 210 nm. | ||
Final ligation, Trp (formyl) deprotection, Cys desulphurization and Cys(Acm) removal
Having established a reliable synthetic route to the N-terminal fragment, neoglycopeptide ‘click’ EPO1-2 (21), the final ligation step between 21 and neoglycopeptide ‘click’ EPO3-5 (16) was next investigated (Fig. 5). The glycopeptides ‘click’ EPO1-2 (21) (2 mM) and ‘click’ EPO3-5 (16) (1 mM) were dissolved in ligation buffer containing MPAA (200 mM) and TCEP·HCl (50 mM) and the pH adjusted to 6.8. The ligation was essentially complete after 3 h (Fig. 5b and c) whereupon β-mercaptoethanol/piperidine solution was added to effect in situ removal of the formyl protecting groups on the Trp residues. After 10 min, rapid deformylation yielded the ‘click’ EPO1-5 neoglycoprotein (22), bearing four different neoglycans which was purified by SPE and used without further purification (Fig. 5d and e).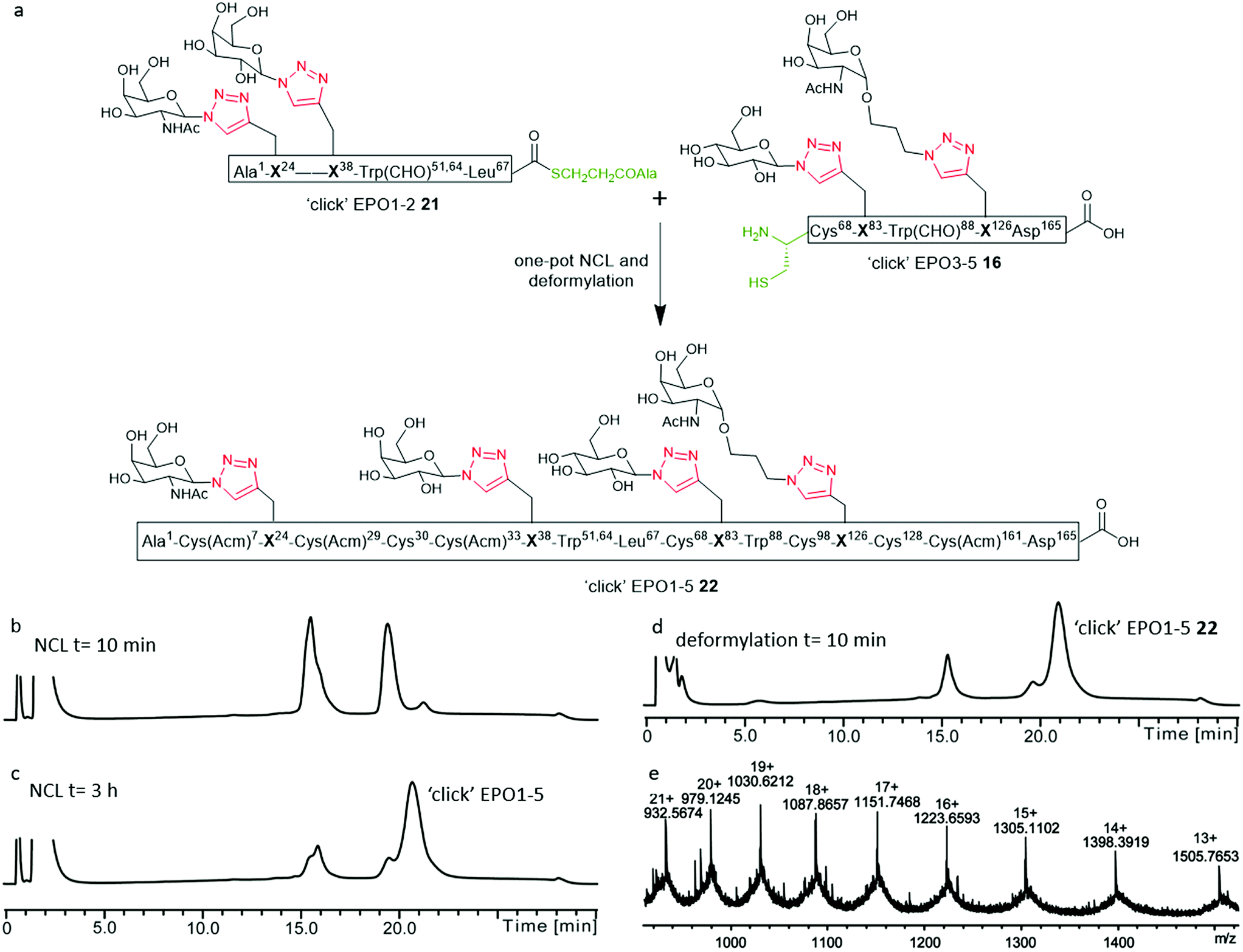 | ||
Fig. 5 (a) NCL and deformylation to access ‘click’ EPO1-5 neoglycopeptide 22 bearing four different ‘click’ neoglycans. Reagents and conditions: NCL and deformylation) 6 M GnHCl/0.2 M Na2HPO4, MPAA (200 mM), TCEP·HCl (50 mM), pH 6.8, 3 h; then β-mercaptoethanol (400 μL) and piperidine (250 μL), 10 min; SPE using C4 semi-prep column and lyophilization; (b–d) analytical LC monitoring of the ligation between ‘click’ EPO1-2 (21) and ‘click’ EPO3-5 (16); (b) t = 10 min of NCL; (c) t = 3 h, the NCL reaction was essentially complete to yield the ligation product ‘click’ EPO1-5 [Cys(Acm)7,29,33,161][Cys30,68,98,128][Trp(CHO)51,64,88]; (d) t = 10 min of deformylation to yield ‘click’ EPO1-5 [Cys(Acm)7,29,33,161][Cys30,68,98,128] (22); (e) mass spectrum of ‘click’ EPO1-5 (22) purified by SPE only (observed: [M + 14H]14+ = 1398.39 Da, calculated: 1398.45 Da. Deconvoluted mass observed: 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 564.63 Da, calculated: 19564.34 Da). The analytical monitoring was carried out using an analytical column (Phenomenex Jupiter C4, 300 Å, 50 mm × 2.0 mm; 5 μm) employing a gradient of 5–65% buffer B over 30 min (buffer A = 0.1% TFA in H2O; buffer B = 0.1% TFA in acetonitrile) at 210 nm. 564.63 Da, calculated: 19564.34 Da). The analytical monitoring was carried out using an analytical column (Phenomenex Jupiter C4, 300 Å, 50 mm × 2.0 mm; 5 μm) employing a gradient of 5–65% buffer B over 30 min (buffer A = 0.1% TFA in H2O; buffer B = 0.1% TFA in acetonitrile) at 210 nm. | ||
Upon successful synthesis of ‘click’ EPO1-5 (22), it was imperative that the cysteines at positions 30, 68, 98 and 128 undergo desulfurization to the native alanine residues without compromising sensitive functional groups in the assembled neoglycoprotein. To effect global desulfurisation, we employed conditions originally reported by Seitz et al.42 (see ESI† for full experimental details). Employing these conditions, modified from the work of Danishefsky et al.,15 avoided metal-based desulfurisation reactions with Pd/Al2O3 or RANEY® nickel that are often plagued by side reactions or low yields. ‘click’ EPO1-5 polypeptide 22 (2 mM) was dissolved in buffer (6 M GnHCl/0.2 M Na2HPO4) containing TCEP·HCl (100 mM), 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) (40 mM) and glutathione (20 mM). The reaction mixture was heated to 40 °C and after 3 h MS analysis revealed that all four cysteines had been quantitatively converted to alanine without any detectable side products (Fig. S11†). The reaction mixture was loaded onto a C4 semi-prep column and washed with 5% aqueous MeCN until all MPAA was removed. Elution with 50% aqueous MeCN followed by lyophilization afforded the crude desulfurised ‘click’ EPO1-5 neoglycoprotein product 23 which was used without further purification. In addition, the Acm protecting groups of cysteines at positions 7, 29, 33 and 161 must be removed for disulfide bond formation upon successful folding. To avoid potential side reactions of mercury(II) with Trp, AgOAc was employed to effect Acm removal (full experimental details in ESI, for reaction monitoring see Fig. S12†). Upon completion (6 h), reverse phase HPLC purification afforded pure ‘click’ EPO1-5[Cys7,29,33,161][Ala30,68,98,128] neoglycoprotein 24 (see Fig. 6a and b) in 21% yield over four steps (from ligation between 21 and 16).
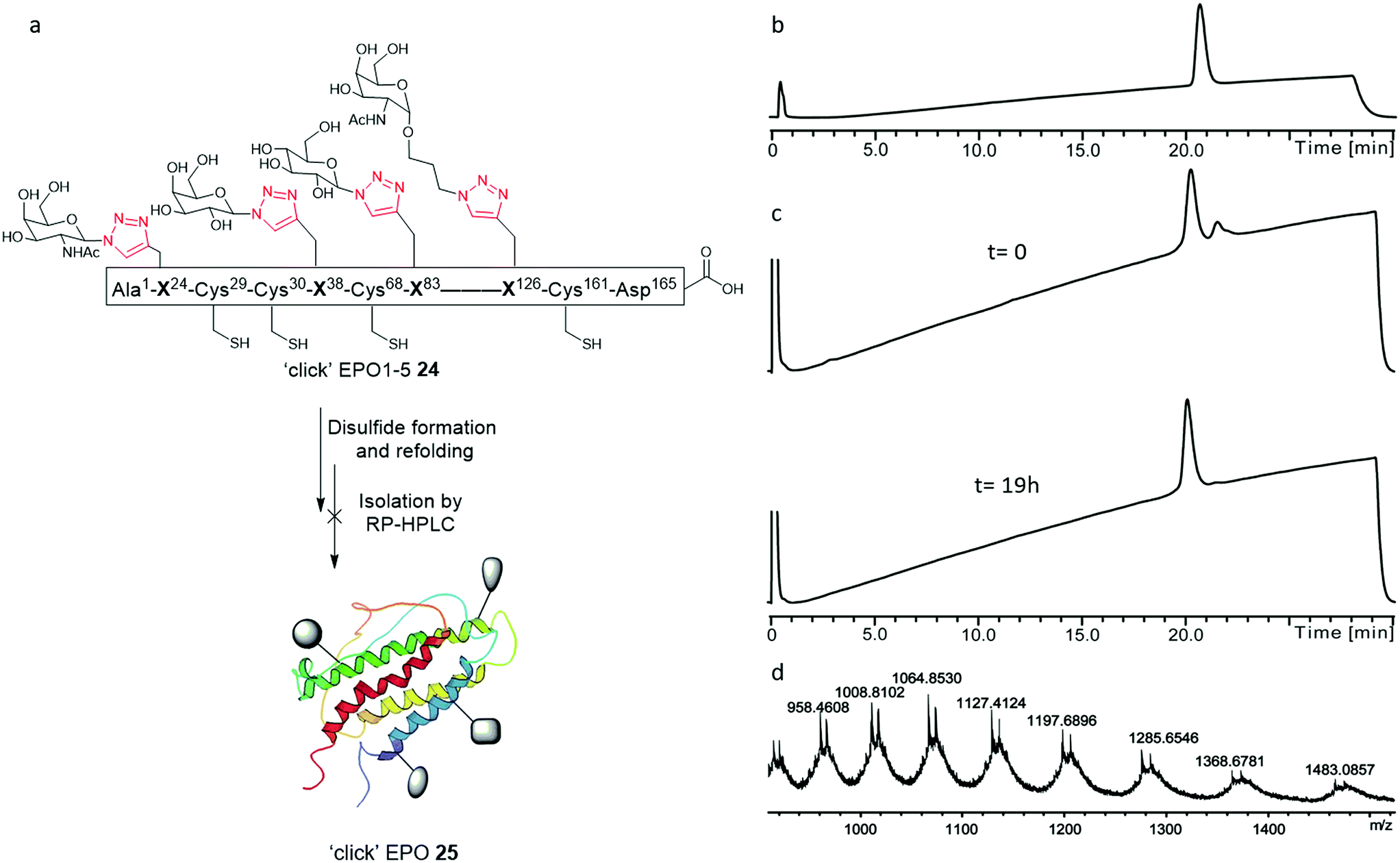 | ||
| Fig. 6 (a) Graphical representation of attempted disulfide formation and protein refolding from the unfolded ‘click’ EPO1-5 neoglycoprotein 24; (b and c) analytical RP-HPLC chromatograms employing an analytical column (Phenomenex Jupiter C4, 300 Å, 50 mm × 2.0 mm; 5 μm) and a gradient of 5–65% buffer B over 30 min (buffer A= 0.1% TFA in H2O; buffer B= 0.1% TFA in acetonitrile); (b) RP-HPLC of pure ‘click’ EPO1-5[Cys7,29,33,161][Ala30,68,98,128] neoglycoprotein 24; (c) analytical LC monitoring of the refolding reaction at 210 nm. Upper spectra: t = 0 h, Lower spectra: t = 19 h; (d) mass spectrum after 19 h of folding. The left of the two peaks corresponded to the correctly folded protein ‘click’ EPO (25) (deconvoluted mass: 19147.68 Da, calculated: 19147.77 Da). The right of the two peaks in the mass spectrum corresponded to the partially folded protein adduct. | ||
Attempted protein folding
With the purified linear ‘click’ EPO1-5 (24) in hand, disulfide formation and concomitant protein folding was attempted to obtain the final, folded ‘click’ EPO (25) (Fig. 6). The procedure adopted for the folding of both PEGylated EPO18 and [lysine24,38,83] human EPO 11 was followed. The linear ‘click’ EPO1-5 polypeptide 24 was first fully denatured by dissolving the lyophilised peptide in a solution of 6 M Gn·HCl (to a concentration of 0.1 mg mL−1 in 100 mM Tris buffer, pH = 8.5). The unfolded polypeptide was then subjected to a three-step dialysis (to a final buffer of 10 mM Tris alone) to remove the denaturant, in the presence of a cysteine–cystine redox system (see ESI† for full experimental details). Unfortunately, application of the redox folding conditions to our ‘click’ EPO1-5 neoglycoprotein 24 resulted in the formation of a mixture of two products, which were inseparable by analytical RP-HPLC. The mixture consisted of (as analyzed by direct infusion ESI-MS), the desired correctly folded product ‘click’ EPO 25 and a partially folded product containing one disulfide bond, a mixed cysteine disulfide, and a cysteine residue with a free thiol (Fig. 6d). Further dialysis in subsequent folding buffers GN·HCl (1 M and 100 mM Tris (pH = 8.0) for 24 h, then 10 mM Tris (pH = 7.0) for 24 h) revealed no significant change in MS profile but the amount of protein present was reduced significantly (as observed by reduced analytical RP-HPLC peak intensity), implying considerable aggregation of misfolded products. Disappointingly, all attempts to isolate the desired correctly folded ‘click’ EPO 25 were unsuccessful.The in vivo activity of EPO depends heavily upon not only resistance to biological degradation, but also solubility and avoiding protein–protein aggregation.20,43,44 To help understand the results of our folding experiments and gain further insight into the relationship between glycosylation and folding, we produced a table to summarise the structure and folding of the most notable synthetic EPO proteins reported in the literature (Table 1). By removal of three key sialyloligosaccharides in a stepwise manner, studies of the Kajihara group20 demonstrated that the in vivo hematopoietic activity and half-life of EPO increases upon increasing number of negatively charged sialyloligosaccharides. Folding yields were also governed by the pattern of glycosylation. The three negatively charged sialyloligosaccharides, particularly those at Asn38 and Asn83, covering hydrophobic areas of protein appear crucial for avoiding protein–protein aggregation and thereby enable the native protein folding to proceed readily.20 In favour of this hypothesis, the polymer-modified EPO reported by Kochendoerfer and co-workers,18 bearing two polymer chains with a total of eight negative charges, demonstrated the most potent in vivo activity to date. EPO analogues devoid of glycosylation, in which the three N-linked glycosylation sites are replaced by lysine, maintain solubility and in vitro activity, presumably by means of the newly endowed cationic charge repulsions.10,11,44 In fact, such an analogue was shown to fold more rapidly than glycosylated EPO.44 While more complex oligosaccharides or mimetics are likely crucial for tuning the required in vivo pharmacokinetics, it appears the solubility and correct folding of EPO merely depends upon introduction of charge repulsions at the designated sites. It is therefore unsurprising to find that our proof of principle ‘click’ EPO1-5 neoglycoprotein 24, in which all four neoglycans are neutral, proved somewhat prone to aggregation and misfolding. Our results suggest that the additional polarity introduced by tetraglycosylation alone is insufficient to elicit favourable physiochemical properties and implementation of glycans bearing a formal charge is crucial to success. Interestingly, while merely introducing a net charge of −2 at position 83 can provide successful folding, incorporation of this charge in position 126 proved less successful (Table 1). In fact, introduction of a sialyloligosaccharide in position 83 also proved superior in reducing hydrophobicity than its concurrent introduction to both position 24 and 38.20 While incorporating charge is almost certainly essential, the variety of mutations tolerated at the designated glycosylation sites by functional synthetic EPO proteins is remarkable and lends itself to the imagination of the synthetic chemist. At present, most researchers have focused upon sialyloligosaccharides, however, the opportunity to introduce positively charged glycans such as GlcN or GalN through ‘click’ chemistry presents an unexplored strategy and likely avoids the synthetic complications arising from the lability of sialyl linkages.
| Synthetic EPO | Glycan/position | Net chargea | Folding protocolb | Folding yield | Ref | |||
|---|---|---|---|---|---|---|---|---|
| 24 | 38 | 83 | 126 | |||||
| a Net charge introduced at positions 24, 38, 83 and 126 only. b Folding protocol 1 = the polypeptide was firstly denatured in a degassed solution of GnHCl (6 M, 5.8 mL) and 100 mM tris (pH = 8.5). The resulting ‘click’ EPO stock solution (0.1 mg mL−1) was then dialyzed against an initial degassed first folding solution containing guanidine (3 M), tris (100 mM), L-cysteine (4 mM) and L-cystine (0.5 mM) adjusted to pH 8.5. After 18 h, the first folding solution was replaced by a second folding solution of guanidine (1 M) and tris (100 mM) adjusted to pH 8.0, and the dialysis allowed to proceed for a further 21 h. The EPO solution was then transferred into a third folding solution of tris (10 mM) adjusted to pH 7.0 and dialyzed for 26 h. After the last dialysis, the ‘click’ EPO solution was concentrated to approx. 2 mL and purification by RP-HPLC was attempted; folding protocol 2 = the folding procedure was altered to begin with 8 M GnHCl at pH 7.5, the subsequent steps follow protocol 1. In some cases, minor variance in the times or temperature at each step are made by some authors. c Yield could not be determined as the correctly folded product could not be isolated from the partially folded adduct. d Residue mutated from native sequence. e Biantennary dodecasacharide = (Sia-Gal-GluNAc-Man)2-Man-GluNAc-GluNAc(Fuc). f No data specified, presumably folding attempted as per consensus glycoform. g No yield reported but peptide reported to aggregate significantly compared to consensus form. h Sequence mutated E21 → A and Q78 →A. i Biantennary dodecasaccharide = (NeuNAc-Gal-GlcNAc-Man)2-Man-GlcNAc-GlcNAc. j Approximation based on peak area. k Sequence mutated Q78→A. l Folding yields were improved by 18–25% when performed with 0.01 mg mL−1 peptide concentration, due to minimizing formation of misfolded oligomers. | ||||||||
| ‘click’ EPO1–165 | GalNAc-‘click’ | Gal-‘click’ | Glc-‘click’ | GalNAcα1-O-(CH2)3-‘click’ | 0 | 1 | N/Ac | This work |
| SEP-PEGylated EPO1–166 | Anionic tetraantennary polymer | Asn | Asn | Anionic tetraantennary polymer | −8 | 1 | 25–40% | 18 |
| Lys24,38,83 EPO1–165 | N/A-Lysd | N/A-Lysd | Lysd | Ser | +3 | 1 | 26% | 11 |
| Consensus EPO1–166 | Biantennary dodecasaccharidee | Biantennary dodecasaccharidee | Biantennary dodecasaccharidee | Sia-Man-Sia(Man) | −8 | 1 | 37% | 17 |
| Glycotruncated consensus EPO1–166 | (GluNAc)2 | (GluNAc)2 | (GluNAc)2 | Sia-Man-Sia(Man) | −2 | 1f | N/Ag | 17 |
| Disialyl83 EPO1–166h | Asn | Asn | Biantennary dodecasaccharidei | Ser | −2 | 2 | 65%j | 45 |
| Disialyl24,38,83 EPO1–166k | Biantennary dodecasaccharidei | Biantennary dodecasaccharidei | Biantennary dodecasaccharidei | Ser | −6 | 2 | 68%j,l | 20 |
| Disialyl38,83 EPO1–166k | Asn | Biantennary dodecasaccharidei | Biantennary dodecasaccharidei | Ser | −4 | 2 | 58%j,l | 20 |
| Disialyl24,83 EPO1–166k | Biantennary dodecasaccharidei | Asn | Biantennary dodecasaccharidei | Ser | −4 | 2 | 43%j,l | 20 |
| Disialyl24,38 EPO1–166k | Biantennary dodecasaccharidei | Biantennary dodecasaccharidei | Asn | Ser | −4 | 2 | 49%j,l | 20 |
| Disialyl83 EPO1–166k | Asn | Asn | Biantennary dodecasaccharidei | Ser | −2 | 2 | 37%j,l | 20 |
Conclusion
Despite significant advancements in the chemical synthesis of native and modified glycosylated protein structures over recent decades, the reality remains that the success of their total chemical synthesis is highly sequence dependant. The design of a flexible synthetic approach, such as that presented here, can however allow for multiple synthetic routes to be explored, overcoming inherent road blocks such as peptide insolubility, inefficient ligations and peptide purification/recovery. By designing flexibility into our synthetic strategy we have fulfilled our primary objective, namely to develop a synthetic platform to assemble ‘click’ neoglycoprotein EPO analogues. The inherent flexibility of our synthetic platform will likely enable the synthesis of a variety of biologically active ‘click’ neoglycoprotein EPO glycoforms in the future. Our results highlight the power of methodologies employing combined NCL and CuAAC ‘click’ chemistry in the context of EPO, a challenging chemical synthesis, particularly with the simplicity conferred by the array of one-pot reactions this strategy enables. EPO, given its synthetic complexity and the relatively unexplored biological significance of the varying glycoforms, is likely to remain a testing ground, at least in the foreseeable future. The failed folding of our ‘click’ neoglycoprotein EPO, in conjunction with previous reports, demonstrate the significance of introducing charge repulsions to otherwise hydrophobic areas of the EPO protein to assist protein folding by the prevention of aggregation. While glycosylation has long been postulated to be critical in guiding protein folding, the relative importance of the glycan structure or charge state is not well understood. Our results strongly suggest the significance of glycan structure and highlight the role that the biologically specified charged oligosaccharides play in preventing aggregation of native and synthetic EPO proteins.Conflicts of interest
There are no conflicts of interest to declare.References
- S. E. Graber and S. Krantz, Annu. Rev. Med., 1978, 29, 51–66 CrossRef CAS PubMed.
- J. W. Eschbach, J. C. Egrie, M. R. Downing, J. K. Browne and J. W. Adamson, N. Engl. J. Med., 1987, 316, 73–78 CrossRef CAS PubMed.
- J. Bohlius, J. Wilson, J. Seidenfeld, M. Piper, G. Schwarzer, J. Sandercock, S. Trelle, O. Weingart, S. Bayliss and B. Djulbegovic, J. Natl. Cancer Inst., 2006, 98, 708–714 CrossRef CAS PubMed.
- M. A. Recny, H. Scoble and Y. Kim, J. Biol. Chem., 1987, 262, 17156–17163 CAS.
- H. Sasaki, B. Bothner, A. Dell and M. Fukuda, J. Biol. Chem., 1987, 262, 12059–12076 CAS.
- N. Kawasaki, M. Ohta, S. Hyuga, M. Hyuga and T. Hayakawa, Anal. Biochem., 2000, 285, 82–91 CrossRef CAS PubMed.
- P. Lai, R. Everett, F.-F. Wang, T. Arakawa and E. Goldwasser, J. Biol. Chem., 1986, 261, 3116–3121 CAS.
- J. C. Cheetham, D. M. Smith, K. H. Aoki, J. L. Stevenson, T. J. Hoeffel, R. S. Syed and J. Egrie, Nat. Struct. Biol., 1998, 5, 861–866 CrossRef CAS PubMed.
- H. Zhan, B. Liu, S. W. Reid, K. H. Aoki, C. Li, R. S. Syed, C. Karkaria, G. Koe, K. Sitney and K. Hayenga, Protein Eng., 1999, 12, 505–513 CrossRef CAS PubMed.
- R. S. Syed, S. W. Reid, C. Li and J. C. Cheetham, Nature, 1998, 395, 511 CrossRef CAS PubMed.
- S. Liu, B. L. Pentelute and S. B. Kent, Angew. Chem., Int. Ed., 2012, 51, 993–999 CrossRef CAS PubMed.
- M. N. Fukuda, H. Sasaki, L. Lopez and M. Fukuda, Blood, 1989, 73, 84–89 CAS.
- G. Ashwell and J. Harford, Annu. Rev. Biochem., 1982, 51, 531–554 CrossRef CAS PubMed.
- M. Goto, K. Akai, A. Murakami, C. Hashimoto, E. Tsuda, M. Ueda, G. Kawanishi, N. Takahashi, A. Ishimoto and H. Chiba, Nat. Biotechnol., 1988, 6, 67–71 CrossRef CAS.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- P. Wang, B. Aussedat, Y. Vohra and S. J. Danishefsky, Angew. Chem., 2012, 124, 11739–11743 CrossRef.
- P. Wang, S. Dong, J.-H. Shieh, E. Peguero, R. Hendrickson, M. A. Moore and S. J. Danishefsky, Science, 2013, 342, 1357–1360 CrossRef CAS PubMed.
- G. G. Kochendoerfer, S.-Y. Chen, F. Mao, S. Cressman, S. Traviglia, H. Shao, C. L. Hunter, D. W. Low, E. N. Cagle and M. Carnevali, Science, 2003, 299, 884–887 CrossRef CAS PubMed.
- K. Hirano, D. Macmillan, K. Tezuka, T. Tsuji and Y. Kajihara, Angew. Chem., 2009, 121, 9721–9724 CrossRef.
- M. Murakami, T. Kiuchi, M. Nishihara, K. Tezuka, R. Okamoto, M. Izumi and Y. Kajihara, Sci. Adv., 2016, 2, e1500678 CrossRef PubMed.
- D. J. Lee, P. W. R. Harris and M. A. Brimble, Org. Biomol. Chem., 2011, 9, 1621–1626 RSC.
- D. J. Lee, K. Mandal, P. W. Harris, M. A. Brimble and S. B. Kent, Org. Lett., 2009, 11, 5270–5273 CrossRef CAS PubMed.
- D. J. Lee, S.-H. Yang, G. M. Williams and M. A. Brimble, J. Org. Chem., 2012, 77, 7564–7571 CrossRef CAS PubMed.
- D. Lim, M. A. Brimble, R. Kowalczyk, A. J. A. Watson and A. J. Fairbanks, Angew. Chem., Int. Ed., 2014, 53, 11907–11911 CrossRef CAS PubMed.
- M. A. Brimble, N. Miller and G. M. Williams, in Amino Acids, Peptides and Proteins in Organic Chemistry, 2011, DOI:10.1002/9783527631827.ch11.
- N. Miller, G. M. Williams and M. A. Brimble, Int. J. Pept. Res. Ther., 2010, 16, 125–132 CrossRef CAS.
- J. M. Wojnar, D. J. Lee, C. W. Evans, K. Mandal, S. B. Kent and M. A. Brimble, in Click Chemistry in Glycoscience, 2013, DOI:10.1002/9781118526996.ch10.
- S. Hotha and S. Kashyap, J. Org. Chem., 2006, 71, 364–367 CrossRef CAS PubMed.
- S. I. van Kasteren, H. B. Kramer, H. H. Jensen, S. J. Campbell, J. Kirkpatrick, N. J. Oldham, D. C. Anthony and B. G. Davis, Nature, 2007, 446, 1105–1109 CrossRef CAS PubMed.
- N. Miller, G. M. Williams and M. A. Brimble, Org. Lett., 2009, 11, 2409–2412 CrossRef CAS PubMed.
- D. Lim, M. A. Brimble, R. Kowalczyk, A. J. Watson and A. J. Fairbanks, Angew. Chem., Int. Ed., 2014, 53, 11907–11911 CrossRef CAS PubMed.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
- P. Kumar, A. H. El-Sagheer, L. Truong and T. Brown, Chem. Commun., 2017, 53, 8910–8913 RSC.
- L. Raibaut, N. Ollivier and O. Melnyk, Chem. Soc. Rev., 2012, 41, 7001–7015 RSC.
- S. Shang, Z. Tan and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5986–5989 CrossRef CAS PubMed.
- N. Ollivier, J. Vicogne, A. Vallin, H. Drobecq, R. Desmet, O. El Mahdi, B. Leclercq, G. Goormachtigh, V. Fafeur and O. Melnyk, Angew. Chem., Int. Ed., 2012, 51, 209–213 CrossRef CAS PubMed.
- E. Boll, J.-P. Ebran, H. Drobecq, O. El-Mahdi, L. Raibaut, N. Ollivier and O. Melnyk, Org. Lett., 2015, 17, 130–133 CrossRef CAS PubMed.
- K. Tanaka, C. Kageyama and K. Fukase, Tetrahedron Lett., 2007, 48, 6475–6479 CrossRef CAS.
- D. C. Kennedy, C. S. McKay, M. C. B. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993–18001 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS PubMed.
- C. Haase, H. Rohde and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 6807–6810 CrossRef CAS PubMed.
- A. Fernández-Tejada, J. Brailsford, Q. Zhang, J.-H. Shieh, M. A. Moore and S. J. Danishefsky, in Protein Ligation and Total Synthesis I, Springer, 2014, pp. 1–26 Search PubMed.
- D. D. Banks, J. Mol. Biol., 2011, 412, 536–550 CrossRef CAS PubMed.
- M. Murakami, R. Okamoto, M. Izumi and Y. Kajihara, Angew. Chem., Int. Ed., 2012, 51, 3567–3572 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03409e |
| This journal is © The Royal Society of Chemistry 2019 |