
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree to tango requires a site-specific substitution: heterotrimetallic molecular precursors for high-voltage rechargeable batteries†
Haixiang
Han
 a,
Zheng
Wei
a,
Alexander S.
Filatov
b,
Jesse C.
Carozza
a,
Melisa
Alkan
c,
Andrey Yu.
Rogachev
c,
Andrey
Shevtsov
d,
Artem M.
Abakumov
d,
Chongin
Pak
e,
Michael
Shatruk
e,
Yu-Sheng
Chen
f and
Evgeny V.
Dikarev
*a
a,
Zheng
Wei
a,
Alexander S.
Filatov
b,
Jesse C.
Carozza
a,
Melisa
Alkan
c,
Andrey Yu.
Rogachev
c,
Andrey
Shevtsov
d,
Artem M.
Abakumov
d,
Chongin
Pak
e,
Michael
Shatruk
e,
Yu-Sheng
Chen
f and
Evgeny V.
Dikarev
*a
aDepartment of Chemistry, University at Albany, SUNY, Albany, NY 12222, USA. E-mail: edikarev@albany.edu
bDepartment of Chemistry, University of Chicago, Chicago, IL 60637, USA
cDepartment of Chemistry, Illinois Institute of Technology, Chicago, IL 60616, USA
dSkolkovo Institute of Science and Technology, Moscow 143026, Russia
eDepartment of Chemistry and Biochemistry, Florida State University, Tallahassee, FL 32306, USA
fNSF's ChemMatCARS, Center for Advanced Radiation Source, The University of Chicago, Argonne, IL 60439, USA
First published on 16th October 2018
Abstract
Design of heterotrimetallic molecules, especially those containing at least two different metals with close atomic numbers, radii, and the same coordination number/environment is a challenging task. This quest is greatly facilitated by having a heterobimetallic parent molecule that features multiple metal sites with only some of those displaying substitutional flexibility. Recently, a unique heterobimetallic complex LiMn2(thd)5 (thd = 2,2,6,6-tetramethyl-3,5-heptanedionate) has been introduced as a single-source precursor for the preparation of a popular spinel cathode material, LiMn2O4. Theoretical calculations convincingly predict that in the above trinuclear molecule only one of the Mn sites is sufficiently flexible to be substituted with another 3d transition metal. Following those predictions, two heterotrimetallic complexes, LiMn2−xCox(thd)5 (x = 1 (1a) and 0.5 (1b)), that represent full and partial substitution, respectively, of Co for Mn in the parent molecule, have been synthesized. X-ray structural elucidation clearly showed that only one transition metal position in the trinuclear molecule contains Co, while the other site remains fully occupied by Mn. A number of techniques have been employed for deciphering the structure and composition of heterotrimetallic compounds. Synchrotron resonant diffraction experiments unambiguously assigned 3d transition metal positions as well as provided a precise “site-specific Mn/Co elemental analysis” in a single crystal, even in an extremely difficult case of severely disordered structure formed by the superposition of two enantiomers. DART mass spectrometry and magnetic measurements clearly confirmed the presence of heterotrimetallic species LiMnCo(thd)5 rather than a statistical mixture of two heterobimetallic LiMn2(thd)5 and LiCo2(thd)5 molecules. Heterometallic precursors 1a and 1b were found to exhibit a clean decomposition yielding phase-pure LiMnCoO4 and LiMn1.5Co0.5O4 spinels, respectively, at the relatively low temperature of 400 °C. The latter oxide represents an important “5V spinel” cathode material for the lithium ion batteries. Transmission electron microscopy confirmed a homogeneous distribution of transition metals in quaternary oxides obtained by pyrolysis of single-source precursors.
Introduction
An insertion of additional 3d transition metals into the parent heterobimetallic assembly (conventionally known as doping or substitution method) to prepare heteromultimetallic compounds is widely recognized as an effective approach to better-performing energy-related materials.1–4 One field that has especially benefited from application of this technique is the development of new rechargeable battery materials.5–8 It is believed that the most important factors with respect to the evaluation of a promising cathode material, such as capacity, reversibility, and working potential, are directly related to a synergy between transition metal constituents.5,9–12The discovery of LiMn2O4 spinel oxide is one of the cornerstones in the exploration of new electrochemically-active materials.10,13–16 This positive electrode material has been widely adopted by the battery industry due to its good overall electrochemical performance, environmental friendliness, and low production costs.11,17 Research-wise, it provides the basis for continued scientific endeavours into the design of new spinel oxides with improved characteristics that can greatly expand their practical applications.7,18,19 A number of studies have demonstrated that Li–Mn spinels exhibit great flexibility in accommodating a variety of dopant metals, such as Cr, Fe, Co, Ni, and Zn.7,19–25 Among the wide range of these transition metal-doped spinel materials, the insertion of Co into the LiMn2O4 framework is particularly interesting.26,27 Based on the LiMn2O4 archetype, [LiMn3+Mn4+O4], the trivalent Mn3+ sites can be partially or fully substituted by the Co3+ cations yielding LiMn2−xCoxO4 spinels with x ≤ 1.28–30 Among the latter oxides with different Mn/Co ratios, the LiMn1.5Co0.5O4 composition (x = 0.5) stands out as the most important with respect to the practical applications as a cathode material.31 It is known to exhibit the most balanced electrochemical properties, especially reversibility and energy density.26,27 Specifically, the addition of the Co4+/Co3+ redox couple endows LiMn1.5Co0.5O4 with a high working potential, rendering this oxide into an exclusive class of cathode materials: the so-called “5V spinels”.7,32–34
The preparation of quaternary oxides is known to suffer from several drawbacks. Formation of the thermodynamically-favoured binary and tertiary intermediates may result in irremovable impurities and/or nonstoichiometric products.20,35,36 On top of that comes an often-overlooked problem of homogeneous distribution of transition metals in the product oxide at the microstructural level.37–39 In the particular case of the Mn-based spinel compounds, the high-temperature preparation routes should be avoided.40–42 The elevated temperatures not only create difficulties in obtaining nanosize materials with regular morphology and a narrow range of the particle size distribution,43–45 but also can cause decomposition of spinel into a monoclinic Li2MnO3 phase.46–48 Therefore, there is a great need for alternative synthetic approaches that allow one to avoid the above problems. One of the ways to overcome the difficulties in preparation of quaternary oxide materials is the use of molecular precursors that contain all necessary metals in the proper ratio and decompose at lower temperatures to yield phase-pure nanosize target materials.49–51 The intimate mixing of different metals within heterotrimetallic precursors ensures the homogeneity of metal distribution in the target products as well as facilitates a rapid interaction between the constituent elements of the molecule upon thermolysis to circumvent the formation of unwanted intermediates.52,53
Design of heterotrimetallic molecular precursors, especially those containing at least two different metal atoms with close atomic numbers, radii, and coordination preferences is not a trivial task. It is certainly helpful to have a model heterobimetallic parent molecule featuring multiple metal sites with only some of those displaying a substitutional flexibility. Several years ago, we introduced the first molecular single-source precursor for the spinel cathode material LiMn2O4.51 The highly volatile and soluble trinuclear complex LiMn2(thd)5 (thd = 2,2,6,6-tetramethyl-3,5-heptanedionate) has been shown to yield phase-pure LiMn2O4 spinel oxide upon thermal decomposition in the solid state/gas phase. Herein we report our strategy to utilize the parent structure of heterobimetallic Li–Mn diketonate as a model to design heterotrimetallic molecular precursors for the Co-containing oxides. While heterometallic compound with the Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn:Co = 1:1:1 ratio (1a) was the focus of our quest into making a “true” heterotrimetallic molecule, the one with 1:1.5:0.5 composition (1b) was the major target for future practical applications.
:Mn:Co = 1:1:1 ratio (1a) was the focus of our quest into making a “true” heterotrimetallic molecule, the one with 1:1.5:0.5 composition (1b) was the major target for future practical applications.
Design of heterotrimetallic Li–Mn–Co precursor: structural and theoretical considerations
In order to design a heterotrimetallic molecular precursor with the desired Li:Mn:Co ratio of 1:1:1, we chose the parent trinuclear structure of LiMn2(thd)5 (Scheme 1). Reported by us back in 2012,51 this heterometallic molecule remains, to the best of our knowledge, the only structurally characterized molecular precursor for the LiMn2O4 spinel oxide material. As of today, we have been unable to replicate this trinuclear arrangement with other diketonate ligands or to obtain analogues with other divalent metals. In particular, the proposed isomorphous LiCo2(thd)5 complex has not been isolated despite numerous attempts.
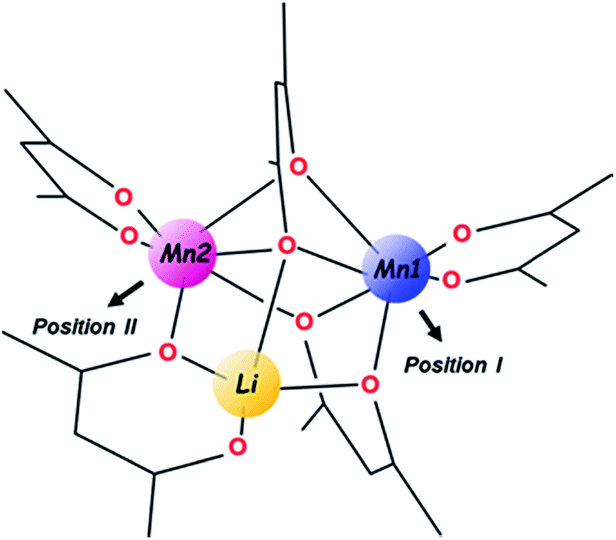 | ||
| Scheme 1 | ||
In the unique structure of LiMn2(thd)5,51 five thd ligands act in chelating (2), chelating–bridging (1) and very unusual purely bridging (2) fashions. Such ligand arrangement ensures tetrahedral coordination for the Li center as well as a distorted octahedral environment for both Mn ions. An important note for further consideration is that the trinuclear molecule contains three chiral metal centres and features two enantiomers in the centrosymmetric crystal structure, namely S-Li,Δ-Mn1,Λ-Mn2 (as shown in Scheme 1) and R-Li,Λ-Mn1,Δ-Mn2. In addition, the ten bulky tBu substituents on diketonate ligands are facing outward of the heterometallic assembly and provide a screening protection for the metal core from being oligomerized or involved in intermolecular interactions of any kind.
For the purpose of this research, the most important feature of the parent structure is the presence of two crystallographically independent Mn positions within the trinuclear molecule. While both of those are high-spin, octahedrally-coordinated MnII ions with one purely chelating diketonate ligand on each, the obvious visual distinction is that the Mn (2) site (position II), is bridged by oxygen from the ligand that chelates the Li center (Scheme 1). A thorough inspection of the two Mn sites in the trinuclear molecule reveals certain numerical differences (ESI, Table S8†) between them: (1) the Mn ion in position II possesses a more distorted octahedral environment than the Mn ion in position I (σ = 14.0 vs. 12.2); (2) the former ion also features longer averaged Mn–O distances (2.21 vs. 2.16 Å), disregarding two chelating bonds that are essentially the same for both.
Taking into account the above structural differences between the two Mn centres, as well as inability to isolate the dicobalt analogue, LiCo2(thd)5, we have wondered if only one of the Mn sites, namely position I, could be preferentially substituted by the Co ion, which has a smaller ionic radius and tends to maintain more rigid octahedral coordination as compared to Mn ion. It should be noted here that the full replacement of Mn at only one specific site will bring about a “true” heterotrimetallic molecule, LiMnCo(thd)5. On the contrary, the substitution at both positions will likely result in a mixture of heterobimetallic (LiMn2(thd)5 and LiCo2(thd)5) and heterotrimetallic species as we have recently demonstrated for the Li2CoNiL6 system.54 In order to assess if the unique trinuclear molecule LiMn2(thd)5 has specific preferences for accommodating cobalt in one of its two transition metal positions, theoretical calculations of thermodynamic stability for the four possible trimetal cores (Fig. 1) were performed using the B2PLYP-D/TZVP/ZORA approach (see ESI† for more details). As can be seen from the results, the parent dimanganese molecule, LiMn2(thd)5 (Fig. 1a), has the lowest stabilization energy (where Estabilization = E(LiMM′(thd)5) − E(Li(thd)) − E(M(thd)2) − E(M′(thd)2)), while its dicobalt analogue, LiCo2(thd)5 (Fig. 1d), is ca. 10 kcal mol−1 higher in energy, thus being the least stable among all systems considered. Most importantly, the comparison between two isomers of the heterotrimetallic LiMnCo(thd)5 molecule (Fig. 1b and c) clearly illustrates the preference for adoption of Co at the position I and also indicates a certain feasibility (the difference of ca. 2 kcal mol−1 with parent LiMn2(thd)5 molecule) for the formation of this particular isomer.
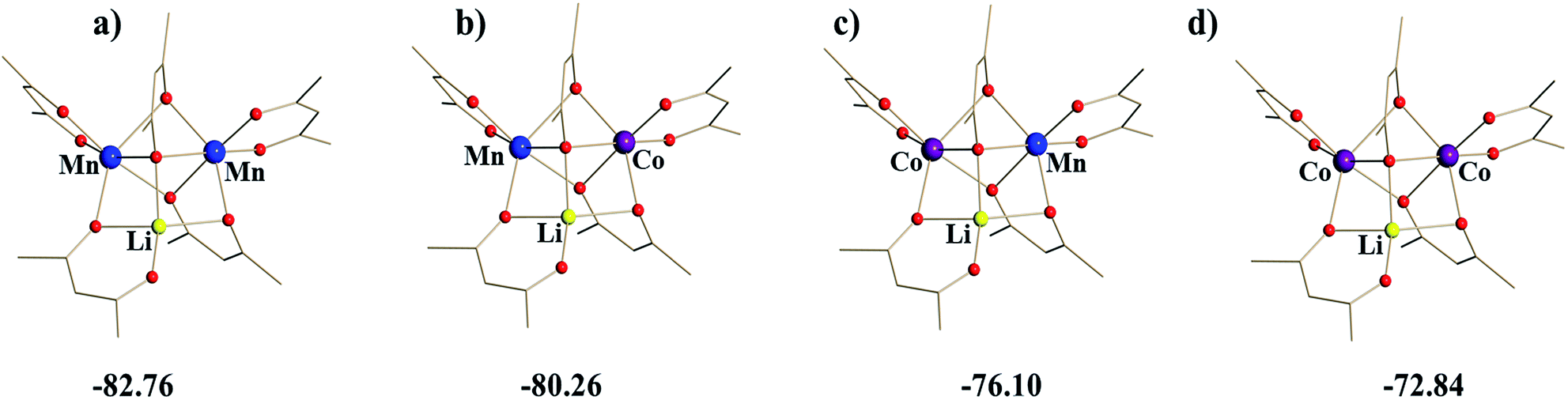 | ||
| Fig. 1 Calculated stabilization energies (kcal mol−1) for four possible arrangements of Mn and Co ions within the trinuclear LiMM′(thd)5 molecule. The tert-butyl groups and hydrogen atoms are omitted for clarity. | ||
Results and discussion
Synthesis and properties
Heterotrimetallic precursors LiMn2−xCox(thd)5 (x = 1 (1a) and 0.5 (1b)) have been prepared by stoichiometric reaction between anhydrous divalent transition metal chlorides and lithium diketonate:| 5Li(thd) + (2 − x) MnCl2 + xCoCl2 → LiMn2−xCox(thd)5 + 4LiCl | (1) |
The reaction can be run both in the solid state and in solution. For the solid state/gas phase process, the starting reagents were sealed in an evacuated ampule and placed in a gradient furnace at 110 °C (note: careful control of the temperature over time is important to avoid product decomposition), allowing crystalline products 1a and 1b to be deposited in the cooler end of the container (5–10 °C lower) with LiCl by-product remaining in the “hot” zone. While the products can be obtained in crystalline form by using this approach, it requires a long time to achieve completion, about 6 and 4 weeks for 1a and 1b, respectively. Compared to the solid state method, the solution approach is faster and can be conveniently performed on a significantly larger scale. Given the decomposition temperature (ca. 115 °C) for both products, refluxing non-coordinating toluene (b.p. 111 °C) was used as a reaction medium. Depending on the amounts of starting reagents, it takes between 3–4 weeks for the synthesis of 1a and 2–3 weeks for 1b to be isolated in essentially pure form. The purple microcrystalline products can be separated from LiCl by filtration based on their different solubilities in toluene. Heterometallic precursors are highly volatile at the temperatures above 70 °C. Complex 1a obtained from solution can be further purified by quantitative sublimation in evacuated ampules at as low as 90 °C.
It should be mentioned that the reaction progress in all cases can be visually controlled: the presence of pink Co(thd)2 in the crystalline products clearly indicates an incomplete reaction as well as the higher than targeted Mn:Co ratio in the heterometallic compound. Elemental analysis of crystalline products confirmed the Li:Mn:Co metal ratios as 1:1:1 (1a) and 1:1.5:0.5 (1b). Phase purity of the bulk samples obtained from the solution method has also been confirmed by the Le Bail fit of X-ray powder diffraction patterns (ESI, Fig. S1 and S2, Tables S1 and S2†).
Apparently, at some point the substitution of Co for Mn proceeds according to eqn (2):
| (1 − x/2)LiMn2(thd)5 + x/2 Li(thd) + xCo(thd)2 → LiMn2−xCox(thd)5 | (2) |
We have tried to run such reactions directly, only to find out that it proceeds extremely slowly, being far from completion even after 2 months at 110 °C. In addition, it requires prior synthesis of LiMn2(thd)5 and Co(thd)2 reagents.
Heterotrimetallic products 1a and 1b appear to be very similar to the parent bimetallic LiMn2(thd)5 diketonate in terms of volatility, solubility, and air/moisture-sensitivity. The only noticeable differences are the colour (dark-purple and purple for 1a and 1b, respectively, vs. bright yellow for LiMn2(thd)5) and slightly (by 5 °C) higher decomposition temperature for the heterotrimetallic compounds. The solid, especially crystalline, products can be conveniently handled in open air in the course of characterization/decomposition studies. Complexes 1a and 1b are readily soluble in all common solvents, with the solutions being quite sensitive to open air. The NMR (1H and 7Li) spectra of both products are silent in non-/weakly-coordinating solvents, such as chloroform or acetone, indirectly confirming the retention of the heterometallic core in solution (ESI, Fig. S12–S17†). On the other hand, strongly-coordinating solvents, such as DMSO, force dissociation of the heterometallic assembly into homometallic M(thd)2(sol)2 and Li(thd)(sol)2 species, as evidenced by the appearance of diamagnetic component signals in the NMR spectra (ESI, Fig. S18 and S19†).
It should be stressed here again that all our attempts to prepare heterobimetallic LiCo2(thd)5 analogue (or even detect it as a minor product) have failed regardless of synthetic methods, experimental conditions, and starting reagents utilized. Analysis of the isolated products by X-ray single crystal/powder diffraction and mass spectrometry techniques did not provide any evidence for the existence of the dicobalt compound; the reaction mixtures were always found to contain only Co(thd)2 and Li(thd) homometallic diketonates.
Crystal structure
Single crystal X-ray investigations of heterotrimetallic precursors 1a and 1b confirm that they are isomorphous to LiMn2(thd)5 and crystallize in the centrosymmetric triclinic space group with very close unit cell parameters (ESI, Table S9†). Similar to the parent compound, the crystal structures of 1a and 1b are rather complex, containing two crystallographically independent molecules in the unit cells. While one of those molecules is perfectly ordered (Fig. 2), the other one is severely disordered. As in the parent structure, this disorder can be fully modelled to reveal a superposition of two enantiomers: S-Li,Δ-Co,Λ-Mn and R-Li,Λ-Co,Δ-Mn (ESI, Fig. S4†), both refined with the site occupancy factor of 0.5 for every atom.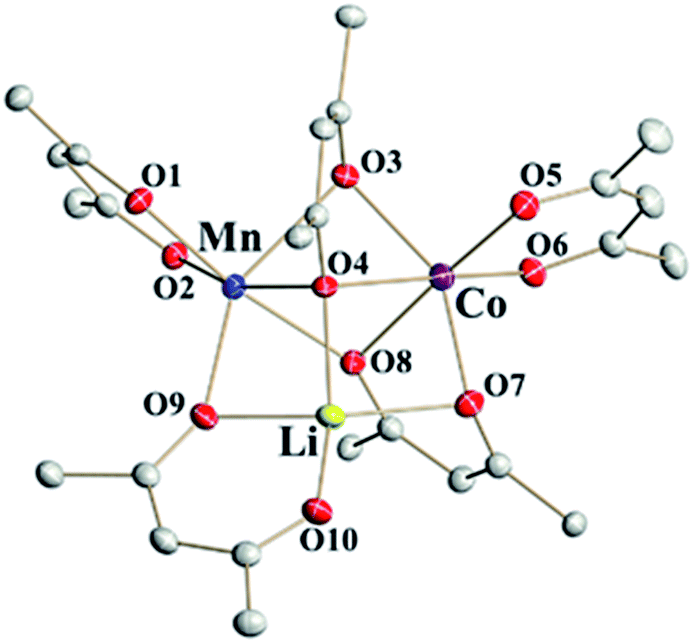 | ||
| Fig. 2 Heterotrimetallic molecule that represents the ordered unit in the structure of LiMnCo(thd)5 (1a). Hydrogen atoms and tert-butyl groups of thd ligands are omitted for clarity. Full view of trimetallic molecules in the structures 1a and 1b can be found in ESI, Fig. S3 and S6.† | ||
As may be expected, the refinement of structure 1a with Co placed either in position I or II, as well as with mixed occupancy Co:Mn = 0.5:0.5 for both positions, did not result in any significant alterations of R-values, standard deviations, or atomic displacement parameters for the metal sites involved (ESI, Table S6†).
However, a careful analysis of M–O distances (possible for ordered units only) in structures 1a and 1b and their comparison to the corresponding bonds in the parent structure of LiMn2(thd)5 revealed an incontestable trend (Table 1). While the distances for the metal in position II remain almost the same as in the all-Mn structure, those for the metal in position I appeared to be significantly shorter. This observation clearly indicates that only position I in the structures of 1a and 1b contains Co (fully or partially), while position II remains occupied solely by Mn in accord with predictions made by theoretical calculations (vide supra). The differences in M–O distances in structure 1b follow the above trend, although not as distinctly as those in structure 1a, which corresponds well to the lower content of Co in the former compound.
| LiMnCo(thd)5 (1a) Δ(M–O), Å | LiMn1.5Co0.5(thd)5 (1b) Δ(M–O), Å | LiMn2(thd)551 M–O, Å |
|||
|---|---|---|---|---|---|
| a Oc – chelating oxygen; Ob-bridging oxygen. | |||||
| Position I | M–Oc | O5 | −0.052 | −0.040 | 2.045(3) |
| O6 | −0.051 | −0.037 | 2.074(2) | ||
| M–Ob | O3 | −0.047 | −0.037 | 2.189(2) | |
| O4 | −0.045 | −0.037 | 2.304(2) | ||
| O7 | −0.066 | −0.048 | 2.157(3) | ||
| O8 | −0.039 | −0.032 | 2.187(2) | ||
| Position II | M–Oc | O1 | −0.001 | −0.003 | 2.079(2) |
| O2 | −0.001 | −0.003 | 2.059(2) | ||
| M–Ob | O3 | −0.013 | −0.011 | 2.335(2) | |
| O4 | 0.013 | 0.002 | 2.304(2) | ||
| O8 | −0.012 | −0.011 | 2.210(2) | ||
| O9 | 0.003 | 0.003 | 2.162(3) | ||
The above observations imply that the structure of LiMnCo(thd)5 (1a) consists of “true” heterotrimetallic molecules as shown in Fig. 2, while the structure of LiMn1.5Co0.5(thd)5 (1b) should contain a statistical mixture of heterotrimetallic LiMnCo(thd)5 and heterobimetallic LiMn2(thd)5 units (ESI, Fig. S6†). However, since this conclusion can be drawn upon consideration of the ordered trinuclear units only, other characterization methods should be employed for analysis of the “whole” structure.
Resonant diffraction investigation
Due to the close atomic numbers of Mn and Co, as well as similar radii of their divalent ions, single crystal X-ray diffraction data cannot be used for unambiguous assignment of 3d transition metal positions, let alone the precise refinement of Mn:Co ratios for the mutually occupied positions in both ordered and disordered units of structures 1a and 1b. In order to resolve this problem, X-ray resonant diffraction investigation, which is based on application of wavelengths near-edge of anomalous X-ray scatterers, was carried out to identify the presence of Mn/Co at particular metal sites and to quantitatively determine their individual occupancy factors at the mixed-metal sites in the structure of LiMn1.5Co0.5(thd)5 (1b). Manganese and cobalt exhibit significant differences in the anomalous dispersion factors at their X-ray absorption edges, while the dispersion factors of other elements remain constant. Thus, the employment of the specific wavelength at Mn/Co K-edges allows for a site-specific elemental analysis as well as for quantitative estimation of occupancies and associated relative errors. A total of seven datasets at seven different wavelengths (three at or near Mn K-edge, three at or near Co K-edge, and one away from both absorption edges) were collected during the resonant diffraction experiment (ESI, Fig. S7 and S8†).
For the ordered crystallographic unit in the structure of 1b, the metal site occupation pattern can be effectively visualized by inspecting the difference Fourier electron density maps that display deep holes for the respective crystallographic metal positions (Fig. 3). It has been unambiguously found that in the ordered crystallographically independent molecule, both of the transition metal sites contain Mn, while only one of those (position I) exhibits Co K-edge absorption, thus indicating the presence of Co. In order to minimize the solid-state effects neglected for calculations of dispersion factors, the data sets collected on the lower-energy side of the respective absorption edges and at 30 keV were used for the quantification of metal occupancies in the ordered unit of structure 1b. It was confirmed that Mn fully occupies position II (s.o.f. = 0.995(1)), while position I contains both Mn and Co in a nearly equiatomic ratio of 0.476(3)/0.524(3). Thus, the structural analysis revealed that this unit (c) is built upon the intergrowth of two different molecules, LiMn2(thd)5 (a) and LiMnCo(thd)5 (b), in a 1:1 ratio (Fig. 4) to maintain the composition LiMn1.5Co0.5(thd)5.
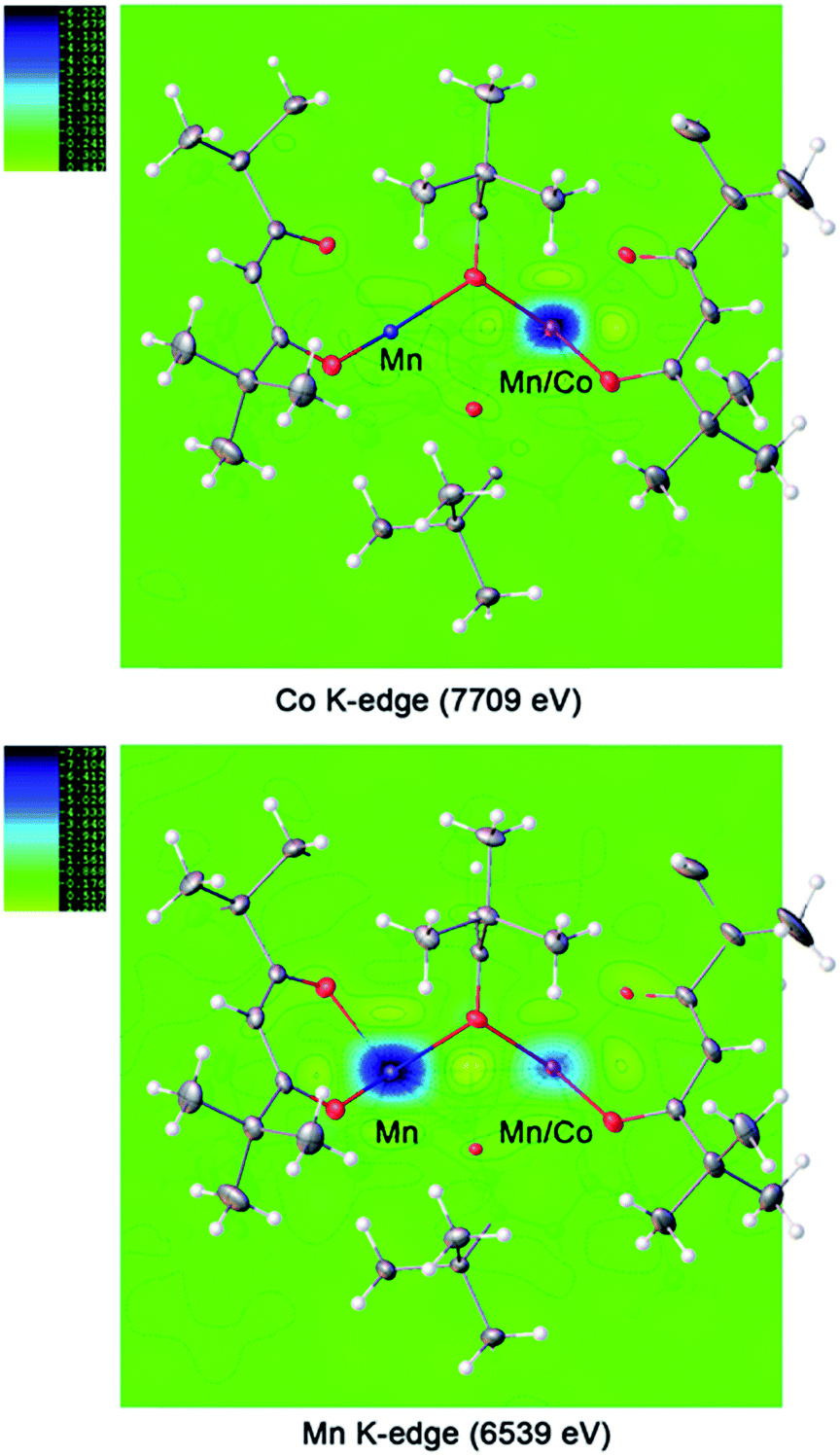 | ||
| Fig. 3 Difference Fourier electron density maps at absorption K-edges of Co and Mn for the ordered trinuclear units in the structure of LiMn1.5Co0.5(thd)5 (1b). | ||
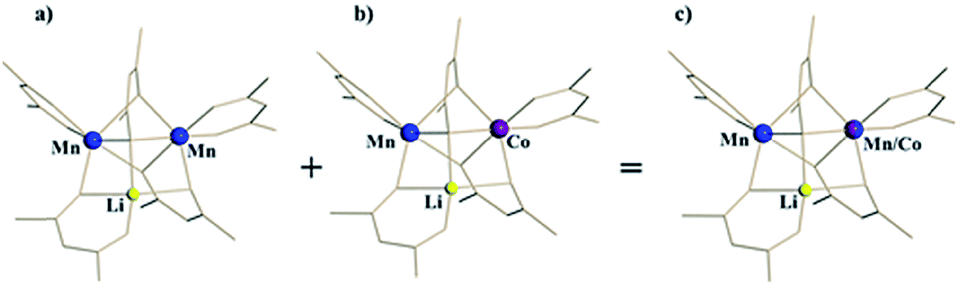 | ||
| Fig. 4 Ordered unit (c) in the crystal structure of LiMn1.5Co0.5(thd)5 (1b) as a superposition of heterobimetallic LiMn2(thd)5 (a) and heterotrimetallic LiMnCo(thd)5 (b) molecules in 1:1 ratio. | ||
For the disordered unit, the modelling appears significantly more challenging. Based on our refinement of the parent structure LiMn2(thd)5,51 the reasonable model for a disordered unit (d) can be achieved by refining it as a superposition of molecule c (S,Δ,Λ) and its enantiomer ɔ (R,Λ,Δ) taken in 1:1 ratio (Fig. 5). Refinement of occupancy factors for metal positions in two individual molecules that comprise the disordered unit yields 0.500(1) and 0.496(1) for Mn and 0.241(3)/0.259(3) for mixed Mn/Co sites, respectively (occupancies for mixed Mn/Co sites of two disordered molecules were constrained to be equal). Thus, the total composition of the disordered unit is essentially the same as that of the ordered one. Overall, these results are in excellent agreement with the formulation of 1b as LiMn1.5Co0.5(thd)5, as determined by elemental analysis, unambiguously confirming the substitution of Co for Mn in position I only.
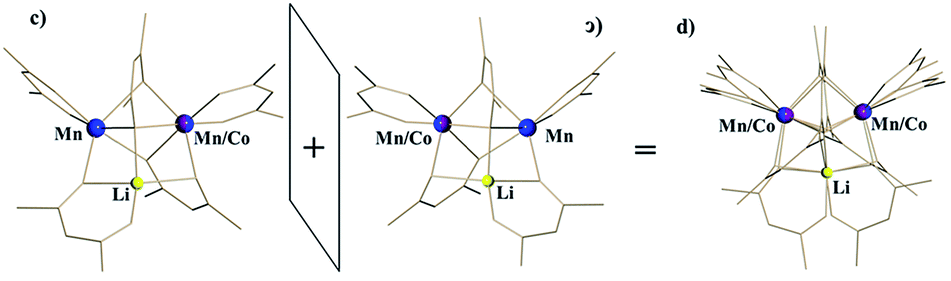 | ||
| Fig. 5 Disordered unit (d) in the crystal structure of LiMn1.5Co0.5(thd)5 (1b) as a superposition of two enantiomers (S,Δ,Λ-c and R,Λ,Δ-ɔ). | ||
Direct analysis in real time (DART) mass spectrometry investigation
Presumably, mass spectrometry is an ideal method to provide accessible, fast, and unambiguous assessment of the presence of heterotrimetallic molecules vs. the mixture of heterobimetallic counterparts or all of the above. It should work well when the two transition metals are well-separated in terms of their atomic masses and isotope distribution patterns, unlike the recently reported case of the Li–Co–Ni system.54 However, for successful application of mass spectrometry it is important that heterometallic molecules exhibit a certain degree of structural integrity in maintaining heterometallic ions at experimental conditions and do not undergo excessive fragmentation/recombination processes in the mass spectrometer. To this end, the Direct Analysis in Real Time (DART) mass spectrometry has already demonstrated3,49,54,55 great promise in retention of heterometallic assemblies due to its soft ionization characteristics, allowing for the effective analysis of the “gas-phase structure”, as well as for identifying the chemical compositions based on the detected ionic species and their isotope distribution patterns.In the positive mode mass spectrum of the solid precursor LiMnCo(thd)5 (1a), its heterotrimetallic character is unambiguously revealed by the presence of two parent ions, [M + H]+ (M = LiMnCoL5; L = thd; meas/calcd = 1037.5812/1037.5877) and [M − L]+ (meas/calcd = 853.4409/853.4414), along with their characteristic isotope distribution patterns (Fig. 6). Importantly, there are no visible peaks that can be assigned to the corresponding heterobimetallic counterparts LiMn2L5 and LiCo2L5 (ESI, Fig. S20, S23, and Table S10†). These observations provide a conclusive evidence that the title complex 1a consists solely of heterotrimetallic trinuclear molecules LiMnCo(thd)5.
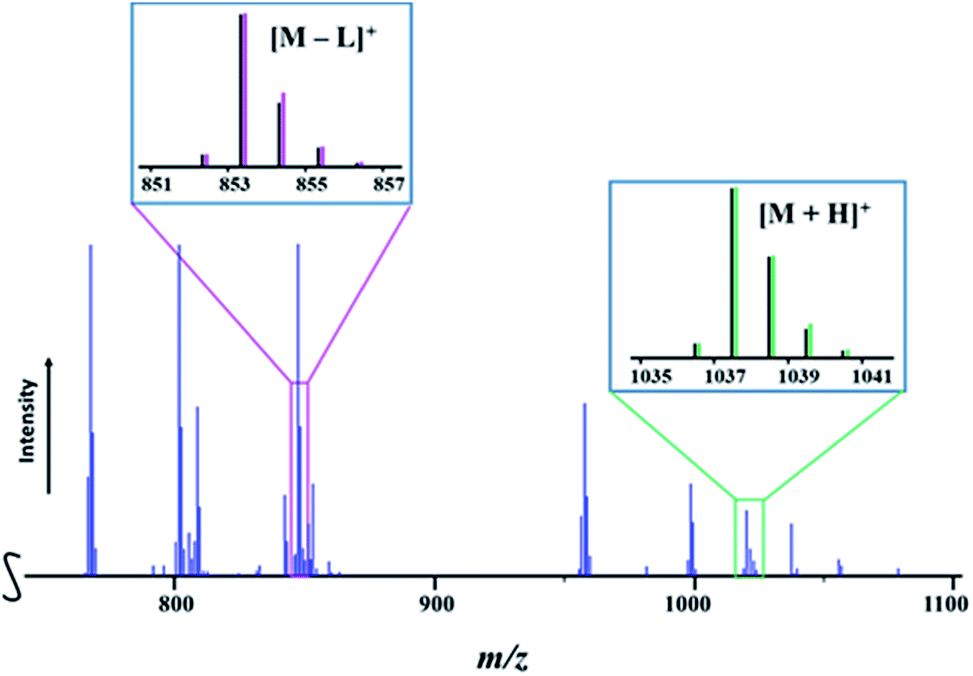 | ||
| Fig. 6 Fragment of the positive ion DART mass spectrum of solid LiMnCo(thd)5 (1a). The isotope distribution patterns for the [M + H]+ and [M − L]+ (M = LiMnCo(thd)5, L = thd) ions are shown as insets. Black and pink/green lines are experimental and simulated patterns, respectively. The full spectrum and peak assignments can be found in ESI, Fig. S20 and Tables S10–S12.† | ||
According to the results of resonant diffraction analysis, precursor LiMn1.5Co0.5(thd)5 (1b) contains heterotrimetallic LiMnCo(thd)5 and heterobimetallic LiMn2(thd)5 molecules co-crystallized in 1:1 ratio. Thus, one should observe the peaks in mass spectrum that represent both corresponding ions. While the heterotrimetallic peak [M − L]+ (M = LiMnCoL5; L = thd; meas/calcd = 853.4409/853.4413) was clearly identified (Fig. 7), the corresponding heterobimetallic [LiMn2L4]+ pattern was found to be severely overlapped (ESI, Table S13†) with one of the major fragmentation products [MnCoL4 + H]+. The latter ion was also encountered in the mass spectrum of 1a, where its experimental isotope distribution pattern fits well with the simulated one (ESI, Table S12†), indicating the absence of the [LiMn2L4]+ ion. On the contrary, the experimental isotope distribution pattern of [MnCoL4 + H]+ ion in the spectrum of 1b does not match the simulated one, especially considering the intensities of the peaks at m/z = 849, 850, and 851, which are significantly higher than calculated values. After a careful analysis, the individual intensities were successfully extracted as 19% and 81% for the [LiMn2L4]+ and [MnCoL4 + H]+ ions, respectively, and the satisfactory fit between calculated and experimental patterns was obtained (ESI, Fig. S22 and Table S16†). Thus the DART mass spectrometry investigation also confirms that heterometallic precursor 1b contains both LiMnCo(thd)5 and LiMn2(thd)5 molecules. As in the mass spectrum of 1a, no evidence for the presence of LiCo2(thd)5 molecule was found (ESI, Fig. S23†).
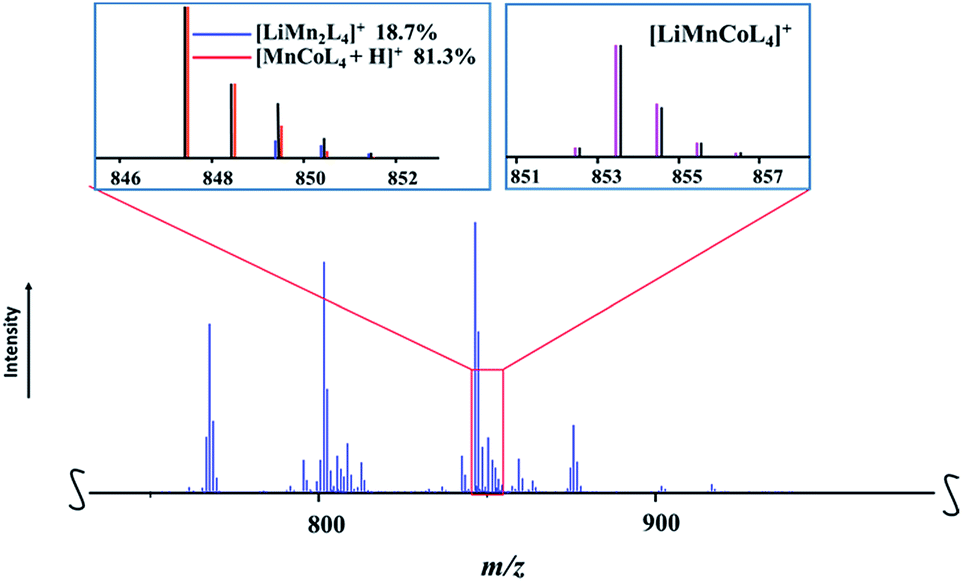 | ||
| Fig. 7 Fragment of the positive ion DART mass spectrum of solid LiMn1.5Co0.5(thd)5 (1b). The isotope distribution pattern for the [LiMnCoL4]+ (L = thd) ion and overlapped pattern for [LiMn2L4]+ and [MnCoL4 + H]+ ions are shown as insets. Black and red/blue/pink lines are experimental and simulated patterns, respectively. The full spectrum and peak assignments can be found in ESI, Fig. S21 and S22, Tables S13–S16.† | ||
Magnetic measurement of heterometallic complexes
A polycrystalline sample of LiMnCo(thd)5 (1a) was used to investigate its magnetic behavior in comparison to that of parent LiMn2(thd)5. Both complexes exhibit a gradual decrease in the product of magnetic susceptibility (χ) by temperature (T) upon cooling (Fig. 8). Fitting the linear part of inverse susceptibility for LiMn2(thd)5 to the Curie–Weiss law, 1/χ = (T − θ)/C, resulted in the Curie constant (C) equal to 9.46(4) emu K mol−1 and the Weiss constant (θ) equal to −36(1) K (R2 = 9996). While the value of C is slightly higher than 8.75 emu K mol−1 expected for two non-interacting high-spin (S = 5/2) Mn2+ ions, the negative Weiss constant and the gradual decrease in the χT product indicate antiferromagnetic exchange coupling between the Mn2+ centres. The temperature dependence of χT was modelled with an isotropic Heisenberg–Dirac–Van Vleck Hamiltonian, Ĥ = −2JŜ1Ŝ2, and the fit to the experimental data (Fig. 8a) yielded the exchange coupling constant J = −2.53(4) cm−1 and the Lande factor g = 2.022(2) (R2 = 9970). The g-factor seems to be somewhat high for the magnetically isotropic d5 Mn2+ ion, but this might be due to the slight error in the mass measurement of LiMn2(thd)5 crystals that were contained in a sealed NMR tube to avoid oxidation of the sample.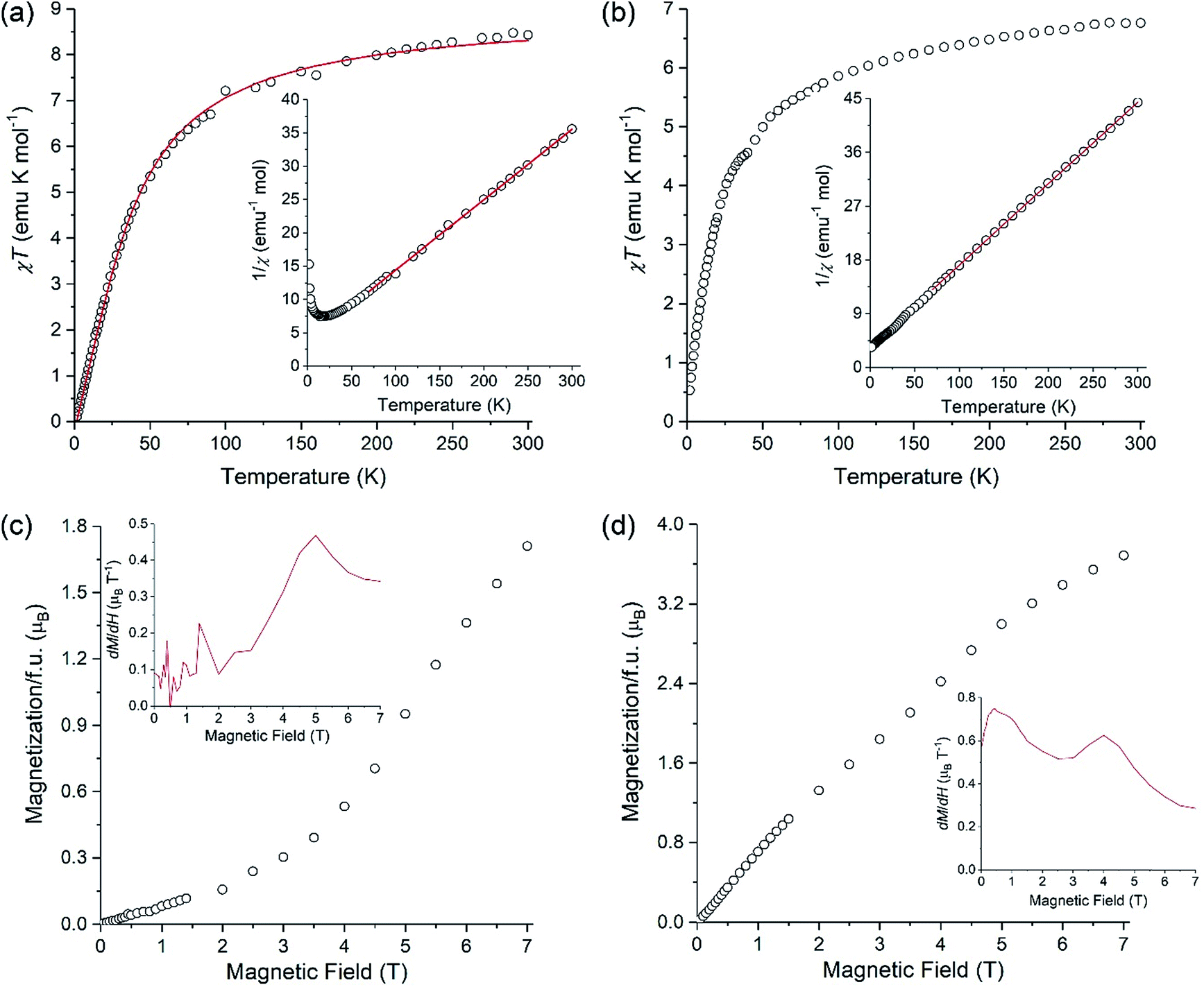 | ||
| Fig. 8 Temperature dependencies of χT measured at applied magnetic field of 0.1 T and field dependencies of magnetization measured at 1.8 K for LiMn2(thd)5 (a and c) and LiMnCo(thd)5 (b and d). The solid red line in (a) shows the theoretical fit. The insets in (a) and (b) show the Curie–Weiss fit (solid red lines) for the temperature dependences of 1/χ, while the insets in (c) and (d) display the first derivative of magnetization over applied magnetic field. | ||
For the LiMnCo(thd)5 (1a) the Curie–Weiss fit gave C = 7.34(2) emu K mol−1 and θ = −25.6(5) K. The Curie constant exceeds the spin-only value of 6.25 emu K mol−1 expected for the non-interacting Mn2+ (S = 5/2) and Co2+ (S = 3/2) ions, which appears reasonable, given the well-known tendency for the Co2+ ion to exhibit the orbital contribution to its magnetic moment and substantial spin–orbit coupling. While the negative Weiss constant and the decrease in the χT product with temperature suggest antiferromagnetic exchange coupling within the dimers, the situation is complicated by the fact that the Curie–Weiss fit also absorbs the spin–orbit coupling and zero-field splitting effects, which will effectively make the θ value appear more negative.
Related to the preceding discussion, the question about chemical composition of trinuclear units in LiMnCo(thd)5 (1a) is critical to understanding the magnetic behaviour of this complex. In this vein, an important observation can be made on the low-temperature behaviour of the χT product. For the parent molecule LiMn2(thd)5, the χT value tends to zero (Fig. 8a), as can be expected for the Stotal = 0 ground state resulting from the mutual cancellation of two antiferromagnetically coupled S = 5/2 centers. A similar behaviour would be expected for LiMnCo(thd)5 (1a) if its crystal structure contained only heterobimetallic LiMn2(thd)5 and LiCo2(thd)5 molecules. This is not what is observed experimentally: the χT product of LiMnCo(thd)5 (1a) decreases to a finite value of ∼0.5 emu K mol−1 at 1.8 K (Fig. 8b). The consequence can be seen in the temperature dependence of 1/χ, which for LiMn2(thd)5 diverges at lower temperature, due to the stabilization of the Stotal = 0 ground state (Fig. 8a, inset), while for LiMnCo(thd)5 (1a) the 1/χ curve tends to a constant value (Fig. 8b, inset).
Additional support for the heterotrimetallic nature of LiMnCo(thd)5 (1a) was obtained by field-dependent magnetization measurements performed at 1.8 K. In the case of LiMn2(thd)5, the magnetization exhibits a slow linear growth, as expected for the antiferromagnetically coupled homometallic (Mn) dimer, but a steeper growth can be seen at fields above 4 T (Fig. 8c). Taking into account the rather small exchange constant of −2.53(4) cm−1, the observed behavior is explained by the field-induced population of the Zeeman-split excited states with non-zero Stotal values. If the sample of LiMnCo(thd)5 (1a) contained only heterobimetallic species, one would assume that its field-dependent magnetization should be reflected by the sum of magnetizations of the individual LiMn2(thd)5 and LiCo2(thd)5 molecules, since the dipolar intermolecular coupling should be negligible due to the bulkiness of the thd ligands. Although the heterobimetallic LiCo2(thd)5 complex is not available to verify this hypothesis, the field-dependent magnetization curve for LiMnCo(thd)5 (1a) does not reveal a steep increase above 4 T (Fig. 8d), as it was observed for LiMn2(thd)5. The magnetization measured for 1a exhibits a nearly linear increase from 0 to 7 T, and the higher magnetization value observed at 7 T, as compared to the value found for LiMn2(thd)5, is consistent with the heterotrimetallic nature of LiMnCo(thd)5 (1a) and non-zero spin ground state in such molecule. To examine the field-dependent magnetization curves of these two samples more carefully, we show their derivatives as insets in Fig. 8c and d, respectively. The dM/dH plot for LiMnCo(thd)5 (1a) reveals a feature at ∼5 T, similar to that seen in the dM/dH plot for LiMn2(thd)5 at ∼4.5 T. Thus, a weak inflection is still present in the M vs. H plot of 1a, suggesting that the sample might contain a small amount of heterobimetallic LiMn2(thd)5 molecules. Such effect might be due to the Mn:Co ratio in the sample slightly exceeding the parity. Overall, however, the observed low-temperature magnetic behaviour of LiMnCo(thd)5 (1a) is consistent with the presence of heterotrimetallic molecules in its crystal structure.
Thermal decomposition of heterometallic precursors
Investigation of thermal behaviour of heterometallic precursors LiMnCo(thd)5 (1a) and LiMn1.5Co0.5(thd)5 (1b) revealed that both compounds exhibit clean, low-temperature decomposition patterns, yielding phase-pure LiMnCoO4 and LiMn1.5Co0.5O4 spinel oxides. The degradation of precursors starts around 115 °C, while fast weight loss occurs in a single step around 148–150 °C and ends around 275–280 °C (ESI, Fig. S24†). The total weight loss is 83.13% and 82.24% that corresponds well to theoretical values of 82.2% and 82.1% for precursors 1a and 1b, respectively. The traces of target products become visible in the X-ray powder patterns at around 350 °C along with a number of unidentified peaks, suggesting the incomplete decomposition process. The phase-pure oxides can be obtained at temperatures as low as 400 °C (Fig. 9). The crystallinity of the decomposition residues can be greatly improved upon elevating the annealing temperature to 550 °C (ESI, Fig. S25–S30†), and the unit cell parameters of thermolysis products (Table 2), calculated from the Le Bail fit, correspond well to the literature data. On the other hand, an appearance of red crystalline Li2MnO3 can be visually observed above 550 °C and further confirmed by X-ray powder diffraction analysis, indicating the subsequent decomposition of spinel phases. | ||
| Fig. 9 X-ray powder diffraction patterns of LiMnCoO4 (a), LiMn1.5Co0.5O4 (b), and LiMn2O4 (c)51 obtained from the thermal decomposition of the corresponding heterometallic precursors at 450 °C. | ||
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) of spinel oxides obtained by thermal decomposition of heterometallic precursors 1a and 1b with literature data
m) of spinel oxides obtained by thermal decomposition of heterometallic precursors 1a and 1b with literature data
The crystal structure and compositional homogeneity of the spinels obtained at 550 °C have been investigated with electron diffraction (ED), transmission electron microscopy (TEM), high angle annular dark field scanning TEM (HAADF-STEM), and energy dispersive X-ray (EDX) analysis using an aberration-corrected Titan G3 electron microscope operated at 200 kV and equipped with Super-X EDX system. Electron diffraction patterns of LiMnCoO4 and LiMn1.5Co0.5O4 (Fig. 10 and S31 of ESI,† respectively) revealed an F-centred cubic unit cell with a ≈ 8.1 Å, characteristic of the spinel type structure. The hk0, h + k = 4n, h, k – even, reflection condition visible in the [001] ED pattern confirms the space group Fdm.
 | ||
| Fig. 10 Electron diffraction patterns of LiMnCoO4 obtained by thermal decomposition of heterometallic precursor 1a at 550 °C. | ||
The TEM images in Fig. 11a and S30a† show that the spinels crystallize in a form of truncated octahedral crystals with the size ranging between 50–200 nm for LiMnCoO4 and 100–300 nm for LiMn1.5Co0.5O4. High-resolution [011] HAADF-STEM images of LiMnCoO4 and LiMn1.5Co0.5O4 (Fig. 11b and S32b,† respectively) reveal well-ordered spinel structures for both decomposition residues. The bright and faint dots in these images stand for the projections of the columns of the 16d positions occupied by the transition metal cations. The difference in brightness occurs because of different projected density of atoms being viewed along [011] direction. The Li and O atoms at the 8a and 32e positions, respectively, are not visible because of their low atomic numbers.
 | ||
| Fig. 11 TEM (a) and HAADF-STEM (b) images of LiMnCoO4 obtained by thermal decomposition of heterometallic precursor 1a at 550 °C. | ||
Importantly, the compositional EDX maps show highly homogeneous distribution of Mn and Co in both LiMnCoO4 and LiMn1.5Co0.5O4 oxides (ESI, Fig. S33 and S34†). Quantitative EDX analysis revealed the Mn:Co atomic ratio to be close to the nominal chemical composition of 0.92(2):1.08(2) for LiMnCoO4. For LiMn1.5Co0.5O4 the Mn:Co ratio of 1.37(3):0.63(3) reflects somewhat lower manganese content as compared to the corresponding heterometallic precursor. The latter can be explained by the fact that LiMn1.5Co0.5(thd)5 (1b) precursor used for the preparation of oxide was shown to contain both heterotrimetallic LiMnCo(thd)5 and heterobimetallic LiMn2(thd)5 molecules that have different volatilities, resulting in some loss of manganese upon thermal decomposition.
Conclusion
In accord with predictions of the theoretical calculations, it has been experimentally confirmed that in the parent trinuclear heterobimetallic molecule LiMn2(thd)5, only one of two manganese sites is sufficiently flexible to be substituted by cobalt. This results in an array of heterotrimetallic solid solutions up to the composition of LiMnCo(thd)5. In this research, we have concentrated on two stoichiometries for heterotrimetallic compounds: Li:Mn:Co = 1:1:1 (1a) and 1:1.5:0.5 (1b). While the former was the major focus of investigation into making a “true” heterotrimetallic molecule, the latter was a specific target for prospective practical applications towards low-temperature preparation of the “5V spinel” cathode material LiMn1.5Co0.5O4.
This study is aligned with our recent quest for a design of heterotrimetallic molecular precursors that contain at least two transition metal atoms with close atomic numbers and radii. We have long stated that the construction of such molecules will ultimately require either different oxidation states for similar metal ions3,58 or different coordination/ligand environment for the isovalent ones. The case reported here provides the first experimental demonstration that site-specific substitution is feasible in some polynuclear heterobimetallic assemblies that feature at least two metal sites with different (yet close) surroundings. This possibility emerges from the fact that one of two parent heterobimetallic molecules does not exist, which directly points to inability to substitute the parent metal atom in both positions.
Several instrumental methods have been found to be handy for deciphering the structure and composition of heterotrimetallic compounds. Synchrotron resonant diffraction experiments allow unambiguous assignment of 3d transition metal positions as well as a precise “elemental analysis” for the Mn and Co content in a single crystal, even in such a difficult case of severely disordered heterometallic molecules. DART mass spectrometry proved to be the most efficient method to analyse the heteromultimetallic molecules. This relatively simple and fast analysis can be successfully executed when there is a sufficient structural integrity of the heterometallic ions in the gas phase as well as when the fragmentation/recombination processes in mass spectrometer can be excluded.
The correct theoretical prediction of the substitution pattern in the parent heterobimetallic LiMn2(thd)5 molecule is noteworthy as it opens a large window of opportunity to explore the design of other heterotrimetallic precursors of the general formula LiMn2−xMx(thd)5. Of special importance are the systems with M = Mg or Ni, which represent single-source precursors for a very special class of spinel cathode materials for the Li-ion batteries.22,25,59,60
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the National Science Foundation is gratefully acknowledged (CHE-1152441 for E. V. D. and CHE-1464955 for M. S.). The authors thank the CRDF Global (FSCX-16-62133-0 for A. M. A.) and (OISE-16-62134-0 for E. V. D.) for funding this collaborative project. A. Yu R. is grateful for the financial support from the Illinois Institute of Technology and partial support from the 381688-FSU/ChemRing/DOD-DOTC grant. NSF's ChemMatCARS Sector 15 is principally supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1346572. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. The authors also thank to Jeff Lengyel (Florida State University) for his assistance with the TGA.References
- P. Venkatesan and J. Santhanalakshmi, Langmuir, 2010, 26, 12225–12229 CrossRef CAS PubMed.
- H. Kuroki, T. Tamaki, M. Matsumoto, M. Arao, K. Kubobuchi, H. Imai and T. Yamaguchi, Ind. Eng. Chem. Res., 2016, 55, 11458–11466 CrossRef CAS.
- C. M. Lieberman, M. C. Barry, Z. Wei, A. Y. Rogachev, X. Wang, J.-L. Liu, R. Clérac, Y.-S. Chen, A. S. Filatov and E. V. Dikarev, Inorg. Chem., 2017, 56, 9574–9584 CrossRef CAS PubMed.
- V. R. Surisetty, A. K. Dalai and J. Kozinski, Ind. Eng. Chem. Res., 2010, 49, 6956–6963 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- C. Yada, A. Ohmori, K. Ide, H. Yamasaki, T. Kato, T. Saito, F. Sagane and Y. Iriyama, Adv. Energy Mater., 2014, 4, 1301416 CrossRef.
- J. S. Thorne, R. A. Dunlap and M. N. Obrovac, J. Electrochem. Soc., 2014, 161, A2232–A2236 CrossRef CAS.
- J. B. Goodenough, J. Power Sources, 2007, 174, 996–1000 CrossRef CAS.
- C. Masquelier, M. Tabuchi, K. Ado, R. Kanno, Y. Kobayashi, Y. Maki, O. Nakamura and J. B. Goodenough, J. Solid State Chem., 1996, 123, 255–266 CrossRef CAS.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 461–472 CrossRef CAS.
- A. M. Abakumov, A. A. Tsirlin, I. Bakaimi, G. Van Tendeloo and A. Lappas, Chem. Mater., 2014, 26, 3306–3315 CrossRef CAS.
- D. K. Kim, P. Muralidharan, H.-W. Lee, R. Ruffo, Y. Yang, C. K. Chan, H. Peng, R. A. Huggins and Y. Cui, Nano Lett., 2008, 8, 3948–3952 CrossRef CAS PubMed.
- J. Molenda, M. Ziemnicki, J. Marzec, W. Zając, M. Molenda and M. Bućko, J. Power Sources, 2007, 173, 707–711 CrossRef CAS.
- G. Rousse, C. Masquelier, J. Rodríguez-Carvajal, E. Elkaim, J. P. Lauriat and J. L. Martínez, Chem. Mater., 1999, 11, 3629–3635 CrossRef CAS.
- M. M. Thackeray, P. J. Johnson, L. A. de Picciotto, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1984, 19, 179–187 CrossRef CAS.
- R. J. Gummow, A. de Kock and M. M. Thackeray, Solid State Ionics, 1994, 69, 59–67 CrossRef CAS.
- T. Takada, H. Hayakawa and E. Akiba, J. Solid State Chem., 1995, 115, 420–426 CrossRef CAS.
- A. Kraytsberg and Y. Ein-Eli, Adv. Energy Mater., 2012, 2, 922–939 CrossRef CAS.
- C. Lai, J. Chen, J. C. Knight, A. Manthiram and A. Navrotsky, ChemPhysChem, 2016, 17, 1942 CrossRef CAS.
- H.-C. Wang and C.-H. Lu, J. Power Sources, 2003, 119–121, 738–742 CrossRef CAS.
- P. Strobel, J. Tillier, A. Diaz, A. Ibarra-Palos, F. Thiery and J. B. Soupart, J. Power Sources, 2007, 174, 910–915 CrossRef CAS.
- P. Strobel, A. Ibarra-Palos, M. Anne, C. Poinsignon and A. Crisci, Solid State Sci., 2003, 5, 1009–1018 CrossRef CAS.
- P. Strobel, A. Ibarra-Palos and C. Poinsignon, Mater. Res. Soc. Symp. Proc., 2003, 756, 225–230 CAS.
- P. Strobel, A. Ibarra Palos, M. Anne and F. Le Cras, J. Mater. Chem., 2000, 10, 429–436 RSC.
- S. L. Zhao, H. Y. Chen, J. B. Wen and D. X. Li, J. Alloys Compd., 2009, 474, 473–476 CrossRef CAS.
- K. Dokko, N. Anzue, M. Mohamedi, T. Itoh and I. Uchida, Electrochem. Commun., 2004, 6, 384–388 CrossRef CAS.
- A. D. Robertson, A. R. Armstrong and P. G. Bruce, J. Power Sources, 2001, 97–98, 332–335 CrossRef CAS.
- M.-s. Zhao, F. Wang and X.-p. Song, Zhongguo Youse Jinshu Xuebao, 2005, 15, 1396–1402 CAS.
- Z. Wang, H. Ikuta, Y. Uchimoto and M. Wakihara, J. Electrochem. Soc., 2003, 150, A1250–A1254 CrossRef CAS.
- K. Mukai and T. Uyama, ACS Omega, 2017, 2, 5142–5149 CrossRef CAS.
- K. Park, D. Yeon, J. H. Kim, J.-H. Park, S. Doo and B. Choi, J. Power Sources, 2017, 360, 453–459 CrossRef CAS.
- P. K. Nayak, E. M. Erickson, F. Schipper, T. R. Penki, N. Munichandraiah, P. Adelhelm, H. Sclar, F. Amalraj, B. Markovsky and D. Aurbach, Adv. Energy Mater., 2017, 8, 1702397 CrossRef.
- F. Zhou, M. Cococcioni, K. Kang and G. Ceder, Electrochem. Commun., 2004, 6, 1144–1148 CrossRef CAS.
- P. T. Barton, Y. D. Premchand, P. A. Chater, R. Seshadri and M. J. Rosseinsky, Chem.–Eur. J., 2013, 19, 14521–14531 CrossRef CAS PubMed.
- Y.-K. Sun, I.-H. Oh and K. Yul Kim, J. Mater. Chem., 1997, 7, 1481–1485 RSC.
- B. Mortemard de Boisse, PhD thesis, Université de Bordeaux, 2014 Search PubMed.
- B. Mortemard de Boisse, D. Carlier, M. Guignard and C. Delmas, J. Electrochem. Soc., 2013, 160, A569–A574 CrossRef CAS.
- M. V. Reddy, G. V. S. Rao and B. V. R. Chowdari, J. Phys. Chem. C, 2007, 111, 11712–11720 CrossRef CAS.
- M. J. Ariza, D. J. Jones, J. Rozière, R. Chitrakar and K. Ooi, Chem. Mater., 2006, 18, 1885–1890 CrossRef CAS.
- M. M. Thackeray, M. F. Mansuetto and C. S. Johnson, J. Solid State Chem., 1996, 125, 274–277 CrossRef CAS.
- M. M. Thackeray, M. F. Mansuetto, D. W. Dees and D. R. Vissers, Mater. Res. Bull., 1996, 31, 133–140 CrossRef CAS.
- S.-T. Myung, K. Amine and Y.-K. Sun, J. Power Sources, 2015, 283, 219–236 CrossRef CAS.
- T. Takada, H. Hayakawa, E. Akiba, F. Izumi and B. C. Chakoumakos, J. Power Sources, 1997, 68, 613–617 CrossRef CAS.
- S.-J. Kim, Y.-W. Lee, B.-M. Hwang, S.-B. Kim, W.-S. Kim, G. Cao and K.-W. Park, RSC Adv., 2014, 4, 11598–11604 RSC.
- M. Bianchini, E. Suard, L. Croguennec and C. Masquelier, J. Phys. Chem. C, 2014, 118, 25947–25955 CrossRef CAS.
- Y. Gao and J. R. Dahn, J. Electrochem. Soc., 1996, 143, 1783–1788 CrossRef CAS.
- M. M. Thackeray, Prog. Solid State Chem., 1997, 25, 1–71 CrossRef CAS.
- H. Han, Z. Wei, M. C. Barry, A. S. Filatov and E. V. Dikarev, Dalton Trans., 2017, 46, 5644–5649 RSC.
- K. M. Fleming, J. C. Pekar and B. D. Leskiw, Rapid Commun. Mass Spectrom., 2016, 30, 2423–2432 CrossRef CAS PubMed.
- A. Navulla, L. Huynh, Z. Wei, A. S. Filatov and E. V. Dikarev, J. Am. Chem. Soc., 2012, 134, 5762–5765 CrossRef CAS PubMed.
- Z. Wei, H. Han, A. S. Filatov and E. V. Dikarev, Chem. Sci., 2014, 5, 813–818 RSC.
- C. Julien, C. Letranchant, S. Rangan, M. Lemal, S. Ziolkiewicz, S. Castro-Garcia, L. El-Farh and M. Benkaddour, Mater. Sci. Eng., B, 2000, 76, 145–155 CrossRef.
- H. Han, Z. Wei, M. C. Barry, J. C. Carozza, M. Alkan, A. Yu. Rogachev, A. S. Filatov, A. M. Abakumov and E. V. Dikarev, Chem. Sci., 2018, 9, 4736–4745 RSC.
- M. C. Barry, C. M. Lieberman, Z. Wei, R. Clérac, A. S. Filatov and E. V. Dikarev, Inorg. Chem., 2018, 57, 2308–2313 CrossRef CAS PubMed.
- J. C. Knight, S. Therese and A. Manthiram, J. Electrochem. Soc., 2015, 162, A426–A431 CrossRef CAS.
- H. Berg and J. O. Thomas, Solid State Ionics, 1999, 126, 227–234 CrossRef CAS.
- C. M. Lieberman, A. S. Filatov, Z. Wei, A. Yu. Rogachev, A. M. Abakumov and E. V. Dikarev, Chem. Sci., 2015, 6, 2835–2842 RSC.
- H. Kobayashi, Y. Arachi, H. Kageyama and K. Tatsumi, J. Mater. Chem., 2004, 14, 40–42 RSC.
- C. Johnson, A. Gutierrez, P. Senguttuvan, S. Lapidus, S. Kim and T. T. Fister, ECS Meeting Abstracts, 2016, vol. MA2016-03, p. 1098 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, synthetic details, X-ray powder diffraction analysis, IR and mass spectra, phase analysis of thermal decomposition traces of heterometallic precursor. CCDC 1844130 and 1844131. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03816c |
| This journal is © The Royal Society of Chemistry 2019 |