
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformer-specific [1,2]H-tunnelling in captodatively-stabilized cyanohydroxycarbene (NC–![[C with combining umlaut]](https://www.rsc.org/images/entities/h2_char_0043_0308.gif) –OH)†
–OH)†
André K.
Eckhardt
,
Frederik R.
Erb
and
Peter R.
Schreiner
 *
*
Institute of Organic Chemistry, Justus Liebig University, Heinrich-Buff-Ring 17, 35392 Giessen, Germany. E-mail: prs@uni-giessen.de
First published on 31st October 2018
Abstract
We report the gas-phase preparation of cyanohydroxycarbene by high-vacuum flash pyrolysis of ethyl 2-cyano-2-oxoacetate and subsequent trapping of the pyrolysate in an inert argon matrix at 3 K. After irradiation of the matrix with green light for a few seconds singlet trans-cyanohydroxycarbene rearranges to its cis-conformer. Prolonged irradiation leads to the formation of cyanoformaldehyde and isomeric isocyanoformaldehyde. Cis- and trans-cyanohydroxycarbene were characterized by matching matrix IR and UV/Vis spectroscopic data with ab initio coupled cluster and TD-DFT computations. Trans-cyanohydroxycarbene undergoes a conformer-specific [1,2]H-tunnelling reaction through a 33.3 kcal mol−1 barrier (the highest penetrated barrier of all H-tunnelling reactions observed to date) to cyanoformaldehyde with a half-life of 23.5 ± 0.5 d; this is the longest half-life reported for an H-tunnelling process to date. During the tunnelling reaction the cis-conformer remains unchanged over the same period of time and the Curtin–Hammett principle does not apply. NIR irradiation of the O–H stretching overtone does not enhance the tunnelling rate via vibrational activation. Push–pull stabilisation of hydroxycarbenes through σ- and π-withdrawing groups therefore is even more stabilizing than push–push substitution.
Introduction
Carbenes are reactive intermediates with a divalent carbon atom whose valence shell contains only six electrons, with triplet methylene (H–![[C with combining umlaut]](https://www.rsc.org/images/entities/char_0043_0308.gif) –H) as its simplest representative.1 In general, carbenes have a triplet electronic ground state according to Hund's rule but this can be affected through substitution.1 Second-period substituents with lone pairs attached to the electron deficient carbene centre lead to singlet ground states. For example, cyanocarbene (1, H––CN) is an electronic ground state triplet molecule that can be photochemically interconverted by UV-light irradiation to its isomer isocyanocarbene (2, H––NC) with a singlet ground state as reported by Maier et al.2 Further irradiation leads to cyclisation of 2 to singlet azacyclopropylidene (3) that can be photochemically re-opened to triplet 1 (Scheme 1); dicyanocarbene shows similar photochemical behaviour.3
–H) as its simplest representative.1 In general, carbenes have a triplet electronic ground state according to Hund's rule but this can be affected through substitution.1 Second-period substituents with lone pairs attached to the electron deficient carbene centre lead to singlet ground states. For example, cyanocarbene (1, H––CN) is an electronic ground state triplet molecule that can be photochemically interconverted by UV-light irradiation to its isomer isocyanocarbene (2, H––NC) with a singlet ground state as reported by Maier et al.2 Further irradiation leads to cyclisation of 2 to singlet azacyclopropylidene (3) that can be photochemically re-opened to triplet 1 (Scheme 1); dicyanocarbene shows similar photochemical behaviour.3
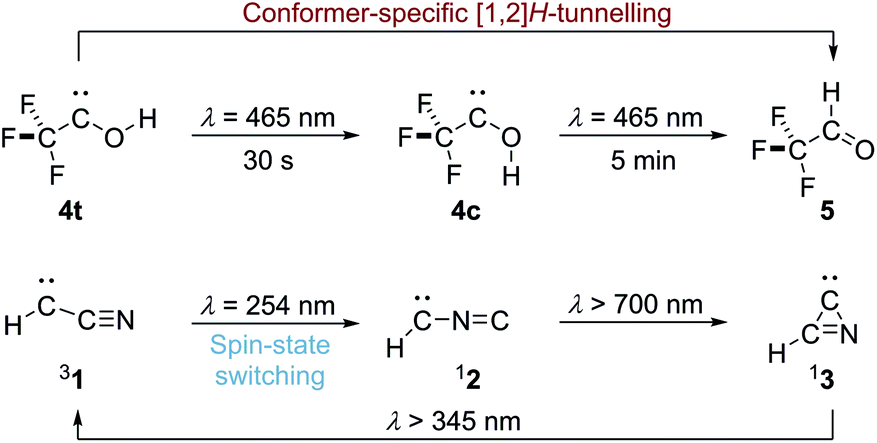 | ||
| Scheme 1 Conformer-specific [1,2]H-tunnelling in singlet trifluoromethyl-hydroxycarbene (4t) and electronic spin-state switching in triplet cyanocarbene (31) to singlet isocyanocarbene (12) under UV light irradiation. | ||
Over the last decade we have prepared various singlet hydroxycarbenes (R––OH) and studied substituent effects on the experimentally observed [1,2]H-tunnelling reactions to the corresponding aldehydes at cryogenic temperatures, leading to the third reactivity paradigm of tunnelling control4,5 next to thermodynamic and kinetic control of chemical reactions. The tunnelling half-life can be extended by introducing π-donors and completely be suppressed by introducing a second lone-pair carrying heteroatom substituent. Recently we demonstrated conformer-specific [1,2]H-tunnelling in trifluoromethylhydroxycarbene 4 (Scheme 1).6 For the first time an energetically less favoured cis-conformer 4c was photochemically produced from trans-conformer 4t. While 4t undergoes [1,2]H-tunnelling to trifluoroacetaldehyde 5, 4c remains unchanged over the same period of time. In contrast to parent hydroxymethylene 6 the tunnelling half-life of 4t is extended from around 2 h in the parent system to 7 d due to the strong σ-electron-withdrawing –CF3 group.
Our initial idea was to combine the reactivity of a cyano- and hydroxyl-group attached to a carbene centre in one molecule, namely hitherto unreported cyanohydroxycarbene (7), and study the photochemical isomerisation of the cyano group vs. [1,2]H-tunnelling of the hydroxyl group. The reactivity of singlet push–pull stabilized alkoxycyanocarbenes was already studied in cyclopropanation reactions.7 The effect of a π-electron withdrawing substituent on [1,2]H-tunnelling in hydroxycarbenes has never been studied and is elaborated in the following. In radical chemistry the stabilisation of radicals by the combined action of an electron donating and electron withdrawing substituent is known as the captodative effect.8 If this effect operates for 7, it should also be stabilized and its tunnelling half-life extended relative to other hydroxycarbenes. Such findings would help in developing and sharpening concepts regarding the appearance of tunnelling and how it can be controlled through substitution.
Results and discussion
Our strategy for the preparation of 7 was high-vacuum flash pyrolysis (HVFP) of ethyl 2-cyano-2-oxoacetate (2-cyano-2-oxo-acetic acid ethyl ester, 10). Similar to our protocol for the gas-phase preparation of carbonic acid,9 we used ester 10 for the preparation of 2-cyano-2-oxo acetic acid (11), an α-ketocarboxylic acid, which have proven to be suitable precursors for several hydroxycarbenes by thermal CO2 extrusion (Scheme 2).4 Ester 10 and its deuterated isotopologue were synthesized according to a literature procedure (see ESI† for details).10 Pyrolysis of 10 at 900 °C and subsequent trapping of the pyrolysate at 3 K led to large amounts of CO2 and ethylene (12) in the corresponding matrix isolation spectra, indicating nearly complete consumption of 10 and 11, respectively, and confirms our suggested dissociation pathway (Scheme 2). Two conformers (cis and trans) of 11 were identified in the pyrolysis spectra as well; these can be readily interconverted by NIR irradiation (Fig. S28†).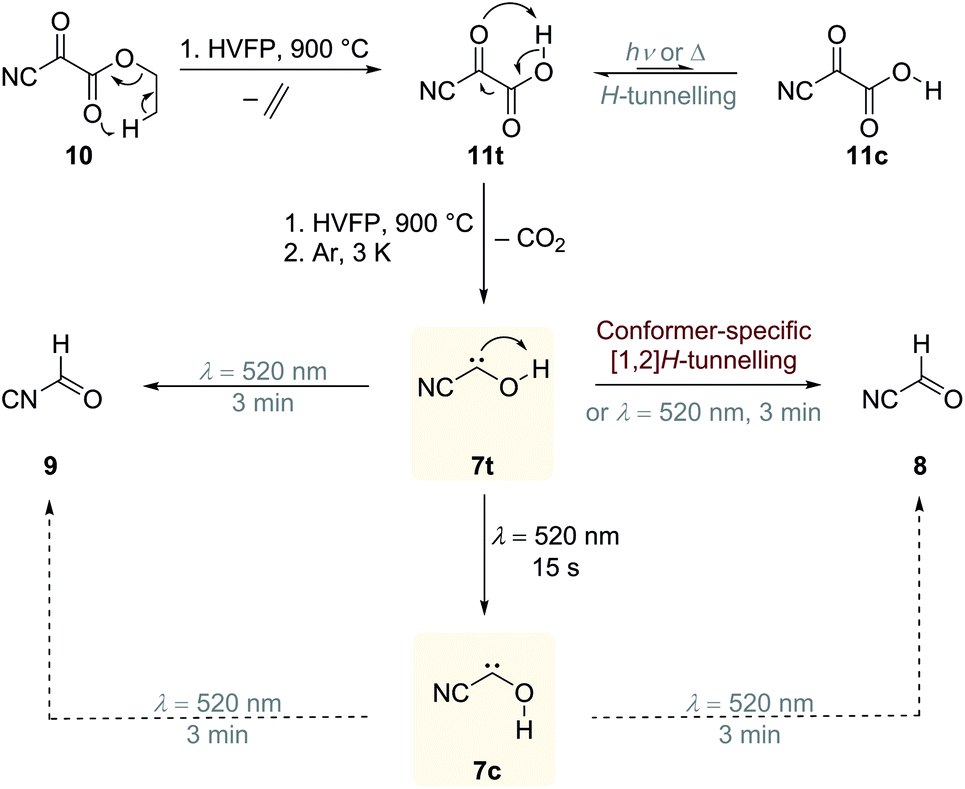 | ||
| Scheme 2 Preparation of cis- and trans-cyanohydroxycarbene 7c and 7t by high-vacuum flash pyrolysis (HVFP) of ethyl 2-cyano-2-oxoacetate (10) at 900 °C. Pyrolysis of 10 first produces α-ketocarboxylic acid 2-cyano-2-oxo acetic acid (11), which further undergoes CO2 extrusion to 7t that is trapped in an argon matrix at 3 K. Carbene 7t can be photochemically interconverted to its cis-conformer 7c. After prolonged irradiation 7t and 7c rearrange to the thermodynamically preferred cyanoformaldehyde (8, formyl cyanide) and isocyanoformaldehyde (9, formyl isocyanide). Only trans-conformer 7t slowly interconverts in a quantum mechanical conformer-specific [1,2]H-tunnelling reaction to 8, while 7c remains unchanged over the same period of time. | ||
Further pyrolysis products were HCN, CO, 8, and 9, as identified by comparison with reference spectra;11–148 has recently been detected in interstellar media.15 In addition, we also observed new unreported signals that we assign to 7t. Irradiation of the matrix immediately after the pyrolysis with green light (λ = 520 nm) for three minutes led to complete disappearance of the signals of 7t while, much to our surprise, the signals of both aldehydes, 8 and 9, increased significantly (Fig. 1). With shorter irradiation times (5–30 s) the IR signals of 7t were not completely depleted but new small signals appeared in the spectrum that we assign to cis-conformer 7c (Fig. 1, red spectrum). Further irradiation led to the disappearance of these signals and again to an increase of 8 and 9. An irradiation time of 15 s immediately after pyrolysis is sufficient for maximal conversion of 7t to 7c. Of course, aldehyde 8 is the [1,2]H-migration product of 7t, while 9 might form from 8 at the cost of the excess energy after irradiation (vide infra).
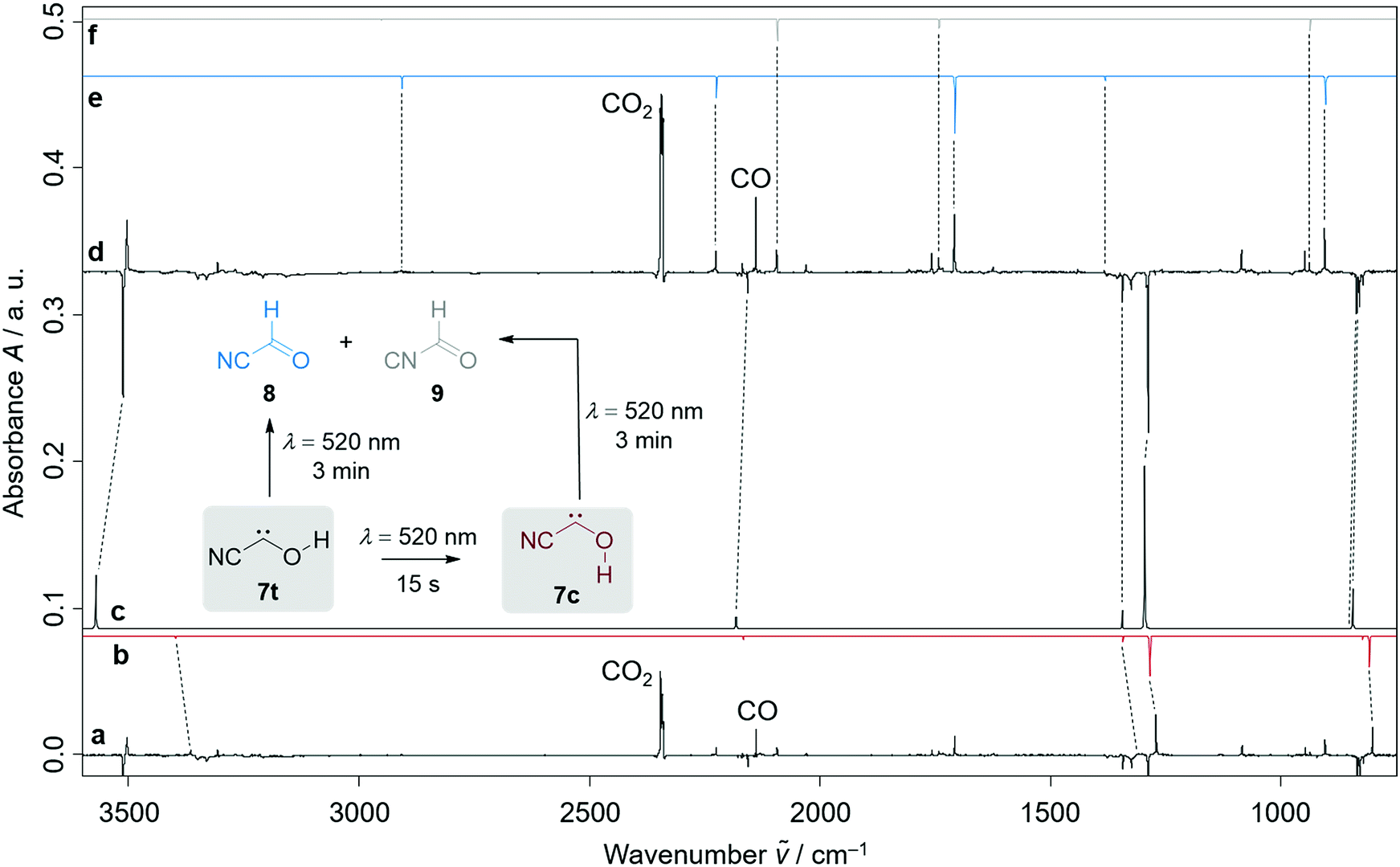 | ||
| Fig. 1 (a): Experimental difference spectrum between the spectra recorded after 15 s irradiation of the matrix with a wavelength of 520 nm and the spectra immediately recorded after 4 h pyrolysis of ethyl 2-cyano-2-oxoacetate (10) at 900 °C; (b and c): computed anharmonic AE-CCSD(T)/cc-pCVQZ vibrational spectra (unscaled) of cis-cyanohydroxycarbene (7c, red, b) and trans-cyanohydroxycarbene (7t, black, c); (d): experimental difference spectrum between the spectra recorded after 3 min irradiation of the matrix with a wavelength of 520 nm and the spectra immediately recorded after 4 h pyrolysis of ethyl 2-cyano-2-oxoacetate (10) at 900 °C; (e and f): experimentally observed vibrational frequencies of isocyanoformaldehyde (9, grey, f) and cyanoformaldehyde (8, blue, e) in combination with computed AE-CCSD(T)/cc-pCVQZ anharmonic intensities. | ||
The strongest bands of 7t are located at 3511.8 and 1287.9 cm−1, respectively. Furthermore, we also identified several weaker signals at 2156.8, 1343.8, 835.4, 829.8 and 821.6 cm−1 (Table S5†). The strongest signals of the cis-conformer 7c are at 1272.2 and 801.9 cm−1. Weaker signals are at 3365.4 cm−1 and 812.3 cm−1 (Table S6†). Our signal assignments of 7c and 7t are based on two IR difference spectra obtained from subtracting the spectra recorded after 15 s and 3 min irradiation of the matrix with a wavelength of 520 nm, respectively, from the spectrum recorded before irradiation of the same matrix in combination with ab initio AE-CCSD(T)/cc-pCVQZ coupled cluster anharmonic frequency computations. The signals of disappearing 7t point downwards while the increasing signals of 7c, 8, and 9 point upwards (Fig. 1).
To confirm the formation of 7t and 7c we performed analogous experiments with 10 deuterated at the ethyl ester function (C2D4-10′) that produces deuterated 7′ in a very clean fashion because there is no exchange of acidic protons as is the case for α-ketocarboxylic acid precursors. The IR signal of the O–D stretching vibration of 7t′ and 7c′ are strongly affected by the isotopic substitution and were observed at 2598.1 cm−1 (exp. shift Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 913.7 cm−1, comp. anh. shift Δ = 909.4 cm−1) and 2498.7 cm−1 (exp. shift Δ = 866.7 cm−1, comp. anharm. shift Δ = 826.7 cm−1), respectively. The C–O–D in-plane and out-of-plane deformation vibrations of 7t′ are shifted to 1050.6 cm−1 (exp. shift Δ = 293.2 cm−1, comp. harm. shift Δ = 301.3 cm−1) and 642.4 cm−1 (exp. shift Δ = 179.4 cm−1, comp. harm. shift Δ = 198.6 cm−1) and for 7c′ to 1021.2 cm−1 (exp. shift Δ = 56.6 cm−1, comp. anh. shift Δ = 42.4 cm−1) and 649.0 cm−1 (exp. shift Δ = 172.8 cm−1, comp. harm. shift Δ= 167.9 cm−1), respectively. The remaining signals remain virtually unchanged (Fig. S27†). A complete band assignment is summarized in the ESI (Tables S7 and S8†).
= 913.7 cm−1, comp. anh. shift Δ = 909.4 cm−1) and 2498.7 cm−1 (exp. shift Δ = 866.7 cm−1, comp. anharm. shift Δ = 826.7 cm−1), respectively. The C–O–D in-plane and out-of-plane deformation vibrations of 7t′ are shifted to 1050.6 cm−1 (exp. shift Δ = 293.2 cm−1, comp. harm. shift Δ = 301.3 cm−1) and 642.4 cm−1 (exp. shift Δ = 179.4 cm−1, comp. harm. shift Δ = 198.6 cm−1) and for 7c′ to 1021.2 cm−1 (exp. shift Δ = 56.6 cm−1, comp. anh. shift Δ = 42.4 cm−1) and 649.0 cm−1 (exp. shift Δ = 172.8 cm−1, comp. harm. shift Δ= 167.9 cm−1), respectively. The remaining signals remain virtually unchanged (Fig. S27†). A complete band assignment is summarized in the ESI (Tables S7 and S8†).
We also proved the formation of 7t using UV/Vis spectroscopy because we experimentally observed a HOMO – LUMO transition at λmax = 467 nm that is typical for hydroxycarbenes5,16,17 in this range (Fig. 2 and S30†). We computed the electronic transitions with time dependent density functional theory at TD-B3LYP/6-311++G(2d,2p) to be at 531.0 nm with an oscillator strength of f = 0.0045 for 7t, in good agreement with experiment. For 7c the HOMO – LUMO transition is computed at 535.5 nm (f = 0.0054). However, we were not able to record a specific UV/Vis spectrum for 7c (see red spectrum after 15 s 520 nm irradiation in Fig. 2) with shorter irradiation times. The similarity of the computed absorptions of 7t and 7c helps us rationalize why both conformers are completely depleted upon prolonged irradiation (blue, orange and grey spectrum in Fig. 2). We assigned the slightly increasing signals at around 300–370 nm to 8 and 9 according to our observations in the IR spectra and through comparison with the literature and computations (Table S3 and S4†).11
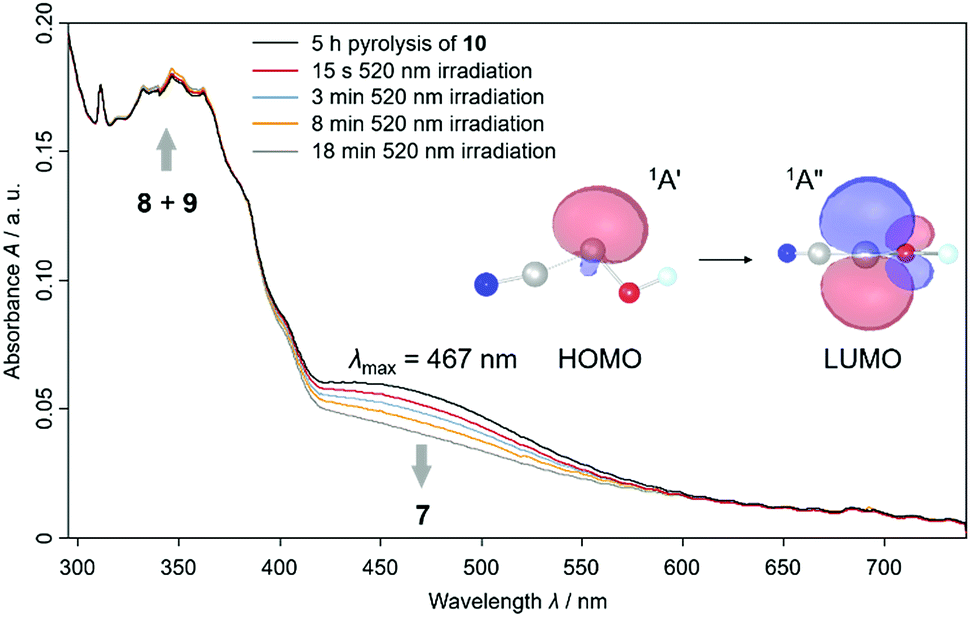 | ||
| Fig. 2 Experimental UV/Vis spectra of the pyrolysate of 2-cyano-2-oxo-acetic acid ethyl ester (10) including cyanohydroxycarbene (7) in argon at 8 K. After 5 h pyrolysis of 10 at 900 °C the black spectrum was recorded. The broad signal of trans-cyanohydroxycarbene (7t) between 400 and 550 nm disappears after prolonged irradiation with a wavelength of 520 nm of the same matrix. We assigned the slightly increasing signals around 350 nm to the photoproducts of 7t, namely cyanoformaldehyde (8) and isocyanoformaldehyde (9). The NBOs for the electronic transition of 7t were computed at the HF/6-311++G(2d,2p) level of theory based on the AE-CCSD(T)/cc-pCVQZ optimized geometry of 7t. HOMO = highest occupied molecular orbital; LUMO = lowest unoccupied molecular orbital; NBO = natural bond orbital. | ||
In the dark the IR signals of 7t disappear with a half-life of around τ = 23.5 ± 0.5 d (temperature independent in the 3–12 K interval) through a quantum mechanical tunnelling (QMT) process as observed for several other hydroxycarbenes.4 The disappearance of the signal of 7t at around 1288 cm−1 over a time range of 47 days is shown in Fig. 3. In contrast to the photochemical interconversion of 7t to 8 and 9, only 8 forms through [1,2]H-tunnelling. Hence, the excess energy after tunnelling through the barrier is efficiently distributed through intramolecular vibrational relaxation (IVR) and removed by the cryogenic surroundings.
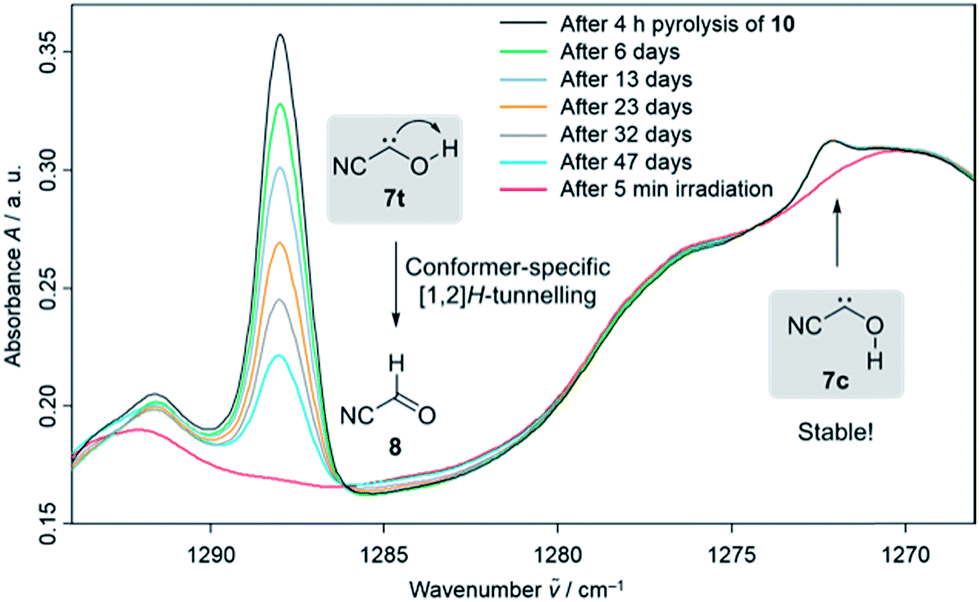 | ||
| Fig. 3 Conformer-specific [1,2]H-tunnelling of trans-cyanohydroxycarbene (7t) with a tunnelling half-life of τ = 23.5 ± 0.5 d over a time period of 47 days monitored by the signal decay at around 1288 cm−1. The amount of the cis-conformer 7c remained unchanged over the same period of time at around 1272 cm−1 (marked with an arrow). After 5 min irradiation of the matrix with a wavelength of 520 nm both carbene signals are completely depleted (red spectrum). | ||
There is not enough excess energy left for subsequent isomerisation of 8 to 9 as in the photochemical interconversion (for details of the potential energy surface, see Fig. 4). The small signal of 7c at around 1272 cm−1 remained unchanged over the same period of time (Fig. 3). Hence, only the trans-conformer 7t undergoes [1,2]H-tunnelling. To the best of our knowledge this is only the second example of a conformer-specific H-tunnelling reaction (which was first observed for 4t).6 As for all other hydroxycarbenes, deuteration of the hydroxyl group suppresses tunnelling completely due to the higher mass. After two weeks in the dark, the matrix spectra with isolated O–D 7t′ were completely unchanged.
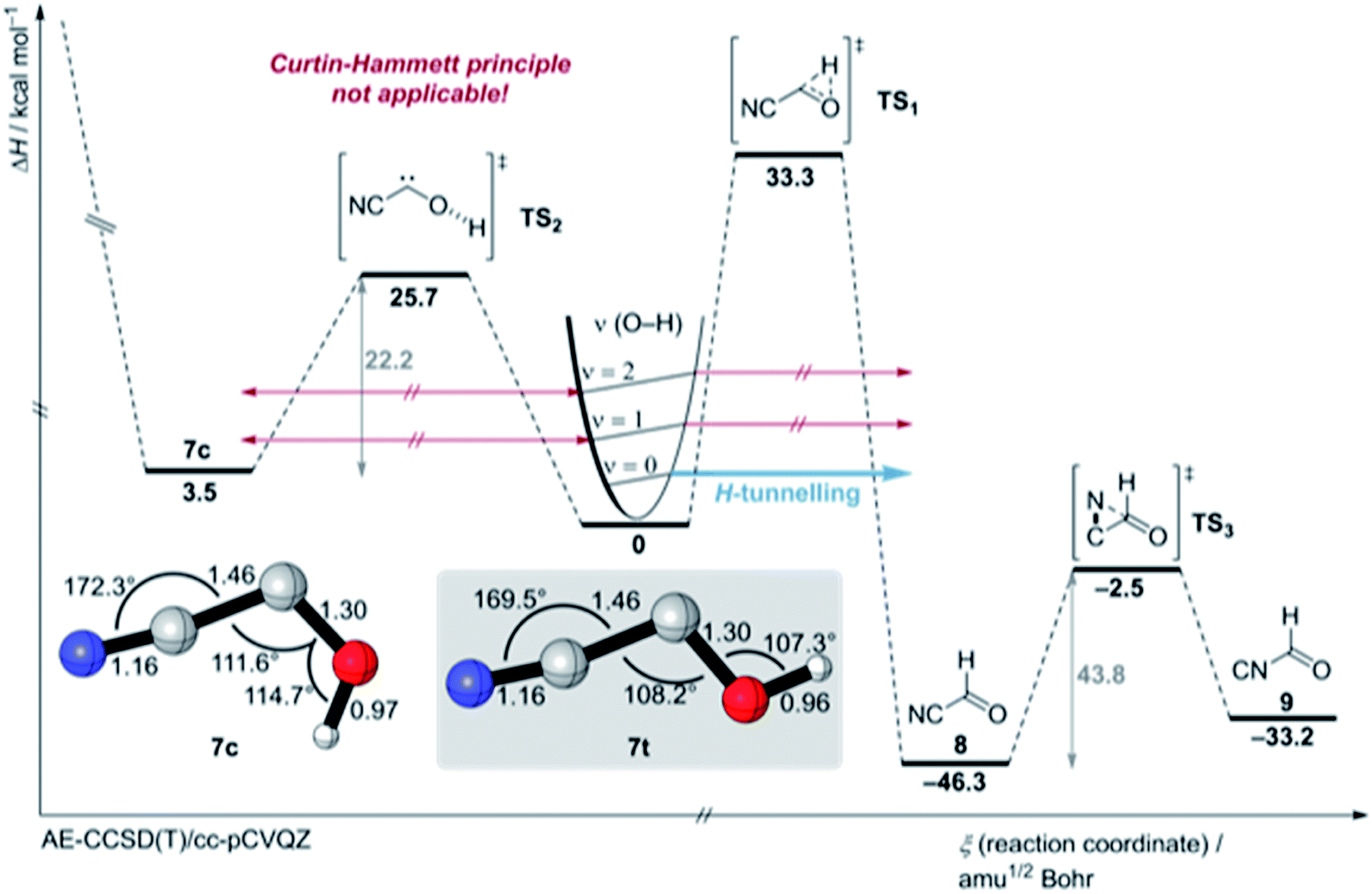 | ||
| Fig. 4 Potential energy surface around cyanohydroxycarbene (7) (colour code: carbon: grey, nitrogen: blue, oxygen: red, hydrogen: white) at the AE-CCSD(T)/cc-pCVQZ + ZPVE level of theory. For an extended version of the PES see Fig. S45 and S46.† | ||
We also attempted to stimulate [1,2]H-tunnelling in 7t through vibrationally activated tunnelling (VAT)18,19 (the [1,2]H-tunnelling half-life from an excited vibrational mode is computed to be around 2 h (Table S13†)) by near infrared (NIR) laser irradiation of the measured O–H stretching vibration overtone of 7t at 6840.2 cm−1 (1461.95 nm ≙ 19.6 kcal mol−1) (Fig. 4 and S21†). However, the spectrum remained unchanged after 2 h irradiation. We did not even observe an enrichment of the energetic less favoured 7c isomer because we irradiated with a photon energy less than the computed torsional barrier for trans–cis isomerisation (25.7 kcal mol−1; see PES in Fig. 4). Hence, excitation of the O–H stretching overtone is ineffective for VAT in hydroxycarbenes and the excitation of the C–O–H deformation vibration seems much more effective from a theoretical point of view because this vibration or “reactive mode” pushes the system towards the tunnelling product 8. Unfortunately, the signal for this specific overtone could not be detected experimentally due to its very low intensity. Also, IVR is likely to be much too fast (ps timescale20) and too inefficient for the excitation of the overtone of the C–O–H deformation vibrational mode (υ = 2). Furthermore, the torsional barrier for trans–cis isomerisation in 7 is one of the highest known barriers for intramolecular OH torsion in comparison to familiar acids,21,22 alcohols23 or hydroxycarbenes.6
We computed the most important part of the potential energy surface (PES) around 7 at the AE-CCSD(T)/cc-pVCQZ and an extended PES version at the B3LYP/6-311++G(2d,2p) + harmonic zero-point vibrational energies (ZPVE) level of theory (Fig. 4, S45 and S46†). Cis- and trans-7 are planar Cs symmetric molecules that have closed-shell singlet 1A′ electronic ground states. There is only one C1 point group triplet state isomer 37 at AE-CCSD(T)/cc-pVCQZ that is much higher in energy (T0 = 20.4 kcal mol−1). The singlet triplet gap in 7t is the smallest in comparison to all other prepared hydroxycarbenes, in which the triplet state typically lies around 25 kcal mol−1 higher in energy than the singlet electronic ground state. The C–O bond length of 7t and 7c of 1.30 Å is much shorter than a typical C–O bond (e.g., 1.41 Å in methanol23) due to π-type stabilisation of the electron-deficient carbene centre through the oxygen p lone pair (Fig. S37 and S41†). A Wiberg bond index of approximately 1.22 is computed for the C–O-bond in 7t and 7c, indicative of zwitterionic and polarized allene-type resonance structures (Fig. S43 and S44†).1 A natural resonance theory (NRT) analysis highly favours the zwitterionic resonance structures over the neutral and allenic species (Fig. S43 and S44†).
In contrast to 4, in which both conformers are almost equal in energy, 7t and 7c differ by 3.5 kcal mol−1. The reaction barrier for the observed conformer-specific [1,2]H-tunnelling reaction of 7t to 8 is 33.3 kcal mol−1 (TS1, Cs) and therefore 2.6 kcal mol−1 higher than in the tunnelling reaction of 4t (Fig. 5). Both conformers of 7 are connected by a 25.7 kcal mol−1 high C1 transition state TS2 in the forward (7t → 7c) and 22.2 kcal mol−1 in the reverse (7c → 7t) reaction. Hence, interconversion and pre-equilibration of 7t to 8 is not given at cryogenic temperatures and the Curtin–Hammett principle is not applicable.6 We did not locate a transition state directly connecting 7c and 8 or 9, respectively.
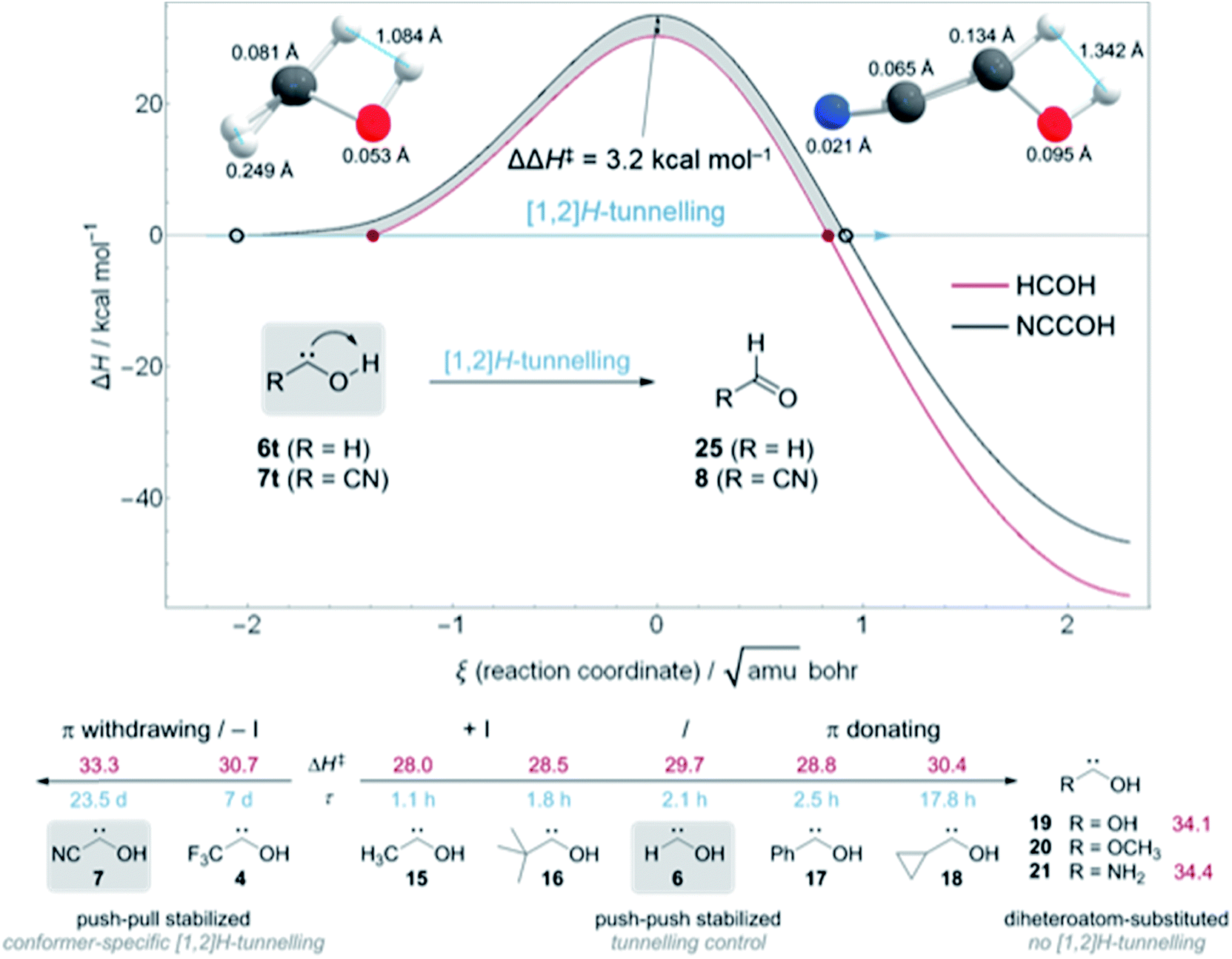 | ||
| Fig. 5 Top: Comparison of the computed ZPVE-corrected reaction barriers for [1,2]H-tunnelling in hydroxymethylene (6t, red) and cyanohydroxycarbene (7t, black) at B3LYP/cc-pVTZ. The barrier for 7t is 3.2 kcal mol−1 higher and much wider than in parent hydroxymethylene; the difference between the two barriers is depicted by the grey area between the two reaction profiles. The two structures on the top (colour code: carbon, grey; oxygen, red; hydrogen, white) show the atomic motions along the reaction coordinate between the two turning points (depicted as red disks and black circles) at the zero-energy level. Bottom: Matrix-isolated hydroxycarbenes, their experimentally determined tunnelling half-lives τ and computed tunnelling barriers for the [1,2]H-shift to the corresponding aldehydes ΔH‡ (in kcal mol−1) at the coupled cluster level of theory with at least a triple-ζ basis set. Push–pull stabilized hydroxycarbenes show conformer-specific [1,2]H-tunnelling while for most of the push–push stabilized hydroxycarbenes (15, 16, and 18) the third reactivity paradigm tunnelling control is applicable. Second-period dihetereo-atom--substituted hydroxycarbenes do not show [1,2]H-tunnelling although the reaction barriers are only slightly higher than those of cyano-hydroxycarbene (7). | ||
Aldehyde 8 is favoured by 13.1 kcal mol−1 over 9; both isomers are connected by a C1 transition structure TS3 with a 43.8 kcal mol−1 barrier in the forward (8 → 9) and a 30.7 kcal mol−1 barrier in the reverse (9 → 8) direction. Both isomers can be interconverted photochemically; a tunnelling isomerisation reaction was experimentally not observed due to the very high and broad reaction barrier and the involvement of heavy atom movement along the reaction path that makes tunnelling less likely.11,24 The transition states (TS5 and TS10) for the isomerisation of the nitrile group in 7 (not shown here, for an extended version of the PES see Fig. S45 and S46†) are computed to 44.9 and 48.1 kcal mol−1, respectively, and these are in the same energetic region as transition state TS4 for the dissociation of 7c into HCN and CO (46.1 kcal mol−1).
We computed the tunnelling half-life of 7t with the semiclassical one-dimensional Wentzel–Kramers–Brillouin (WKB) tunnelling model25–27 at the AE-CCSD(T)/cc-pCVQZ//MP2/cc-pVTZ level of theory to τ = 77 d, in good agreement with the experimental result (see ESI† for details). For deuterated 7t′ the tunnelling half-life is computed to several thousand years. We also carried out multidimensional instanton28 and canonical variational transition state theory in combination with small curvature tunnelling correction (CVT/SCT)29,30 computations at B3LYP/cc-pVTZ, which describes the PES around 7 almost as well as the high level coupled cluster computations (see ESI† for further details). With the instanton approach the tunnelling half-life is determined to τ ≈ 40 d in an almost perfect agreement with experiment (Table S12†).31 To the best of our knowledge, the experimentally observed H-tunnelling process here passes through the highest reaction barrier (33.3 kcal mol−1) resulting in the longest hitherto observed H-tunnelling half-life (23.5 ± 0.5 d) reported to date.32
We computed the ZPVE-corrected reaction barriers for [1,2]H-tunnelling in 6t to formaldehyde (25) and 7t to 8 at B3LYP/cc-pVTZ (Fig. 5, Top) and compared the different barrier shapes with each other. The barrier for the isomerisation of 7t is 3.2 kcal mol−1 higher in energy and the overall reaction almost 8 kcal mol−1 less exothermic than in 6t → 25, resulting in a much wider barrier according to the Bell–Evans–Polanyi principle. The difference between the two reaction profiles is depicted as the grey area in Fig. 5. Tunnelling through this area increases the half-life of 7t to almost 24 d in contrast to 2.1 h in 6t. Due to the much wider barrier the tunnelling distances of every atom along the reaction coordinate change. We compared the atomic motion along the reaction arc of 6t and 7t between the two classical turning points (depicted as red disks and black circles in Fig. 5 (Top)) at the zero-energy level that already includes the attempt energy for tunnelling because the reaction barriers are fully ZPVE corrected in which the reactive mode is already included. Note that the distances of atom movement along the reaction arc slightly differ from the Euclidean distance of the two turning point geometries because the reaction coordinate is multidimensional. The two structures on the top in Fig. 5 clearly show that every atom participates in the tunnelling process and not only the transferred hydrogen, which, of course, contributes the most.31,33 Due to the smaller barrier width for the isomerisation of 6t the transferred hydrogen only moves 1.084 Å in contrast to the 1.342 Å in 7t. The tunnelling distances of the heavier oxygen and carbene carbon atom are almost doubled in 7t in comparison to 6t.
Fig. 5 (Bottom) summarizes the QMT reactivity of hydroxycarbenes prepared to date. Additional π-donors (+M) increase the tunnelling half-life of parent 616 from 2.1 h to 2.5 h in phenylhydroxycarbene17 (17) and almost 18 h in cyclopropylhydroxycarbene34 (18) through push–push stabilisation.1 With an inductive donating (+I) substituent, like a methyl group in methylhydroxycarbene5,3315 or tert-butyl group in tert-butylhydroxycarbene3516 the tunnelling half-lives slightly decrease to 1.1 and 1.8 h, respectively, in comparison to 6. However, the second substituent in 15, 16, and 18 leads to the appearance of tunnelling control in which the product through the higher but narrower barrier forms, irrespective of the kinetic preference.4,5,34,35 [1,2]H-tunnelling is completely suppressed in diheteroatom-substituted hydroxycarbenes dihydroxycarbene36 (19), methoxyhydroxycarbene36 (20), and aminohydroxycarbene (21).4 Even in the simplest aminocarbene, aminomethylene, [1,2]H-tunnelling was not observed due to the strong π-donation of the nitrogen atom.37
On the other hand, electron-withdrawing groups seem to stabilize hydroxycarbenes even more than non-heteroatom electron donating substituents through push–pull stabilisation (Fig. 5, bottom left).1 The tunnelling half-life with an inductive electron withdrawing (−I) CF3-group in 4 is 7 d and therefore almost 80 times larger than in parent 6. In this study we determined the tunnelling half-life of 7 with a π withdrawing cyano group to 23.5 ± 0.5 d, i.e., 269 times larger than in 6.
Hence, π-withdrawing groups (−M) seem to have a stronger effect on the tunnelling half-lives than −I substituents. Even the energetically less favourable cis-conformer 4c and 7c were experimentally observed, likely due to push–pull stabilisation of the electron deficient carbene centre. These findings led to the notion of conformer-specific [1,2]H-tunnelling6 for which we presented a second example here.
Conclusions
We present the first spectroscopic characterisation of cyanohydroxycarbene by matrix isolation IR and UV/Vis spectroscopy. The energetically less preferred cis-conformer can be photochemically generated from the trans conformer. The trans-conformer undergoes conformer specific [1,2]H-tunnelling from the vibrational ground state to cyanoformaldehyde through a 33.3 kcal mol−1 high but narrow barrier with a half-life of 23.5 ± 0.5 d (computed 40–80 d) while the cis-conformer remains virtually unchanged over the same period of time. Based on our experimental results and ab initio AE-CCSD(T)/cc-pCVQZ computations, both conformers are Cs symmetric singlet ground state carbenes that are connected by a high rotational barrier (25 kcal mol−1) so that interconversion at cryogenic temperatures is suppressed and the Curtin–Hammett principle is not applicable. Both carbene conformers can be photochemically isomerised through prolonged irradiation to cyanoformaldehyde and isocyanoformaldehyde. Vibrationally activated tunnelling by means of near infrared excitation of the O–H stretching overtone of trans-cyanohydroxycarbene and photochemical isomerisation of cyanohydroxycarbene to isocyanohydroxycarbene was experimentally not observed.In general, hydroxycarbenes can be stabilized through electron donating and withdrawing substituents. Hereby, the half-lives slightly increase through π-donating groups (push–push stabilisation) and significantly extend through σ-donation (+I) and, as demonstrated here, even better through π-withdrawing groups (push–pull stabilisation, captodative effect). Tunnelling is completely suppressed through a strong second π-donating substituent such as heteroatoms bearing lone pairs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. K. E. thanks the Fonds der Chemischen Industrie for a scholarship. The authors thank Dr Dennis Gerbig and Henrik Quanz (both JLU Giessen) for setting up the NIR-Laser.Notes and references
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- G. Maier, H. P. Reisenauer and K. Rademacher, Chem.–Eur. J., 1998, 4, 1957–1963 CrossRef CAS.
- G. Maier, H. P. Reisenauer and R. Ruppel, Eur. J. Org. Chem., 2003, 2003, 2695–2701 CrossRef.
- P. R. Schreiner, J. Am. Chem. Soc., 2017, 139, 15276–15283 CrossRef CAS PubMed.
- P. R. Schreiner, H. P. Reisenauer, D. Ley, D. Gerbig, C. H. Wu and W. D. Allen, Science, 2011, 332, 1300–1303 CrossRef CAS PubMed.
- A. Mardyukov, H. Quanz and P. R. Schreiner, Nat. Chem., 2017, 9, 71–76 CAS.
- R. A. Moss, T. Zdrojewski, K. Krogh-Jespersen, M. Włostowski and A. Matro, Tetrahedron Lett., 1991, 32, 1925–1928 CrossRef CAS.
- H. G. Viehe, Z. Janousek, R. Merenyi and L. Stella, Acc. Chem. Res., 1985, 18, 148–154 CrossRef CAS.
- H. P. Reisenauer, J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2014, 53, 11766–11771 CrossRef CAS PubMed.
- O. Achmatowicz and J. Szymoniak, Tetrahedron, 1982, 38, 1299–1302 CrossRef CAS.
- K. Banert, J. R. Fotsing, M. Hagedorn, H. P. Reisenauer and G. Maier, Tetrahedron, 2008, 64, 5645–5648 CrossRef CAS.
- D. J. Clouthier and D. Moule, J. Am. Chem. Soc., 1987, 109, 6259–6261 CrossRef CAS.
- W. Lewis-Bevan, R. D. Gaston, J. Tyrrell, W. D. Stork and G. L. Salmon, J. Am. Chem. Soc., 1992, 114, 1933–1938 CrossRef CAS.
- M. Gronowski, P. Eluszkiewicz and T. Custer, J. Phys. Chem. A, 2017, 121, 3263–3273 CrossRef CAS PubMed.
- J. R. Anthony, J. M. Hollis, F. J. Lovas, D. S. Wilmer, P. R. Jewell and D. S. Meier, Astrophys. J., Lett., 2008, 675, L85 CrossRef.
- P. R. Schreiner, H. P. Reisenauer, F. C. Pickard IV, A. C. Simmonett, W. D. Allen, E. Matyus and A. G. Csaszar, Nature, 2008, 453, 906–909 CrossRef CAS PubMed.
- D. Gerbig, H. P. Reisenauer, C. H. Wu, D. Ley, W. D. Allen and P. R. Schreiner, J. Am. Chem. Soc., 2010, 132, 7273–7275 CrossRef CAS PubMed.
- M. Pettersson, E. M. S. Maçôas, L. Khriachtchev, R. Fausto and M. Räsänen, J. Am. Chem. Soc., 2003, 125, 4058–4059 CrossRef CAS PubMed.
- E. M. Greer, K. Kwon, A. Greer and C. Doubleday, Tetrahedron, 2016, 72, 7357–7373 CrossRef CAS.
- S. Perry David, G. A. Bethardy and X. Wang, Ber. Bunsen-Ges. Phys. Chem., 1995, 99, 530–535 CrossRef.
- M. Pettersson, E. M. S. Maçôas, L. Khriachtchev, J. Lundell, R. Fausto and M. Räsänen, J. Chem. Phys., 2002, 117, 9095–9098 CrossRef CAS.
- E. M. S. Maçôas, L. Khriachtchev, M. Pettersson, R. Fausto and M. Räsänen, J. Am. Chem. Soc., 2003, 125, 16188–16189 CrossRef PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, 0, S1–S19 RSC.
- W. T. Borden, WIREs Comput. Mol. Sci., 2016, 6, 20–46 CrossRef CAS.
- G. Wentzel, Z. Phys., 1926, 38, 518–529 CrossRef.
- H. A. Kramers, Z. Phys., 1926, 39, 828–840 CrossRef.
- L. Brillouin, C. R. Acad. Sci., 1926, 183, 24–26 Search PubMed.
- J. B. Rommel, T. P. M. Goumans and J. Kästner, J. Chem. Theory Comput., 2011, 7, 690–698 CrossRef CAS PubMed.
- A. Kuppermann and D. G. Truhlar, J. Am. Chem. Soc., 1971, 93, 1840–1851 CrossRef CAS.
- R. T. Skodje, D. G. Truhlar and B. C. Garrett, J. Phys. Chem., 1981, 85, 3019–3023 CrossRef CAS.
- J. Kästner, Chem.–Eur. J., 2013, 19, 8207–8212 CrossRef PubMed.
- H. Rostkowska, L. Lapinski and M. J. Nowak, Phys. Chem. Chem. Phys., 2018, 20, 13994–14002 RSC.
- A. K. Eckhardt, D. Gerbig and P. R. Schreiner, J. Phys. Chem. A, 2018, 122, 1488–1495 CrossRef CAS PubMed.
- D. Ley, D. Gerbig, J. P. Wagner, H. P. Reisenauer and P. R. Schreiner, J. Am. Chem. Soc., 2011, 133, 13614–13621 CrossRef CAS PubMed.
- D. Ley, D. Gerbig and P. R. Schreiner, Chem. Sci., 2013, 4, 677–684 RSC.
- P. R. Schreiner and H. P. Reisenauer, Angew. Chem., Int. Ed., 2008, 47, 7071–7074 CrossRef CAS PubMed.
- A. K. Eckhardt and P. R. Schreiner, Angew. Chem., Int. Ed., 2018, 57, 5248–5252 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, detailed spectra and signal assignments, computational details coordinates, and absolute energies of all computed structures. See DOI: 10.1039/c8sc03720e |
| This journal is © The Royal Society of Chemistry 2019 |