DOI:
10.1039/C8SC03514H
(Edge Article)
Chem. Sci., 2019,
10, 743-751
Elucidating the molecular mechanisms of Criegee-amine chemistry in the gas phase and aqueous surface environments†
Received
7th August 2018
, Accepted 24th October 2018
First published on 24th October 2018
Abstract
There is now an evolving body of evidence suggesting that high molecular weight oligomers from the ozonolysis of alkenes play an important role in new particle formation in the atmosphere. Using high-level quantum chemical calculations and Born Oppenheimer Molecular Dynamics (BOMD) simulations, we suggest that the reactions of anti-substituted Criegee intermediates with amine, especially dimethylamine, could lead to oligomers, which may comprise an unexplored fraction of organic nitrogen-based aerosols in urban polluted environments. The quantum chemical calculations suggest that the barrier for a given Criegee-amine reaction in the gas phase decreases with increase in methyl substitution on the amine to such an extent that the dimethylamine reactions of CH2OO and anti-CH3CHOO occur barrierlessly. The BOMD simulation results suggest that at the air–water interface, which represents a unique reaction medium in the atmosphere, the anti-CH3CHOO–methylamine reaction occurs via multiple mechanisms, which are distinctly different from that in the gas phase. An important implication of these results is that the Criegee-amine chemistries may account for an appreciable fraction of aerosol particles in California's central valley, New York City and Paris areas where significant amounts of nitrogen-based aerosol particles have been detected, but their precise details are still not well understood. Alternatively, these chemistries could also serve as a potential source of the hydroxyl radical and hydrogen peroxide under tropospheric conditions.
Introduction
Understanding new particle formation (NPF) from low-volatile gas-phase precursors, which accounts for ∼50% of the aerosol number production in the troposphere, is a significant challenge.1–3 NPF in the atmosphere occurs in two distinct stages: nucleation of gaseous precursors to form a critical nucleus and subsequent growth of the critical nucleus into a larger mass. Species and mechanisms involved in NPF need to be well established to accurately assess the influence of aerosols on urban visibility, human health, and global climate.4 While sulfuric acid (H2SO4) has been frequently implicated in NPF events,5–7 there is now evidence suggesting that other atmospheric species, such as nitrogenous bases and highly oxidized organic compounds, also play an important role in aerosol formation.8–12 For example, an appreciable amount of fine particulate matter (PM2.5) in the Eastern United States is made up of ammonium nitrate.13–16 Alkylaminium ions and carboxylate ions are detected in the mass spectra of nanoparticles collected during NPF events in Hyytiala, Finland.17 Measurements in Tecamac, Mexico also predict an important role of organic species in particle growth.12 Recently, Arquero et al.18 performed experiments to examine the role of oxalic acid in NPF from vapor phase methanesulfonic acid, methylamine (CH3NH2), and H2O. The addition of water to the mixture of oxalic acid and CH3NH2 was found to enhance particle formation by an order of magnitude. The particle formation and initial growth processes over coastal regions are shown to be dominated by iodine oxoacids and iodine oxide vapors.19,20
Ammonia (NH3) and alkylamines are emitted from various sources, including biological processes in the ocean, animal husbandry, agricultural fertilizers, biomass burning and industrial emissions.21 Laboratory experiments have shown that amines are more efficient than NH3 in enhancing particle formation.22–26 For example, experiments using the CLOUD chamber at CERN have demonstrated that dimethylamine ((CH3)2NH) concentrations exceeding three parts per trillion by volume enhance the NPF rate by more than 3 orders of magnitude relative to that seen with NH3.22 Although NH3 and alkylamines are highly volatile, both participate readily in multiphase reactions with organic species, which impact the aerosol nucleation and growth processes.8,9 The role of multiphase reactions of basic species such as acid–base neutralization, interaction with carbonyls, and particle-phase oxidation reactions in the growth of secondary organic aerosols (SOA) in the atmosphere is well documented in the literature.27 For example, recent quantum chemical calculations28,29 suggest that though amines are at least an order of magnitude less abundant than NH3, their gas-phase complexes with inorganic acids (e.g., methanesulfonic acid and nitric acid) are relatively more abundant. Furthermore, the Born–Oppenheimer Molecular Dynamics (BOMD) simulations suggest that the NH3/amine-induced acid–base chemistries at the air–water interface occur on the time scale of femtoseconds (fs) to picoseconds (ps).28,29 The time scale of these interfacial chemistries is found to depend upon the nature of an acid as well as that of an alkylamine. Though a variety of species have been shown to play a role in NPF events, the involvement of Criegee-amine chemistries in the nucleation and growth of SOA is yet to be considered. Criegee intermediates are transient species that are formed in olefin ozonolysis.30 They impact the tropospheric budgets of OH radicals, organic acids, hydroperoxides, nitrates, sulfates and particulate matter.31,32 In the troposphere, Criegee intermediates participate in various unimolecular and bimolecular chemistries.31,32 Though various bimolecular Criegee reactions have been studied experimentally and theoretically,31,32 the Criegee-amine reactions are yet to be explored both in the gas phase as well as at the air–water interface. Interestingly, the bimolecular reactions of Criegee intermediates with peroxyl and hydroperoxyl radicals in the context of NPF events have been previously discussed.33–35 However, Criegee-amine chemistries have never been explored before from that perspective. It is important to mention here that the Criegee-NH3 reaction has been studied before.36,37 Unfortunately, this reaction was suggested to be tropospherically irrelevant based on computational kinetic analysis.
Herein we apply quantum chemical calculations and BOMD simulations to study the Criegee-amine chemistries in the gas phase and at the air–water interface. The air–water interface is a simplistic representative of aqueous aerosols, such as clouds, fog, thin films, water microdroplets or aqueous sea salt particles and plays an important role in atmospheric and environmental processes.38–46 Many chemical reactions at the aqueous surface proceed faster and sometimes via different mechanisms than the corresponding processes in bulk water or the gas phase, as illustrated by oxidation processes of trace gases by ozone and singlet oxygen in water film surfaces,36 and hydration and hydrosulfidation of Criegee intermediates on the water droplet.42,45 The BOMD simulations capture bond forming and bond breaking events and thus may play a crucial role in establishing the initial stages of the Criegee-amine-based NPF events in the troposphere. The results from our gas-phase calculations suggest that methyl substituents in amines tune the barrier for a Criegee-amine reaction to such an extent that the Criegee-dimethylamine reaction occurs barrierlessly in the case of CH2OO and anti-CH3CHOO. These results not only help in identifying key reactivity determinants of Criegee-amine chemistries, but also point to the fact that existing atmospheric models need to be revised to incorporate facile Criegee-amine reactions. The BOMD simulation data reveal that the anti-CH3CHOO–CH3NH2 reaction at the air–water interface occurs on a picosecond (ps) time scale. Unlike the gas phase reaction, the interfacial anti-CH3CHOO–CH3NH2 reaction follows multiple stepwise mechanisms. This adds to an emerging library of the interfacial Criegee processes that occur on a ps time scale.40,42,44–48
Methods
(i) Gas-phase electronic structure calculations
All quantum chemical calculations reported here were performed using the Gaussian 09 (ref. 49) suite of programs for electronic structure and property calculations. The gas-phase reactions of the simplest Criegee intermediates (CH2OO), anti-CH3CHOO, syn-CH3CHOO and (CH3)2COO with NH3, CH3NH2, and (CH3)2NH were examined. The stationary points on all the reaction profiles were fully optimized at the M062X50/aug-cc-PVTZ51 level of theory. The energetics of these gas-phase reactions were further improved by performing single-point energy calculations at the CCSD(T)52/aug-cc-PVTZ level, for which the M062X/aug-cc-PVTZ optimized geometries were used. Harmonic vibrational frequency analysis at the M062X/aug-cc-PVTZ theoretical level was performed to confirm the authenticity of stationary points in all cases.
(ii) Born–Oppenheimer molecular dynamics simulations
We examined the reaction between anti-CH3CHOO and CH3NH2 at the air–water interface using the BOMD simulations based on the DFT method as implemented in the CP2K53 program package. The selection of anti-CH3CHOO in the present work is driven by the fact that larger Criegee intermediates (>C1) remain dynamically stable on the water surface and are likely to react with non-water trace gases in the troposphere.44 The droplet system is comprised of 191 water molecules, one anti-CH3CHOO molecule and one CH3NH2 molecule. It is important to mention here that if a bimolecular reaction on the water droplet has to be observed, both reactants should not react with the water molecules of the droplet. Otherwise, the probability of that bimolecular reaction would be significantly reduced. In the present work, we considered anti-CH3CHOO and CH3NH2 for a bimolecular reaction at the air–water interface because the Criegee intermediate (anti-CH3CHOO)44 and CH3NH2,54 when adsorbed individually on the water droplet, do not react with surface water molecules. As a result, both these precursors may react with each other on the water droplet. The configuration of the water droplet was first obtained by performing classical MD simulations for 5 ns with COMPASS force field, followed by 5 ps BOMD simulations. Subsequently, the anti-CH3CHOO–CH3NH2 complex was placed on the surface of a water droplet and the entire system was studied using the BOMD simulation, where the atomic forces were calculated within the DFT framework. To avoid the effects of initial configurations, we considered 25 different starting geometries for the anti-CH3CHOO–CH3NH2 complex on the water droplet, in which the distance between anti-CH3CHOO and CH3NH2 was varied from 2.0 to 3.7 Å and the complex was placed on different parts of the water droplet. The radius of the water droplet in our system was ∼10.5 Å. A cubic simulation box of side 35 Å was used. This translated into the smallest distance of ∼14 Å between the adjacent periodic images of the water droplet. The resulting box size was found to be large enough to eliminate any interaction between the adjacent periodic images. The system was fully relaxed using a DFT method prior to the BOMD simulations, in which the exchange and correlation interaction is treated with the Becke–Lee–Yang–Parr (BLYP)55,56 functional. The Grimme's dispersion correction method57,58 was applied to account for the weak dispersion interactions (BLYP-D3). A double-ζ Gaussian basis set combined with an auxiliary basis set59 and the Goedecker–Teter–Hutter (GTH) norm-conserved pseudopotentials60 were adopted to treat the valence and the core electrons, respectively. An energy cutoff of 280 Rydberg was imposed for the plane wave basis set, while a 40 Rydberg cutoff was used for the Gaussian basis set. The BOMD (BLYP-D3) simulations were carried out in the constant volume and temperature (NVT) ensemble, with the Nose–Hoover chain method for controlling the temperature (300 K) of the system. The integration step is set as 1 fs, which has been previously shown to achieve sufficient energy conservation for the water system.40,42–47
Results and discussion
Criegee-amine interactions in the gas phase
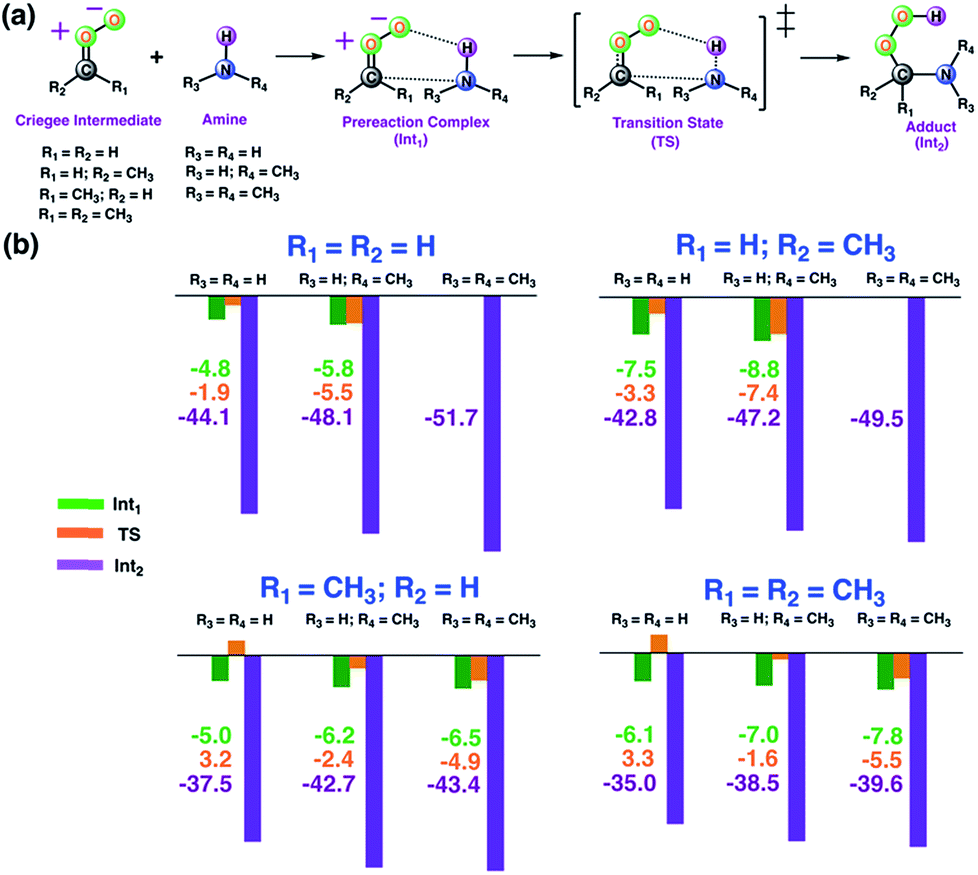
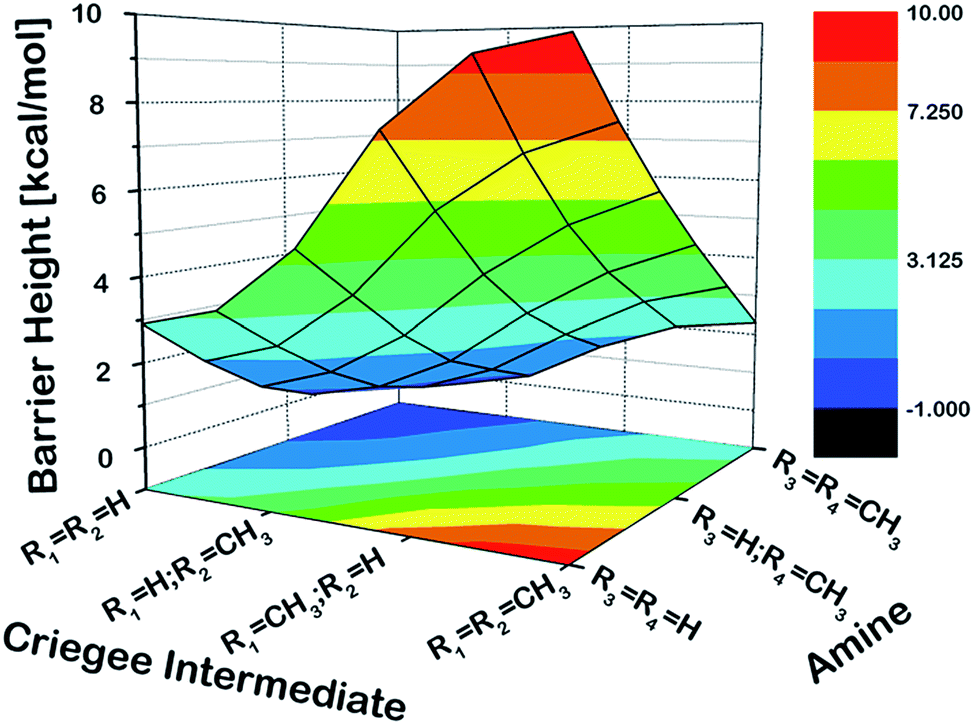
As a first step, we examined the Criegee-amine reactions in the gas phase. Specifically, we studied the gas-phase reactions of CH2OO, anti-CH3CHOO, syn-CH3CHOO and (CH3)2COO with NH3, CH3NH2, and (CH3)2NH (Fig. 1a) at the CCSD(T)/aug-cc-pVTZ//M06-2X/aug-cc-pVTZ level of theory. As pointed out in the Introduction section, although the Criegee-NH3 reaction has been previously studied,36,37 we re-examined the same reaction here in order to facilitate comparative analysis of the Criegee-NH3 and Criegee-amine reactions at the same theoretical footing. The CCSD(T)/aug-cc-pVTZ//M06-2X/aug-cc-pVTZ calculated reaction profiles for all these reactions are given in Fig. 1b. The optimized geometries of key species involved in these reactions are given in Table S1.† The results show that the gas-phase Criegee-NH3/amine reactions are concerted exergonic reactions, which are mediated by prereaction and postreaction complexes. The basic mechanism of the gas-phase reaction involves the heterolytic addition of the polar H–N bonds of NH3/amines across the –COO functionality of Criegee intermediates. The CCSD(T)//M06-2X calculated barrier for the CH2OO–NH3 reaction is 2.9 kcal mol−1, which is in good agreement with previously reported CCSD(T) numbers of 3.99–5.40 kcal mol−1. The CH2OO–NH3 reaction is predicted to be 44.1 kcal mol−1 exothermic, which is again consistent with the recently reported CCSD(T)/CBST and CCSDT(Q)/CBS values of 43.7 and 42.1 kcal mol−1, respectively.37 The calculated barriers for the NH3 reactions of CH2OO and anti-CH3CHOO are smaller than those for the syn-CH3CHOO and (CH3)2COO reactions. This is consistent with the well-established notion that the Criegee reactivity towards the bimolecular reactions is determined by the nature and location of substituents.31,32,40 As we move along the NH3 → CH3NH2 → (CH3)2NH series, the barriers for all the Criegee reactions are consistently lowered. This points to an interesting trend: the barriers for the Criegee-NH3/amine reactions correlate inversely with the number of methyl substituents on a given amine (Fig. 2). The Criegee-(CH3)2NH reactions have the lowest barriers among all the amine reactions studied. Interestingly, the impact of this amine-tuned reactivity of the bimolecular Criegee reactions is so pronounced that both the CH2OO–(CH3)2NH and anti-CH3CHOO–(CH3)2NH reactions occur barrierlessly whereas the barriers for the syn-CH3CHOO–(CH3)2NH and (CH3)2COO–(CH3)2NH are 6.6 and 7.1 kcal mol−1 lower than those for the syn-CH3CHOO–NH3 and (CH3)2COO–NH3, respectively. Furthermore, the exothermicities associated with the Criegee-NH3/amine reactions are consistently enhanced as we move along the NH3 → CH3NH2 → (CH3)2NH series. The Criegee-(CH3)2NH reactions have 4.6–7.6 kcal mol−1 larger exothermicities than the analogous Criegee-NH3 reactions. Considering that the barrierless bimolecular Criegee reactions (e.g., Criegee-HNO3 and Criegee-HCOOH) have been previously shown to have rate constants on the order of ∼10−10 cm3 per s per molecule,61–63 both the CH2OO–(CH3)2NH and anti-CH3CHOO–(CH3)2NH reactions are likely to have at least similar rate constants.
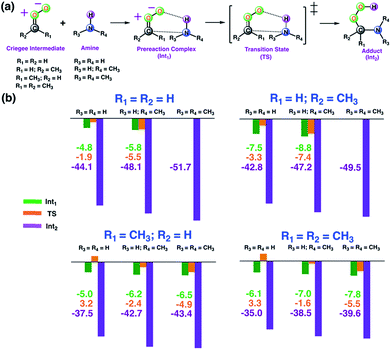 |
| | Fig. 1 (a) General description of the gas-phase Criegee-amine reactions studied here. (b) The CCSD(T)/aug-cc-pVTZ//M06-2X/aug-cc-pVTZ calculated energies (in kcal mol−1 units) of key stationary points for the gas-phase Criegee-amine reactions at 298.15 K and 1 atm. | |
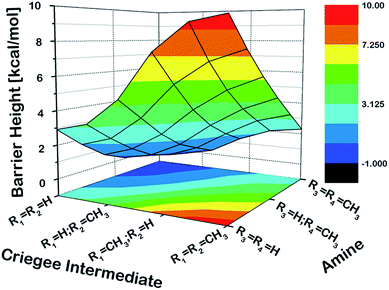 |
| | Fig. 2 The three-dimensional plot showing the CCSD(T)/aug-cc-pVTZ//M06-2X/aug-cc-pVTZ calculated barrier heights (kcal mol−1) for the gas-phase reactions of various Criegee intermediates with (R3)(R4)NH at 298.15 K and 1 atm. Since the CH2OO–(CH3)2NH and anti-CH3CHOO–(CH3)2NH reactions occur barrierlessly, we have used zero to represent their barrier heights here. | |
Criegee-amine interactions at the air–water interface
Considering that the aqueous surfaces, which are ubiquitous in fogs, cloud waters, microdroplets and aerosols, provide interesting reaction media for atmospheric chemistries,38–41 we next examined the reaction between anti-CH3CHOO and CH3NH2 on a water droplet of 191 water molecules within the framework of the BOMD simulations. The selection of these particular precursors for the interfacial reaction is inspired by the fact that recent studies predict them to remain undissociated on the water droplet,44,54 thus indicating the possibility of an interfacial reaction between them.
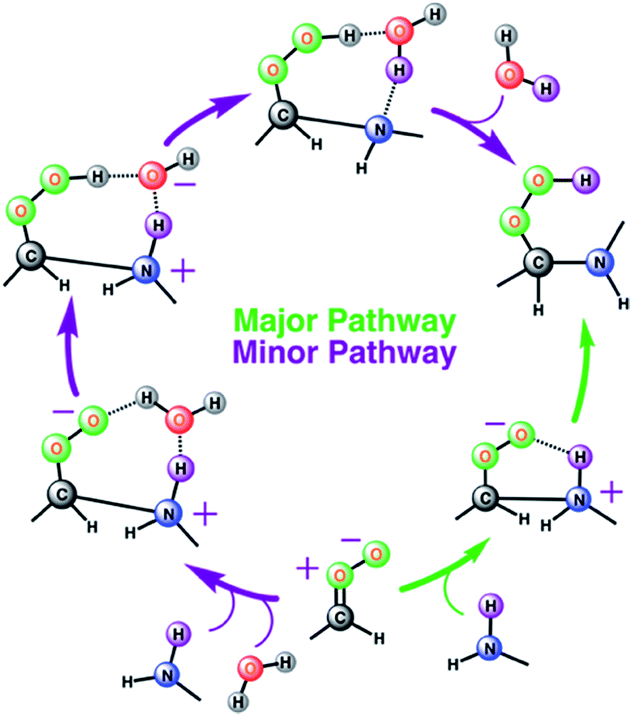
The results from the BOMD simulations suggest that the interfacial anti-CH3CHOO–CH3NH2 reaction occurs on a ps time scale. This adds to a growing library of the interfacial bimolecular Criegee reactions, which occur on the ps time scale.40,42,44–48 Furthermore, the BOMD simulations reveal that the air–water interface and gas-phase mechanisms for the anti-CH3CHOO–CH3NH2 reaction are distinctly different: unlike the gas phase reaction, the reaction on the aqueous surface follows multiple stepwise mechanisms. Both the direct reaction between anti-CH3CHOO and CH3NH2, in which the interfacial water molecules do not directly participate in the reaction, and the interfacial water molecule-mediated addition reaction follow stepwise mechanisms. 25 different simulations were run to probe the mechanism of the anti-CH3CHOO–CH3NH2 reaction on the water droplet. The former pathway is observed in 22 simulations whereas the latter pathway is seen in only 3 simulations. This suggests that the direct reaction between anti-CH3CHOO and CH3NH2 is the dominant chemistry on the water surface whereas the water-mediated reaction is only a minor channel. Details of these addition pathways are provided below:
Direct reaction between anti-CH3CHOO and CH3NH2.
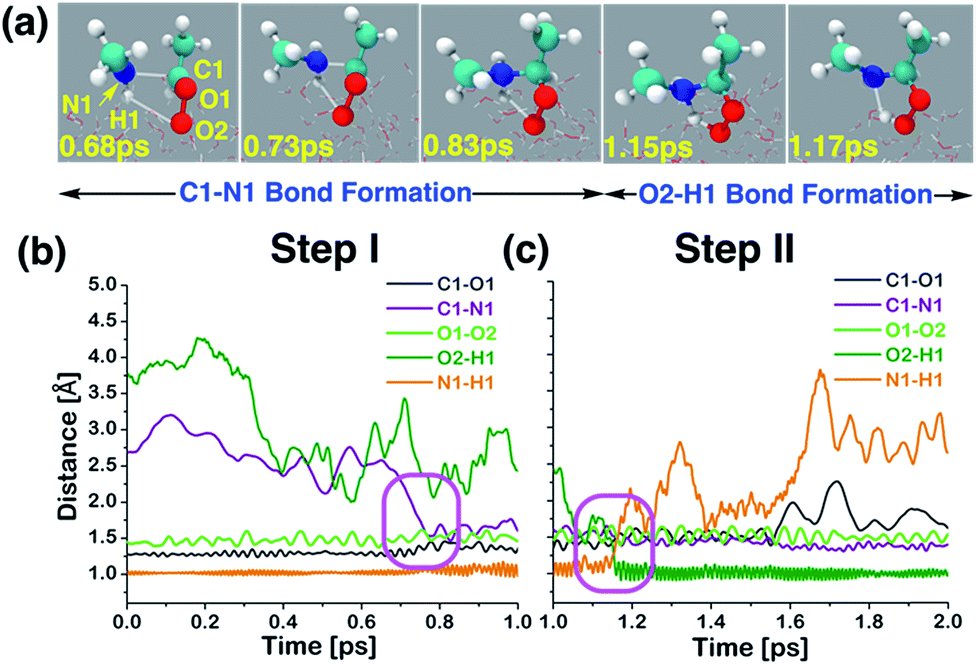
At the air–water interface, the direct reaction between anti-CH3CHOO and CH3NH2 comprises two steps: (i) C1–N1 bond formation (anti-CH3CHOO + CH3NH2 → anti-CH3CHOO−–CH3NH2+) and (ii) N1 → O2 proton transfer (anti-CH3CHOO−–CH3NH2+ → C(CH3)(H)(OOH)(CH3NH)). Details of this direct reaction are shown in Scheme 1, Fig. 3 and Movie S1.† The prereaction-like complex for the C1–N1 bond forming step is formed at 0.68 ps. At that point, the C1–N1 bond is 2.45 Å long. The CH3NH2 is hydrogen-bonded to the anti-CH3CHOO, as indicated by the H1–O2 bond length of 2.81 Å. See Fig. S1† for additional details. The transition state-like complex for the C1–N1 bond formation is observed at 0.73 ps. The C1–N1 bond has now shrunk to 1.93 Å whereas the hydrogen bonding interaction (O2–H1) between anti-CH3CHOO and CH3NH2 is of 2.71 Å length. The C1–O1 and O1–O2 bonds are 1.27 and 1.50 Å long, respectively. At 0.78 ps, the formation of the C1–N1 bond is complete; the C1–N1 is now 1.53 Å long whereas the C1–O1 bond is now converted into a pure single bond of 1.43 Å length. The O2–H1 hydrogen bond and the O1–O2 bond are 2.07 and 1.48 Å long, respectively. This results in the formation of a Zwitterion-type intermediate, anti-CH3CHOO−–CH3NH2+, which remains stable for 0.37 ps. Subsequently, proton transfer from the N1 of CH3NH2 to the O2 of anti-CH3CHOO occurs, resulting in the formation of an adduct, C(CH3)(OOH)(CH3NH). The activation complex for proton transfer is formed at 1.15 ps. At that point, the N1–H1 and O2–H1 bonds are nearly equidistant, i.e., the N1–H1 is 1.78 Å long whereas the O2–H1 bond is 1.26 Å long. The C1–N1 bond (1.51 Å) remains intact. The C1–O1 bond is 1.45 Å long whereas the O1–O2 bond is lengthened from 1.48 Å at 0.78 ps to 1.62 Å at 1.15 ps. The proton transfer is deemed complete at 1.17 ps; the O2–H1 bond is now transformed into a pure covalent bond of 0.91 Å length whereas the N1–H1 bond has become a hydrogen bonding interaction of 1.78 Å length. The C1–O1 and O1–O2 bonds are now 1.55 and 1.44 Å long, respectively. We have run the BOMD simulations up to 20 ps. The adduct (C(CH3)(OOH)(CH3NH)) remains intact at the air–water interface over the simulated time scale (Fig. S2†) where it is hydrogen bonded to the interfacial water molecules. The snapshots of the adduct as a function of time are shown in Fig. 4. The adduct forms 3.3 average number of hydrogen bonds with the surface water molecules. Precisely, O1, O2 and H1 form one hydrogen bond with the interfacial water molecules on average. The methyl groups or N atom hardly forms any hydrogen bonding interaction with the nearby water molecules.
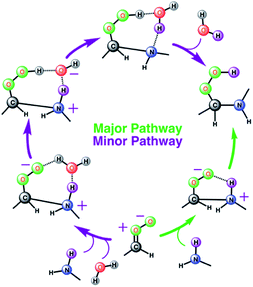 |
| | Scheme 1 Schematic representation of two stepwise pathways predicted from the BOMD simulation-based population analysis of the reaction between anti-CH3CHOO and with methylamine on the water droplet of 191 water molecules. For further details, see also Fig. 3–5. | |
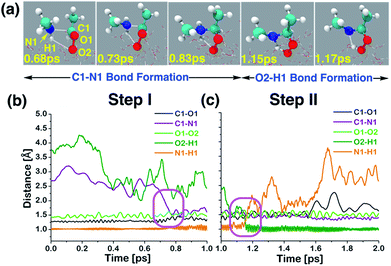 |
| | Fig. 3 (a) Snapshot structures taken from the BOMD simulations, which illustrate the stepwise mechanism for direct adduct formation between anti-CH3CHOO and methylamine (CH3NH2) on the water droplet of 191 water molecules. (b) Time evolution of key bond distances (C1–O1, C1–N1, O1–O2, O2–H1, and N1–H1) involved in C1–N1 bond formation. (c) Time evolution of key bond distances involved in proton transfer from CH3NH2 to the terminal oxygen of anti-CH3CHOO. Pink rectangles illustrate the formation of C1–N1 and O2–H1 bonds. See also Fig. S1.† | |
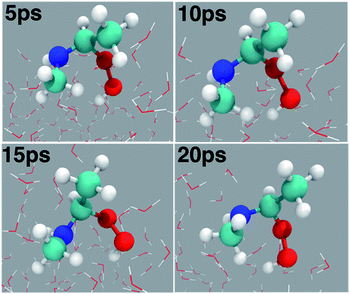 |
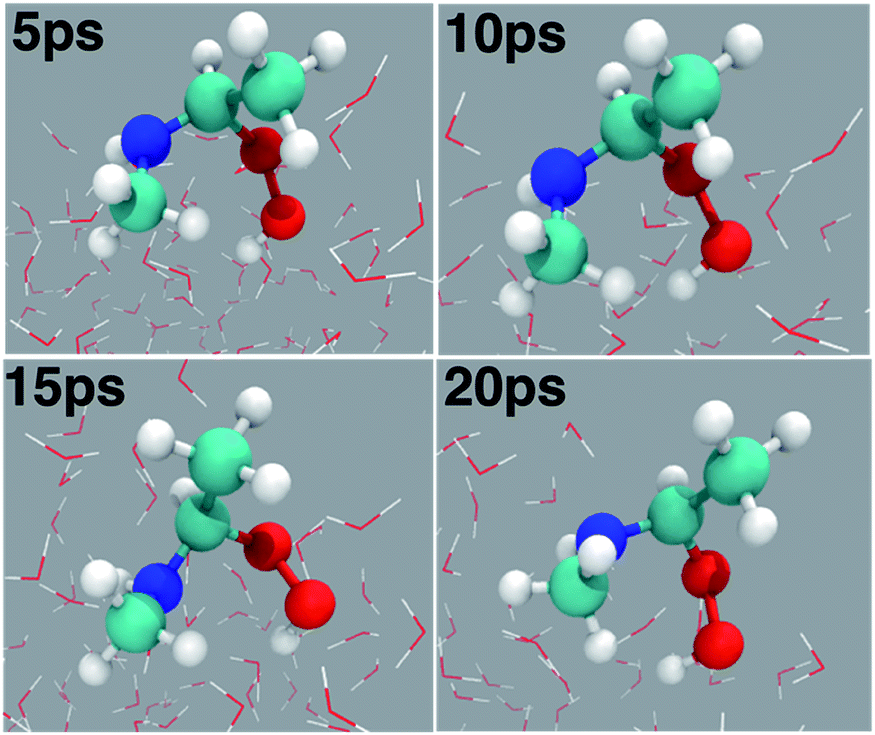
| | Fig. 4 Snapshot structures of the adduct (C(CH3)(OOH)(CH3NH)) taken from the BOMD simulations over the 20 ps time scale. | |
Interfacial water-mediated reaction between anti-CH3CHOO and CH3NH2.
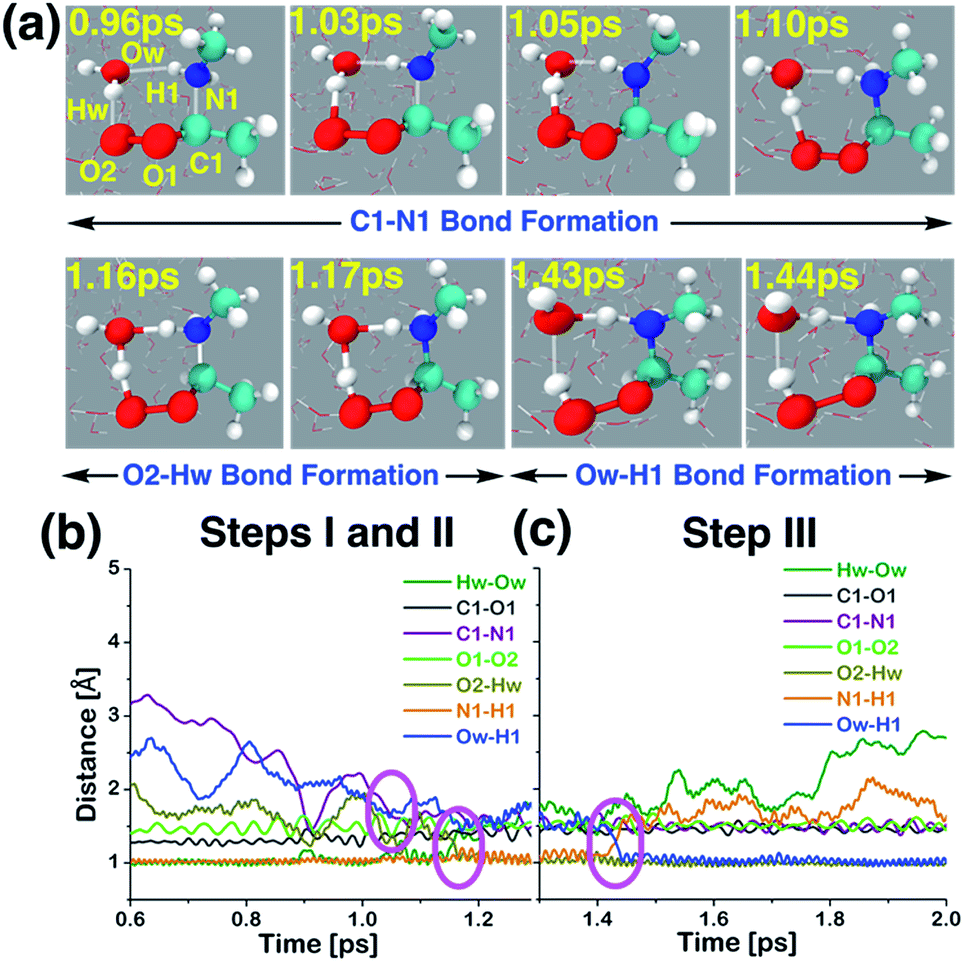
The interfacial water-mediated adduct formation between anti-CH3CHOO and CH3NH2 involves a single water molecule and occurs in three steps: (i) C1–N1 bond formation (anti-CH3CHOO + CH3NH2 + H2O·W190 → anti-CH3CHOO−–CH3NH2+ + H2O·W190), (ii) proton transfer from the interfacial water molecule to the terminal Criegee oxygen (O2) (anti-CH3CHOO−–CH3NH2+ + H2O·W190 → C(CH3)(H)(OOH)(CH3NH2+)(OH−) + W190), and (iii) proton transfer from the N1 of CH3NH2 to the interfacial water molecule (C(CH3)(H)(OOH)(CH3NH2+)(OH−) + W190 → C(CH3)(H)(OOH)(CH3NH) + W191). Here W190 represents 190 water molecules of the aqueous interface used in the BOMD simulations. Details of this multi-step addition reaction are illustrated in Scheme 1, Fig. 5 and Movie S2.† The prereaction complex for the first step is formed at 0.96 ps. The C1–N1 bond has now shrunk from 3.20 Å at 0 ps to 2.10 Å. The C1–O1 and O1–O2 bonds are 1.34 and 1.47 Å long, respectively. At that point, the interfacial water molecule is hydrogen-bonded to the N1 of CH3NH2 and the O2 of anti-CH3CHOO, i.e., Ow–H1 = 2.16 Å; O2–Hw = 1.75 Å. Here Ow and Hw are the oxygen and hydrogen atoms of the interfacial water molecule that are involved in the reaction. See Fig. S3† for additional details. The transition state-like complex for the C1–N1 bond formation is observed at 1.03 ps. The C1–N1 bond is now 1.92 Å long whereas the Ow–H1 and O2–Hw hydrogen bonds are 1.71 and 1.62 Å long, respectively. The C1–O1 and O1–O2 bonds are 1.29 and 1.67 Å long, respectively. The C1–N1 bond formation is deemed complete at 1.05 ps; the C1–N1 bond is now 1.63 Å long. H1 is still attached to N1 (N1–H1 = 1.02 Å), which further supports the formation of the Zwitterion-type anti-CH3CHOO−–CH3NH2+ intermediate. The C1–O1 and O1–O2 bonds are 1.35 and 1.38 Å long, respectively. This intermediate remains stable for 0.11 ps. The shorter lifetime of the Zwitterionic intermediate in the water-mediated reaction may be due to the fact that the water-mediated reactions occur faster due to a proton shuttling mechanism. Proton transfer from the Ow of the interfacial water molecule to the O2 of anti-CH3CHOO−–CH3NH2+ then occurs, which results in the formation of C(CH3)(H)(OOH)(CH3NH2+)(OH−). The transition state-like complex for the Ow → O2 proton transfer is formed at 1.16 ps; Ow–Hw = 1.28 Å and O2–Hw = 1.30 Å. The Ow is still hydrogen-bonded to H1 in C(CH3)(H)(OOH)(CH3NH2+)(OH−), i.e., Ow–H1 = 1.54 Å and N1–H1 = 1.11 Å. The C1–O1 (1.44 Å) and O1–O2 (1.60 Å) bonds are pure single covalent bonds at this stage. The formation of the C(CH3)(H)(OOH)(CH3NH2+)(OH−) intermediate is complete at 1.17 ps as the Ow–Hw bond (1.47 Å) has now changed into a hydrogen bonding interaction whereas the O2–Hw bond (1.04 Å) has become a proper single bond. The hydrogen bond involving interfacial Ow and H1 in C(CH3)(H)(OOH)(CH3NH2+)(OH−) is still intact, i.e., Ow–H1 = 1.53 Å and N1–H1 = 1.11 Å. This intermediate remains stable for ∼0.26 ps and then eventually decomposes into C(CH3)(H)(OOH)(CH3NH) and H2O. In this final step, proton transfer from N1 to Ow takes place, which regenerates the interfacial water molecule by converting (CH3CHOOH)(CH3NH2+)(OH−) into C(CH3)(H)(OOH)(CH3NH)(H2O). The transition state-like complex for the N1 → Ow is formed at 1.43 ps, in which both the N1–H1 and Ow–H1 bonds are equidistant, i.e., N1–H1 = 1.28 Å and Ow–H1 = 1.28 Å. The Ow–Hw bond is now a hydrogen bonding interaction (1.81 Å) whereas the C1–N1 bond (1.57 Å) is still intact. At 1.44 ps, the formation of the interfacial water-mediated C(CH3)(H)(OOH)(CH3NH) adduct is complete. At that point, the Ow–H1 is 1.02 Å long whereas the N1–H1 bond is 1.59 Å long. The time evolution of C1–N1, C1–O1, O1–O2, and O2–Hw further supports the formation of C(CH3)(H)(OOH)(CH3NH).
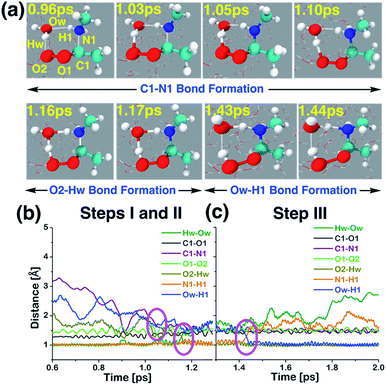 |
| | Fig. 5 (a) Snapshot structures taken from the BOMD simulations, which illustrates the interfacial water-mediated stepwise mechanism for the adduct formation between anti-CH3CHOO and methylamine (CH3NH2) on the water droplet of 191 water molecules. (b) Time evolution of key bond distances involved in two chemical events, which describe the C–N bond formation (C1–N1) between anti-CH3CHOO and CH3NH2, and the proton transfer from the interfacial water molecule to the terminal oxygen of anti-CH3CHOO (i.e., the O2–Hw bond formation), respectively. (c) Time evolution of key bond distances involved in the proton transfer from CH3NH2 to the interfacial water molecule (i.e., the Ow–H1 bond formation). See also Fig. S3.† | |
Atmospheric implications.
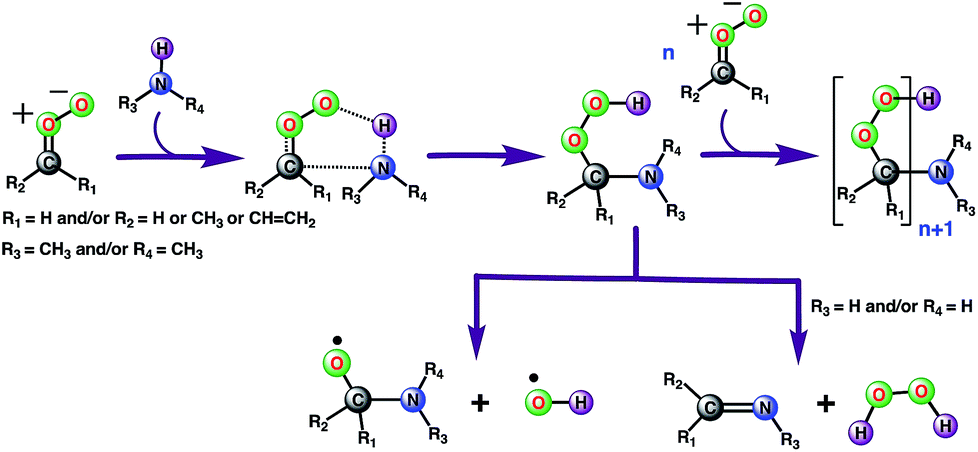
Ozonolysis of alkenes is an important source of SOA in the atmosphere.1–4,64 High-molecular weight oligomers have been recognized as major constituents of SOA from ozonolysis of alkenes.33,34,65–69 However, the identity of these oligomers and their formation mechanisms are yet to be fully established. Our gas-phase calculations and air–water interface inclusive simulations suggest that anti-substituted Criegee intermediates may react with amines, preferably dimethylamine, to form oligomers (Scheme 2). In this mechanism, an amine acts as an agent to initiate oligomerization of Criegee intermediates by reacting with an initial one to form a peroxide functionality, which further reacts with Criegee intermediates. Though the role of bimolecular reactions of Criegee intermediates with carboxylic acids, carbonyls, peroxy and hydroperxy radicals in the particle formation is previously discussed in the literature,31,32 this is the first time the involvement of the Criegee-amine chemistries in aerosol particle formation is being suggested. Though the Criegee-amine reactions in the gas phase may face strong competition from the Criegee-water reaction, these reactions are expected to be favored on the aqueous surfaces, which are ubiquitous in the atmosphere and provide a unique reaction medium. Our recent BOMD simulations suggest that larger Criegee intermediates, >C1 at the aqueous surface do not react with water,44 which makes them available for reactions with other potential precursors. The present BOMD simulations indicate that the N–H bonds in CH3NH2 become sufficiently polar on the water droplet so that it adds to the COO functionality of anti-CH3CHOO and results in C(CH3)(H)(OOH)(CH3NH) over the ps time scale. The reaction between anti-CH3CHOO and CH3NH2 on the water droplet is favored over the reaction between anti-CH3CHOO and H2O because amines are more reactive than water.70 The interfacial water molecules stabilize oligomers originating from the Criegee-amine reactions via hydrogen-bonding and thus, may play a role in lowering their vapor pressures and enhancing the rate of particle formation. Our viewpoint is indirectly supported by laboratory experiments demonstrating that although amines have concentrations at least an order of magnitude lower than that of NH3 in the atmosphere,21 they are more effective than NH3 in enhancing particle formation.22–26 Our results may help in better understanding the aerosol formation in the fog waters of California's central valley, and in polluted urban environments such as New York and Paris where significant nitric acid-amine chemistries have been recently suggested to play a role in the aerosol particle formation in these areas.71–73 In the New York City area, amines are likely emitted from marine sources whereas in Paris, the source of amines is the agricultural activities around the City area. The intense traffic in these urban regions further support the role of the Criegee-amine interactions in the particle formation. Though the nitric acid-amine chemistries have been recently suggested to play a role in the aerosol particle formation in these areas,29 the current results reveal an unexplored source of organic nitrogen.
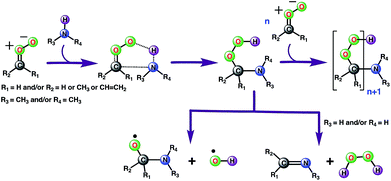 |
| | Scheme 2 Plausible mechanism for the formation of low-volatility oligomers containing stabilized Criegee intermediates as repeat units during the olefin ozonolysis. The oligomer formation involves the sequential addition of Criegee intermediates to amines (preferably dimethylamine because of its greater reactivity). An alternate channel suggests the possibility of the Criegee-amine reaction being a source of the tropospheric hydroxyl radical. | |
Alternatively, the C(CH3)(H)(OOH)(CH3NH) adduct formed from the reaction between anti-CH3CHOO and CH3NH2 in the gas phase may decompose into C(CH3)(H)(CH3NH)(O) and the OH radical (Scheme 2). The bond dissociation energy for the O–OH bond in Criegee-derived hydroperoxides is ∼40 kcal mol−1.74–76 Considering that the adduct in the Criegee-amine reactions is formed with an additional energy of 38.5–51.9 kcal mol−1, the O–OH bond breakage in C(CH3)(H)(OOH)(CH3NH)-type adducts may occur under tropospheric conditions, which makes these Criegee-amine chemistries a potential source of the tropospheric OH radical. The C(CH3)(H)(OOH)(CH3NH) adduct formed from the Criegee-CH3NH2/NH3 reaction may also decompose into H2O2 and C(CH3)(H)(CH3N)/C(CH3)(H)(H2N) due to the presence of two polar N–H bonds in both CH3NH2 and NH3 (Scheme 2). Both OH radical and H2O2 forming paths from bimolecular Criegee reactions have previously been shown to involve similar energetic demands,74–76 and thus, are equally likely to occur in the troposphere. Since the H2O2 forming pathway requires the presence of two polar N–H bonds in a given amine, such a mechanistic option in the case of the Criegee-(CH3)2NH reactions will not be available.
Conclusions
In summary, we have elucidated the molecular mechanisms of the Criegee-amine reactions both in the gas phase and at the air–water interface using quantum chemical calculations and BOMD simulations, respectively. The quantum chemical calculations suggest that the barrier for a Criegee-amine reaction decreases with the extent of methyl substitution in the amine. This barrier lowering is so pronounced that the dimethylamine reactions of CH2OO and anti-CH3CHOO occur barrierlessly. Though the Criegee-ammonia reactions have been found to be tropospherically insignificant, the facile nature of Criegee-dimethylamine reactions suggests that these chemistries may play a role in the new particle forming events under certain conditions and thus, need to be updated in the existing atmospheric models. Alternatively, these reactions could also serve as a potential source of the OH radical and hydrogen peroxide under tropospheric conditions. The air–water interface inclusive BOMD simulations reveal the diverse mechanistic pathways available for the otherwise simple looking addition reaction between anti-CH3CHOO and CH3NH2 on the aqueous surface. The reaction follows a stepwise mechanism, which may or may not be mediated by the interfacial water molecules and occurs on a picosecond time scale. Overall, these results suggest that the Criegee-amine interactions could contribute towards the organic fraction of the aerosol particles in the atmosphere.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the University of Nebraska Holland Computing Center.
References
-
Core Writing Team, Contribution of Working Groups I, II and III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, Climate Change 2007: Synthesis Report, ed. R. K. Pachauri and A. Reisinger, IPCC, Geneva, 2007 Search PubMed.
-
B. J. Finlayson-Pitts and J. N. Pitts Jr, Chemistry of the Upper and Lower Atmosphere-Theory Experiments and Applications, Academic Press, San Diego, 2000 Search PubMed.
-
J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics From Air Pollution to Climate Change, Wiley Interscience, New York, 2006 Search PubMed.
-
Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. IPCC, 2013, Climate Change 2013: The Physical Science Basis, ed. T. F. Stocker, D. Qin, G.-K.Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013 Search PubMed.
- R. J. Weber, G. Chen, D. D. Davis, R. L. Mauldin III, D. J. Tanner, F. L. Eisele, A. D. Clarke, D. C. Thornton and A. R. Bandy, J. Geophys. Res., 2001, 106, 24107 CrossRef CAS.
- R. J. Weber, J. J. Marti, P. H. McMurry, F. L. Eisele, D. J. Tanner and A. Jefferson, Chem. Eng. Commun., 1996, 151, 53 CrossRef CAS.
- R. J. Weber, J. J. Marti, P. H. McMurry, F. L. Eisele, D. J. Tanner and A. Jefferson, J. Geophys. Res., 1997, 102, 4375 CrossRef CAS.
- R. Zhang, A. Khalizov, L. Wang, M. Hu and W. Xu, Chem. Rev., 2012, 112, 1957 CrossRef CAS.
- R. Zhang, G. Wang, S. Guo, M. L. Zamora, Q. Ying, Y. Lin, W. Wang, M. Hu and Y. Wang, Chem. Rev., 2015, 115, 3803 CrossRef CAS PubMed.
- R. Y. Zhang, I. Suh, J. Zhao, D. Zhang, E. C. Fortner, X. X. Tie, L. T. Molina and M. J. Molina, Science, 2004, 304, 1487 CrossRef CAS PubMed.
- R. Zhang, L. Wang, A. F. Khalizov, J. Zhao, J. Zheng, R. L. McGraw and L. T. Molina, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17650 CrossRef CAS PubMed.
- J. N. Smith, M. J. Dunn, T. M. VanReken, K. Iida, M. R. Stolzenburg, P. H. McMurry and L. G. Huey, Geophys. Res. Lett., 2008, 35, L04808 Search PubMed.
-
J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics, Wiley, New York, 1998 Search PubMed.
- A. S. Ansari and S. N. Pandis, Environ. Sci. Technol., 1998, 32, 2706 CrossRef CAS.
- C. L. Blanchard, P. M. Roth, S. J. Tanenbaum, S. D. Ziman and J. H. Seinfeld, J. Air Waste Manage. Assoc., 2000, 50, 2073 CrossRef CAS.
- J. J. West, A. S. Ansari and S. N. Pandis, J. Air Waste Manage. Assoc., 1999, 49, 1415 CrossRef CAS.
- J. N. Smith, K. C. Barsanti, H. R. Friedli, M. Ehn, M. Kulmala, D. R. Collins, J. H. Scheckman, B. J. Williams and P. H. McMurry, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6634 CrossRef CAS.
- K. D. Arquero, R. B. Gerber and B. J. Finlayson-Pitts, Environ. Sci. Technol., 2017, 51, 2124 CrossRef CAS.
- C. D. O'Dowd, J. L. Jimenez, R. Bahreini, R. C. Flagan, J. H. Seinfeld, K. Hämeri, L. Pirjola, M. Kulmala, S. G. Jennings and T. Hoffmann, Nature, 2002, 417, 632 CrossRef.
- M. Sipilä, N. Sarnela, T. Jokinen, H. Henschel, H. Junninen, J. Kontkanen, S. Richters, J. Kangasluoma, A. Franchin, O. Peräkylä, M. P. Rissanen, M. Ehn, H. Vehkamäki, T. Kurten, T. Berndt, T. Petäjä, D. Worsnop, D. Ceburnis, V.-M. Kerminen, M. Kulmala and C. D. O'Dowd, Nature, 2016, 537, 532 CrossRef.
- X. Ge, A. S. Wexler and S. L. Clegg, Atmos. Environ., 2011, 45, 524 CrossRef CAS.
- J. Almeida, S. Schobesberger, A. Kürten, I. K. Ortega, O. Kupiainen-Määttä, A. P. Praplan, A. Adamov, A. Amorim, F. Bianchi, M. Breitenlechner, A. David, J. Dommen, N. M. Donahue, A. Downard, E. Dunne, J. Duplissy, S. Ehrhart, R. C. Flagan, A. Franchin, R. Guida, J. Hakala, A. Hansel, M. Heinritzi, H. Henschel, T. Jokinen, H. Junninen, M. Kajos, J. Kangasluoma, H. Keskinen, A. Kupc, T. Kurtén, A. N. Kvashin, A. Laaksonen, K. Lehtipalo, M. Leiminger, J. Leppä, V. Loukonen, V. Makhmutov, S. Mathot, M. J. McGrath, T. Nieminen, T. Olenius, A. Onnela, T. Petäjä, F. Riccobono, I. Riipinen, M. Rissanen, L. Rondo, T. Ruuskanen, F. D. Santos, N. Sarnela, S. Schallhart, R. Schnitzhofer, J. H. Seinfeld, M. Simon, M. Sipilä, Y. Stozhkov, F. Stratmann, A. Tomé, J. Tröstl, G. Tsagkogeorgas, P. Vaattovaara, Y. Viisanen, A. Virtanen, A. Vrtala, P. E. Wagner, E. Weingartner, H. Wex, C. Williamson, D. Wimmer, P. Ye, T. Yli-Juuti, K. S. Carslaw, M. Kulmala, J. Curtius, U. Baltensperger, D. R. Worsnop, H. Vehkamäki and J. Kirkby, Nature, 2013, 502, 359 CrossRef CAS.
- T. Kurtén, V. Loukonen, H. Vehkamaki and M. Kulmala, Atmos. Chem. Phys., 2008, 8, 4095 CrossRef.
- T. Berndt, F. Stratmann, M. Sipila, J. Vanhanen, T. Petaja, J. Mikkila, A. Gruner, G. Spindler, R. L. Mauldin III, J. Curtius, M. Kulmala and J. Heintzenberg, Atmos. Chem. Phys., 2010, 10, 7101 CrossRef CAS.
- H. Yu, R. McGraw and S.-H. Lee, Geophys. Res. Lett., 2012, 39, 1 CrossRef.
- W. A. Glasoe, K. Volz, B. Panta, N. Freshour, R. Bachman, D. R. Hanson, P. H. McMurry and C. Jen, J. Geophys. Res., 2015, 120, 1933 CAS.
- C. Qiu and R. Y. Zhang, Phys. Chem. Chem. Phys., 2013, 15, 5738 RSC.
- M. Kumar and J. S. Francisco, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12401 CrossRef CAS.
- M. Kumar, H. Li, X. Zhang, X. C. Zeng and J. S. Francisco, J. Am. Chem. Soc., 2018, 140, 6456 CrossRef CAS.
- R. Criegee, Angew. Chem., Int. Ed., 1975, 14, 745 CrossRef.
- D. L. Osborn and C. A. Taatjes, Int. Rev. Phys. Chem., 2015, 34, 309 Search PubMed.
- L. Vereecken, D. R. Glowacki and M. J. Pilling, Chem. Rev., 2015, 115, 4063 CrossRef CAS PubMed.
- A. Sadezky, R. Winterhalter, B. Kanawati, A. Rompp, B. Spengler, A. Mellouki, G. L. Bras, P. Chaimbault and G. K. Moortgat, Atmos. Chem. Phys., 2008, 8, 2667 CrossRef CAS.
- Y. Sakamoto, S. Inomata and J. Hirokawa, J. Phys. Chem. A, 2013, 117, 12912 CrossRef CAS.
- Y. Zhao, L. M. Wingen, V. Perraud, J. Greaves and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2015, 17, 12500 RSC.
- S. Jorgensen and A. Gross, J. Phys. Chem. A, 2009, 113, 10284 CrossRef CAS.
- J. P. Misiewicz, S. N. Elliott, K. B. Moore III and H. F. Schaefer III, Phys. Chem. Chem. Phys., 2018, 20, 7479 RSC.
- R. B. Gerber, M. E. Varner, A. D. Hammerich, S. Riikonen, G. Murdachaew, D. Shemesh and B. J. Finlayson-Pitts, Acc. Chem. Res., 2015, 48, 399 CrossRef CAS.
- A. R. Ravishankara, Science, 1997, 276, 1058 CrossRef CAS.
- J. Zhong, M. Kumar, J. S. Francisco and X. C. Zeng, Acc. Chem. Res., 2018, 51, 1229 CrossRef CAS PubMed.
- D. J. Donaldson and V. Vaida, Chem. Rev., 2006, 106, 1445 CrossRef CAS.
- C. Q. Zhu, M. Kumar, J. Zhong, L. Lei, J. S. Francisco and X. C. Zeng, J. Am. Chem. Soc., 2016, 138, 11164 CrossRef CAS.
- L. Li, M. Kumar, C. Q. Zhu, J. Zhong, J. S. Francisco and X. C. Zeng, J. Am. Chem. Soc., 2016, 138, 1816 CrossRef CAS.
- J. Zhong, M. Kumar, C. Q. Zhu, J. S. Francisco and X. C. Zeng, Angew. Chem., Int. Ed., 2017, 56, 7740 CrossRef CAS.
- M. Kumar, J. Zhong, X. C. Zeng and J. S. Francisco, Chem. Sci., 2017, 8, 5385 RSC.
- M. Kumar, J. Zhong, X. C. Zeng and J. S. Francisco, J. Am. Chem. Soc., 2018, 140, 4913 CrossRef CAS PubMed.
- M. Kumar and J. S. Francisco, J. Phys. Chem. A, 2017, 121, 9421 CrossRef CAS PubMed.
- N. M. Kidwell, H. Li, X. Wang, J. M. Bowman and M. I. Lester, Nat. Chem., 2016, 8, 509 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS.
- J. Noga and R. J. Bartlett, J. Chem. Phys., 1987, 86, 7041 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103 CrossRef CAS.
- R. D. Hoehn, M. A. Carignano, S. Kais, C. Zhu, J. Zhong, X. C. Zeng, J. S. Francisco and and I. Gladich, J. Chem. Phys., 2016, 144, 214701 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B, 1996, 54, 1703 CrossRef CAS.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B, 1998, 58, 3641 CrossRef CAS.
- O. Welz, A. J. Eskola, L. Sheps, B. Rotavera, J. D. Savee, A. M. Scheer, D. L. Osborn, D. Lowe, A. M. Booth, P. Xiao, M. A. H. Khan, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Angew. Chem., Int. Ed., 2014, 53, 4547 CrossRef CAS PubMed.
- E. S. Foreman, K. M. Kapnas and C. Murray, Angew. Chem., Int. Ed., 2016, 55, 10419 CrossRef CAS PubMed.
- P. Raghunath, Y.-P. Lee and M. C. Lin, J. Phys. Chem. A, 2017, 121, 3871 CrossRef CAS PubMed.
- D. Johnson and G. Marston, Chem. Soc. Rev., 2008, 37, 699 RSC.
- T. B. Nguyen, A. P. Bateman, D. L. Bones, S. A. Nizkorodov, J. Laskin and A. Laskin, Atmos. Environ., 2010, 44, 1032 CrossRef CAS.
- S. Inomata, K. Sato, J. Hirokawa, Y. Sakamoto, H. Tanimoto, M. Okumura, S. Tohno and T. Imamura, Atmos. Environ., 2014, 97, 397 CrossRef CAS.
- A. Sadezky, P. Chaimbault, A. Mellouki, A. Rompp, R. Winterhalter, G. Le Bras and G. K. Moortgat, Atmos. Chem. Phys., 2006, 6, 5009 CrossRef CAS.
- M. Ehn, E. Kleist, H. Junninen, T. Petaja, G. Lonn, S. Schobesberger, M. Dal Maso, A. Trimborn, M. Kulmala, D. R. Worsnop, A. Wahner, J. Wildt and T. F. Mentel, Atmos. Chem. Phys., 2012, 12, 5113 CrossRef CAS.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipila, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I. H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurten, L. B. Nielsen, S. Jorgensen, H. G. Kjaergaard, M. Canagaratna, M. Dal Maso, T. Berndt, T. Petaja, A. Wahner, V. M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, Nature, 2014, 506, 476 CrossRef CAS.
- J. E. Perez, M. Kumar, J. S. Francisco and A. Sinha, J. Phys. Chem. A, 2017, 121, 1022 CrossRef CAS.
- Q. Zhang and C. Anastasio, Atmos. Environ., 2001, 35, 5629 CrossRef CAS.
- Y.-L. Sun, Q. Zhang, J. J. Schwab, K. L. Demerjian, W.-L. Chen, M.-S. Bae, H.-M. Hung, O. Hogrefe, B. Frank, O. V. Rattigan and Y.-C. Lin, Atmos. Chem. Phys., 2011, 11, 1581 CrossRef CAS.
- H. Petetin, J. Sciare, M. Bressi, V. Gros, A. Rosso, O. Sanchez, R. Sarda-Esteve, J.-E. Petit and M. Beekmann, Atmos. Chem. Phys., 2016, 16, 10491 Search PubMed.
- J. M. Anglada, P. Aplincourt, J. M. Bofill and D. Cremer, ChemPhysChem, 2002, 2, 215 CrossRef.
- J. M. Anglada, J. González and M. Torrent-Sucarrat, Phys. Chem. Chem. Phys., 2011, 13, 13034 RSC.
- M. Kumar and J. S. Francisco, Angew. Chem., Int. Ed., 2016, 55, 13432 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures providing deeper insights into the adduct formation at the air–water interface and the distance between the center of mass of the adduct and that of the water droplet, optimized geometries of key species involved in all gas-phase reactions, videos of trajectories of the BOMD simulations for the various anti-CH3CHOO–CH3NH2 reaction pathways at the air–water interface. Trajectories of the BOMD simulation for the direct reaction between anti-CH3CHOO and CH3NH2 at the air–water interface (MPG). Trajectories of the BOMD simulation for the interfacial water molecule-mediated reaction between anti-CH3CHOO and CH3NH2 at the air–water interface (MPG). See DOI: 10.1039/c8sc03514h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab