
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of (−)-naltrexone†
Sun
Dongbang
,
Blaine
Pedersen
and
Jonathan A.
Ellman
 *
*
Department of Chemistry, Yale University, Connecticut 06520, USA. E-mail: jonathan.ellman@yale.edu; Tel: +1-203-432-2647
First published on 23rd October 2018
Abstract
(−)-Naltrexone, an opioid antagonist used extensively for the management of drug abuse, is derived from naturally occurring opioids. Herein, we report the first asymmetric synthesis of (−)-naltrexone that does not proceed through thebaine. The synthesis starts with simple, achiral precursors with catalytic enantioselective Sharpless dihydroxylation employed to introduce the stereogenic centers. A Rh(I)-catalyzed C–H alkenylation and torquoselective electrocyclization cascade provides the hexahydro isoquinoline bicyclic framework that serves as the precursor to the morphinan core. The acidic conditions used for Grewe cyclization not only provide the morphinan framework, but also cause a hydride shift resulting in the introduction of the C-6 oxo functionality present in (−)-naltrexone. The C-14 hydroxyl group is installed by an efficient two-step sequence of Pd-mediated ketone to enone dehydrogenation followed by C–H allylic oxidation using Cu(II) and O2, a method that has not previously been reported either for the synthesis or semi-synthesis of opioids. The longest linear sequence is 17 steps, and because the stereogenic centers in the product rely on Sharpless asymmetric dihydroxylation, the route could be used to access either enantiomer of the natural product, which have disparate biological activities. The route also may be applicable to the preparation of opioid derivatives that could not be easily prepared from the more fully elaborated and densely functionalized opioid natural products that have traditionally served as the starting inputs.
Introduction
(−)-Morphine (1) is one of the oldest and most extensively used analgesics which led it, along with its congeners (−)-codeine (2) and (−)-thebaine (3), to garner significant attention from numerous organic chemists as targets for synthesis (Fig. 1).1 Extensive modifications to the naturally occurring opioids have also been investigated in endeavors to increase potency and in vivo efficacy and have led to the discovery of semi-synthetic opioids such as (−)-ketorfanol (4)2 and the extensively prescribed (−)-oxycodone (5).3 However, despite the medical importance of naturally occurring and semi-synthetic opioid agonists, undesired side effects such as addictive properties and the potential for fatal overdoses are enormous societal problems. Therefore, semi-synthetic opioid antagonists were developed to address these growing problems.4 Naltrexone (6) was first patented in 1967 (ref. 5) and is currently an important treatment option for opioid abuse and alcohol dependence.4 The C-14 hydroxyl and N-cyclopropylmethyl substituent are essential structural features that are important for (−)-naltrexone's potency and antagonistic properties.4 | ||
| Fig. 1 Representative naturally occurring opioids (1–3) and semisynthetic opioid agonists (4–5) and an antagonist (6). | ||
The prevalent commercial routes to (−)-naltrexone (6) employ the natural product (−)-thebaine (3) as the starting material. A number of strategies have been developed for exchange of the N-methyl for an N-cyclopropylmethyl group.6
Hudlicky has extensively investigated this step,7 including a particularly efficient alkylation and demethylation sequence of the thebaine derivative oripavine to give 9 (Fig. 2).7c The dienol ether functionality present in (−)-thebaine allows for the straightforward introduction of the C-14 hydroxyl group by oxidative methods. For example, in Hudlicky's sequence, the dienol ether functionality in 9 was epoxidized followed by in situ oxirane ring opening to generate enone 10 followed by hydrogenation to give (−)-naltrexone (Fig. 2).8 Rice has also reported a semi-synthesis of the (+)-enantiomer of naltrexone. His synthesis proceeded through (+)-thebaine, which was prepared in five steps from the natural product (+)-sinomenine as an innovative starting material.9
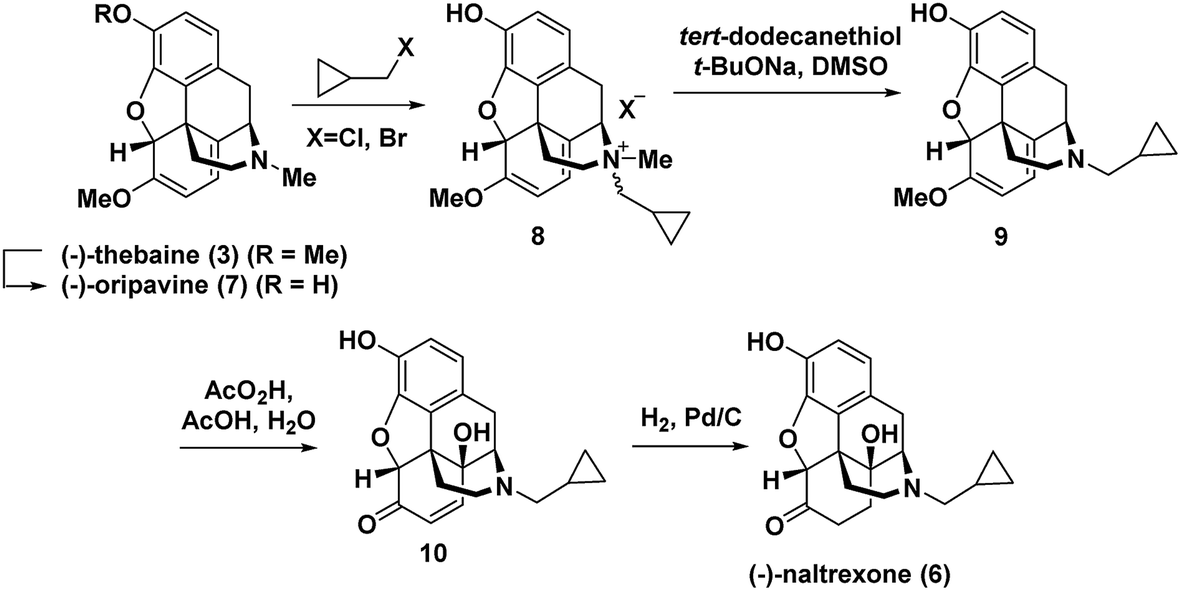 | ||
| Fig. 2 Hudlicky's semi-synthesis of (−)-naltrexone.7c | ||
(−)-Thebaine is currently isolated from opium poppies but is only a minor component of opium (0.3–1.5%), and poppy farming is problematic due to illicit drug activities.10 An alternative synthetic route that bypasses (−)-thebaine and starts with commercially available achiral precursors would provide novel entry to (−)-naltrexone. Moreover, starting from simple inputs should enable the investigation of a variety of analogs not accessible from more fully elaborated and densely functionalized morphinan natural products like (−)-thebaine.11
Recently, we reported the synthesis of the unnatural enantiomer of the opioid agonist (+)-ketorfanol (4) (Fig. 1) using a Rh(I)-catalysed C–H alkenylation and torquoselective 6-π electrocyclization cascade as a key step in the sequence.12 Herein, we further apply this cascade approach to the synthesis of the considerably more complex opioid antagonist (−)-naltrexone (6) (Scheme 1). Imine 11 is efficiently prepared from achiral starting materials with catalytic asymmetric dihydroxylation used to introduce the stereogenic centers. Intramolecular Rh(I)-catalysed alkenylation to give azatriene 12 is followed by in situ torquoselective electrocyclization to set the desired stereochemistry in bicyclic hexahydroisoquinoline 13, which is reduced to obtain 14 as a single diastereomer. Acid treatment provides the tetracyclic morphinan 15 by concomitant removal of the protecting groups, Grewe cyclization and redox neutral conversion of the diol to the desired keto group. The 4-methoxy-3,5-disilyloxy substitution pattern on the phenyl ring of 14 was designed to allow for incorporation of the required aromatic ring oxygen substitution pattern while simultaneously ensuring that Grewe cyclization occurs without the possibility of generating regiosomeric products (vide infra).1f The pentacyclic ether 16 is then obtained from tetracyclic 15 by treatment with Br2 in AcOH according to methods developed for the synthesis of codeine.13
 | ||
| Scheme 1 Approach to (−)-naltrexone from simple, achiral precursors. | ||
For intermediate 16, installation of the hydroxyl group at C-14 would most efficiently be accomplished by dehydrogenation to an enone followed by C–H γ-hydroxylation. However, in syntheses of morphinan natural products such as morphine and codeine, researchers have found that for the efficient dehydrogenation of 6-keto derivatives to enones the nitrogen must be protected by an electron withdrawing carbamoyl or sulphonamide rather than an N-alkyl group as is present in 16.1q,1u,14 Therefore, to enable dehydrogenation and γ-hydroxylation of 16 without the protection of the nitrogen, a Pd-mediated dehydrogenation method and Cu(II)-catalysed O2-mediated allylic C–H oxidation conditions were developed. Alkene hydrogenation with concomitant reductive cleavage of both the triflate and the bromide in 18 gave 19, which upon demethylation provided (−)-naltrexone in 17 steps for the longest linear sequence. Because the stereochemistry is set by asymmetric catalytic dihydroxylation, the same route could equally be employed to prepare the (+)-enantiomer of naltrexone, which has been reported to have antagonist activity toward Toll-like receptor 4.15
Results and discussion
The synthesis commenced with the preparation of imine 11 in seven steps from simple achiral starting materials (Scheme 2). Horner–Wadsworth–Emmons (HWE) reaction of aldehyde 21 and phosphonate 23 afforded 24 in 67% yield. Based upon a report by Takacs,16 we found that use of LiOH as the base in the presence of 4 Å molecular sieves minimized competitive self-condensation of aldehyde 21 and thus provided the most robust and reproducible HWE reaction conditions particularly on larger scales. Highly regio- and enantioselective dihydroxylation with AD-mix-β followed by protection of the diol as an acetonide gave 26 in high overall yield. The benzyl chloride 27, which was coupled with alkyne 26, was readily prepared in 75% overall yield from 4-O-methyl-3,5-dihydroxybenzoic acid, by silylation,17 LiAlH4 reduction to the benzyl alcohol,1af and chlorination with thionyl chloride. For the coupling of benzyl chloride 27 and alkyne 26, the Cu-free conditions developed by Buchwald for the Heck alkynylation of benzyl chlorides proved to be the most effective approach.18 Coupling product 28 was reliably obtained in good yield using Pd(OAc)2 as the precatalyst and X-Phos as the ligand. DIBAL reduction of the ester followed by Dess–Martin periodinane (DMP) oxidation of the resulting alcohol gave aldehyde 29 in 71% overall yield. Condensation with cyclopropylmethylamine then provided 11, which was taken on to the next step without purification. | ||
| Scheme 2 Reactions and conditions: (a) (COCl)2 (1.16 equiv.), DMSO (2.2 equiv.), Et3N (5.0 equiv.), CH2Cl2, −78 → 23 °C; (b) P(OEt)3, neat, 120 °C; (c) LiOH·H2O (1.1 equiv.), MS 4 Å, THF, reflux; (d) K2OsO4·2H2O (1 mol%), (DHQD)2PHAL (5 mol%), K3Fe(CN)6 (3.00 equiv.), K2CO3 (3.00 equiv.), MeSO2NH2 (1.00 equiv.), tBuOH/H2O, 0 °C; (e) 2,2-dimethoxypropane (10 equiv.), TsOH·H2O (10 mol%), CH2Cl2, 0 °C; (f) Pd(OAc)2 (5 mol%), XPhos (15 mol%), Cs2CO3 (1.5 equiv.), dioxane, 65 °C; (g) DIBAL (4.0 equiv.), THF, −78 °C; (h) DMP (1.75 equiv.), pyridine (6.0 equiv.), CH2Cl2, 0 °C; (i) cyclopropylmethylamine (1.2 equiv.), MS 3 Å, PhMe, 23 °C. | ||
Next, imine 11 was treated with 5 mol% of [RhCl(coe)2]2 precatalyst using (pNMe2)PhPEt2 as the ligand with heating in toluene to initiate the Rh(I)-catalysed cascade sequence. This process proceeds by Rh(I)-catalysed C–H activation followed by intramolecular insertion of the alkyne to give azatriene 12, which in situ undergoes rapid electrocyclization to provide 1,2-dihydropyridine 13 (Scheme 3). This cascade process not only forms the desired bicyclic ring system, but also proceeds with high torquoselectivity as enforced by the isopropylidene protected diol to provide the desired diastereomer.12 Without isolation, hexahydroisoquinoline 13 was reduced under mild conditions to give the octahydroisoquinoline 14 as a single stereoisomer in 66% overall yield from imine 11.
 | ||
| Scheme 3 Rh(I) C–H functionalization cascade. Condition and reagents: (a) [RhCl(coe)2]2 (5 mol%), (pNMe2)PhPEt2 (10 mol%), PhMe, 85 °C; (b) NaBH(OAc)3 (5.0 equiv.), AcOH, EtOH, 0 °C. | ||
Treatment of octahydroisoquinoline 14 with dilute H3PO4 and heat afforded morphinan 15 in 66% yield (Scheme 4). Acid treatment to provide morphinan 15 likely proceeds by removal of the silyl and acetonide protecting groups followed by in situ redox neutral conversion of the diol to the keto group via allylic alcohol ionization and a hydride shift to provide 30, which then undergoes Grewe cyclization. Methods for α-bromination and subsequent nucleophilic displacement by an adjacent phenol as developed for the synthesis of codeine13 gave the dihydrobenzofuran 16 in good overall yield. Two equivalents of Br2 were needed because the first equivalent of Br2 was very rapidly consumed by bromination of the highly electron-rich aromatic ring. Bromine substitution did not pose a problem because we anticipated that it could be removed with a global reduction step planned for later in the sequence (vide infra). Prior to installation of the C-14 hydroxyl, the free phenol in 16 was converted to the triflate in 17,17 which would also be removed in the reduction step.
 | ||
| Scheme 4 Synthesis of dehydrogenated enone 31 as the precursor to Installation of C-14. Conditions and reagents: (a) 55% H3PO4, 125 °C; (b) Br2 (2.0 equiv.), AcOH, 23 °C; NaOH(aq), 23 °C; (c) Tf2O (3.3 equiv.), pyridine, 0 °C; (d) Pd(TFA)2 (1.4 equiv.), TFA, DMSO, 80 °C. | ||
C-14 C–H hydroxylation of triflate 17 first required dehydrogenation to enone 31 in order to activate this site for γ oxidation (Scheme 4). Direct dehydrogenation has been reported to proceed with low yield for basic opioids with N-alkyl amine substituents.14 To overcome this challenge in the synthesis of opioids, the amine is typically protected as a sulfonamide or carbamate,1q,au,14 which then requires subsequent protecting group removal and installation of the N-alkyl group. Alternatively, more indirect, longer sequences have been developed to introduce this unsaturation.19 We instead chose to investigate contemporary ketone dehydrogenation approaches that have been reported to be compatible with amine functionality. We first evaluated the IBX-MPO system, but the conversion of 17 to enone 31 was not observed.20 Efficient, catalytic dehydrogenation methods utilizing [Pd(allyl)2Cl]2 with zinc amide bases have recently been reported,21 but only partial conversion to enone 31 along with some allylation at the α-position occurred. Dehydrogenation with Pd(TFA)2 in AcOH using O2 as an external oxidant resulted in significant N-dealkylation.22 This outcome is not surprising because Pd(II) catalysts under oxidizing conditions have been developed for the N-dealkylation of tertiary amines.7a,23 However, by addition of trifluoroacetic acid to protect the amine as the corresponding salt, along with DMSO as a coordinating solvent to stabilize Pd(TFA)2, complete conversion of ketone 17 was observed, with enone 31 isolated in 82% yield.
For the C-14 C–H hydroxylation of enone 31, we looked for inspiration in prior semi-syntheses of oxycodone from morphine and codeine. Unfortunately, neither Co(OAc)3 in acetic acid as reported by Rice24 nor MnO2 in CHCl3 as reported by Sainsbury25 resulted in C-14 hydroxylation of 31. We next investigated a method reported for the direct C-14 C–H hydroxylation of codeinone by metal-catalysed peroxidation with O2 followed by in situ reduction with sodium thiosulfate (entry 1, Table 1).26 Disappointingly, no product was detected presumably because enone 31 is completely insoluble in the aqueous pH 8 buffer used as the solvent.27 This led us to extensively explore organic co-solvents in combination with different oxidation catalysts and reductants. A large number of different co-solvents were first investigated, including DMF, THF, EtOH, DMSO and CH3CN, but little to no product was detected. With pyridine as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 co-solvent with aqueous pH 8 buffer, enone 31 was highly soluble, and while some of the desired C–H hydroxylation product 18 was formed, extensive over-oxidation occurred as determined by LCMS (entry 2). However, by attenuating the basicity of the PBS buffer to pH 7, an improved yield of desired product 18 was obtained (entry 3). Under these conditions, CuSO4 proved to be a superior oxidation catalyst than KMnO4 (entry 4). Use of ketoglutaric acid instead of thiosulfate to reduce the peroxide intermediate was investigated for both KMnO4 (entry 5) and CuSO4 (entry 6). A significant improvement was observed for both catalysts with the highest yield obtained for CuSO4 (entry 6). Finally, varying the ratio of pyridine to PBS buffer resulted in vast differences in reaction rate. While the use of pyridine without any aqueous co-solvent resulted in a slower rate, reaction progress could be more easily monitored and therefore the reaction could be reliably terminated before significant over-oxidation had occurred (entry 7).
:1 co-solvent with aqueous pH 8 buffer, enone 31 was highly soluble, and while some of the desired C–H hydroxylation product 18 was formed, extensive over-oxidation occurred as determined by LCMS (entry 2). However, by attenuating the basicity of the PBS buffer to pH 7, an improved yield of desired product 18 was obtained (entry 3). Under these conditions, CuSO4 proved to be a superior oxidation catalyst than KMnO4 (entry 4). Use of ketoglutaric acid instead of thiosulfate to reduce the peroxide intermediate was investigated for both KMnO4 (entry 5) and CuSO4 (entry 6). A significant improvement was observed for both catalysts with the highest yield obtained for CuSO4 (entry 6). Finally, varying the ratio of pyridine to PBS buffer resulted in vast differences in reaction rate. While the use of pyridine without any aqueous co-solvent resulted in a slower rate, reaction progress could be more easily monitored and therefore the reaction could be reliably terminated before significant over-oxidation had occurred (entry 7).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalysta (2 mol%) | Reductantb (4.5 equiv.) | Solventc | Timed (h) | NMR yielde (%) | |
| sm(31) | pdt(18) | |||||
| a Catalysts were added as 5 mM stock solutions in deionized H2O. b Reductants were added as a 150 mM stock solution in deionized H2O. c All solvents were sparged with O2 prior to reaction. d The reaction was stopped when the product was at maximum yield as determined by LCMS exact ion count. e 1,3,5-Trimethoxybenzene was used as a standard for NMR yields. Remaining percent balance corresponds to unidentified overoxidized or degraded product. | ||||||
| 1 | KMnO4 | Na2S2O3 | PBS pH 8 | 42 | 49 | 0 |
| 2 | KMnO4 | Na2S2O3 | PBS pH 8/pyridine (1:1) |
42 | 0 | 11 |
| 3 | KMnO4 | Na2S2O3 | PBS pH 7/pyridine (1:1) |
42 | 11 | 17 |
| 4 | CuSO4 | Na2S2O3 | PBS pH 7/pyridine (1:1) |
5 | 5 | 31 |
| 5 | KMnO4 | Ketoglutaric acid | PBS pH 7/pyridine (1:1) |
18 | 15 | 33 |
| 6 | CuSO4 | Ketoglutaric acid | PBS pH 7/pyridine (1:1) |
1.5 | 0 | 55 |
| 7 | CuSO4 | Ketoglutaric acid | Pyridine | 48 | 8 | 59 |

After γ-hydroxylation to afford 18, we attempted to remove the aryl bromide and triflate as well as hydrogenate the enone to access 19 in a single step (Scheme 5). While formic acid with Pd(PPh3)4 only reductively cleaved the triflate and bromide, hydrogenation with H2 and Pd/C reduced the double bond and achieved hydrodebromination but without reductive cleavage of the triflate. However, Pearlman's catalyst (Pd(OH)2) under 1 atm of H2 resulted in the complete reduction of the double bond along with the reductive removal of both the bromide and the triflate to give 19 in nearly quantitative yield.28 For this reason, 19 was not purified but rather was directly submitted to final BBr3 mediated demethylation, affording (−)-naltrexone 6 in 67% yield over the two steps.
 | ||
| Scheme 5 Installation of C-14 hydroxylation and endgame synthesis to (−)-naltrexone (6). Conditions and reagents: (a) CuSO4 (2 mol%), ketoglutarate (4.5 equiv.), pyridine, 23 °C, O2; (b) Et3N (10 equiv.), Pd(OH)2 (20 wt%), EtOAc:MeOH = 1:3, H2, 23 °C; (c) BBr3 (5 equiv.), CH2Cl2, −40 → 0 °C. | ||
Conclusions
We have developed a new approach for the synthesis of (−)-naltrexone in 17 linear steps. Starting with commercially available achiral substrates, a bicyclic hexahydroquinoline intermediate was accessed via a Rh(I)-catalyzed C–H alkenylation and torquoselective electrocyclization cascade. Grewe cyclization then provided the morphinan core with a concomitant hydride shift introducing the C-6 oxo functionality present in naltrexone. After formation of the dihydrobenzofuran, Pd-mediated dehydrogenation to the enone followed by allylic C–H oxidation using Cu(II) and O2 introduced the C-14 hydroxyl group. This new route for the asymmetric synthesis of (−)-naltrexone from simple, achiral precursors could provide a means to prepare morphinan derivatives that would be difficult to access through semi-synthesis from opioid natural products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NIH (R35GM122473). We gratefully acknowledge Prof. Timothy Newhouse for helpful discussions.Notes and references
- For selected reviews and publications on total synthesis of opioids: (a) U. Rinner and T. Hudlicky, Top. Curr. Chem., 2012, 309, 33 CrossRef CAS PubMed; (b) J. Zezula and T. Hudlicky, Synlett, 2005, 388 CAS; (c) M. Gates and G. Tschudi, J. Am. Chem. Soc., 1952, 74, 1109 CrossRef CAS; (d) D. Elad and D. Ginsburg, J. Am. Chem. Soc., 1954, 76, 312 CrossRef CAS; (e) R. Grewe and W. Friedrichsen, Chem. Ber., 1967, 100, 1550 CrossRef CAS PubMed; (f) H. C. Beyerman, J. van Berkel, T. S. Lie, L. Maat, J. C. M. Wessels, H. H. Bosman, E. Buurman, E. J. M. Bijsterveld and H. J. M. Sinnige, Recl. Trav. Chim. Pays-Bas, 1978, 97, 127 CrossRef CAS; (g) K. C. Rice, J. Org. Chem., 1980, 45, 3135 CrossRef CAS; (h) W. H. Moos, R. D. Gless and H. Rapoport, J. Org. Chem., 1983, 48, 227 CrossRef CAS; (i) J. D. White, G. Caravatti, T. B. Kline, E. Edstrom, K. C. Rice and A. Brossi, Tetrahedron, 1983, 39, 2393 CrossRef CAS; (j) J. E. Toth, P. R. Hamann and P. L. Fuchs, J. Org. Chem., 1988, 53, 4694 CrossRef CAS; (k) K. A. Parker and D. Fokas, J. Am. Chem. Soc., 1992, 114, 9688 CrossRef CAS; (l) M. A. Tius and M. A. Kerr, J. Am. Chem. Soc., 1992, 114, 5959 CrossRef CAS; (m) C. Y. Hong, N. Kado and L. E. Overman, J. Am. Chem. Soc., 1993, 115, 11028 CrossRef CAS; (n) J. Mulzer, G. Dürner and D. Trauner, Angew. Chem., Int. Ed., 1996, 35, 2830 CrossRef CAS; (o) J. D. White, P. Hrnciar and F. Stappenbeck, J. Org. Chem., 1997, 62, 5250 CrossRef CAS; (p) G. Butora, T. Hudlicky, S. Fearnley, M. Stabile, A. Gum and D. Gonzales, Synthesis, 1998, 665 CrossRef CAS; (q) D. F. Taber, T. D. Neubert and A. L. Rheingold, J. Am. Chem. Soc., 2002, 124, 12416 CrossRef CAS PubMed; (r) B. M. Trost and W. Tang, J. Am. Chem. Soc., 2002, 124, 14542 CrossRef CAS PubMed; (s) B. M. Trost, W. Tang and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 14785 CrossRef CAS PubMed; (t) K. A. Parker and D. Fokas, J. Org. Chem., 2006, 71, 449 CrossRef CAS PubMed; (u) K. Uchida, S. Yokoshima, T. Kan and T. Fukuyama, Org. Lett., 2006, 8, 5311 CrossRef CAS PubMed; (v) A. T. Omori, K. J. Finn, H. Leisch, R. J. Carroll and T. Hudlicky, Synlett, 2007, 2859 CAS; (w) H. Tanimoto, R. Saito and N. Chida, Tetrahedron Lett., 2008, 49, 358 CrossRef CAS; (x) M. Varin, E. Barré, B. Iorga and C. Guillou, Chem.–Eur. J., 2008, 14, 6606 CrossRef CAS PubMed; (y) H. Leisch, A. T. Omori, K. J. Finn, J. Gilmet, T. Bissett, D. Ilceski and T. Hudlicky, Tetrahedron, 2009, 65, 9862 CrossRef CAS; (z) P. Magnus, N. Sane, B. P. Fauber and V. Lynch, J. Am. Chem. Soc., 2009, 131, 16045 CrossRef CAS PubMed; (a a) G. Stork, A. Yamashita, J. Adams, G. R. Schulte, R. Chesworth, Y. Miyazaki and J. J. Farmer, J. Am. Chem. Soc., 2009, 131, 11402 CrossRef CAS PubMed; (a b) H. Koizumi, S. Yokoshima and T. Fukuyama, Chem.–Asian J., 2010, 5, 2192 CrossRef CAS PubMed; (a c) T. Erhard, G. Ehrlich and P. Metz, Angew. Chem., Int. Ed., 2011, 50, 3892 CrossRef CAS PubMed; (a d) V. Varghese and T. Hudlicky, Synlett, 2013, 369 CAS; (a e) J. Li, G.-L. Liu, X.-H. Zhao, J.-Y. Du, H. Qu, W.-D. Chu, M. Ding, C.-Y. Jin, M.-X. Wei and C.-A. Fan, Chem.–Asian J., 2013, 8, 1105 CrossRef CAS PubMed; (a f) M. Geffe and T. Opatz, Org. Lett., 2014, 16, 5282 CrossRef CAS PubMed; (a g) M. Tissot, R. J. Phipps, C. Lucas, R. M. Leon, R. D. M. Pace, T. Ngouansavanh and M. J. Gaunt, Angew. Chem., Int. Ed., 2014, 53, 13498 CrossRef CAS PubMed; (a h) L. Rycek, J. J. Hayward, M. A. Latif, J. Tanko, R. Simionescu and T. Hudlicky, J. Org. Chem., 2016, 81, 10930 CrossRef CAS PubMed; (a i) K. H. Park, R. Chen and D. Y.-K. Chen, Chem. Sci., 2017, 8, 7031 RSC.
- A. Manmade, H. C. Dalzell, J. F. Howes and R. K. Razdan, J. Med. Chem., 1981, 24, 1437 CrossRef CAS PubMed.
- (a) E. Falk, Muench. Med. Wochenschr., 1917, 20, 381 Search PubMed; (b) E. Kalso, J. Pain Symptom Manage., 2005, 29, 47 CrossRef PubMed.
- V. Varghese and T. Hudlicky, A Short History of the Discovery and Development of Naltrexone and Other Derivatives, in Natural Products in Medicinal Chemistry, ed. S. Hanessian, Wiley-VCH, Weinheim, 2014, ch. 6, pp. 225–250 Search PubMed.
- H. Blumberg, I. J. Pachter and Z. Matossian, US Pat., 1967, vol. 3, pp. 332–950.
- For examples of N-demethylation of opioids: (a) J. von Braun, Ber. Dtsch. Chem. Ges., 1900, 33, 1438 CrossRef CAS; (b) K. C. Rice, J. Org. Chem., 1975, 40, 1850 CrossRef CAS PubMed; (c) K. C. Rice and E. L. May, J. Heterocycl. Chem., 1977, 14, 665 CrossRef CAS; (d) J. H. Cooley and E. J. Evain, Synthesis, 1989, 1, 1 CrossRef; (e) R. A. Olofson, J. T. Martz, J. P. Senet, M. Piteau and T. Malfroot, J. Org. Chem., 1984, 49, 2081 CrossRef CAS; (f) A. Coop, J. W. Janetka, J. W. Lewis and K. C. Rice, J. Org. Chem., 1998, 63, 4392 CrossRef CAS; (g) K. McCamley, J. A. Ripper, R. D. Singer and P. J. Scammells, J. Org. Chem., 2003, 68, 9847 CrossRef CAS PubMed; (h) G. Kok, T. D. Asten and P. J. Scammells, Adv. Synth. Catal., 2009, 351, 283 CrossRef CAS; (i) Z. Dong and P. J. Scammells, J. Org. Chem., 2007, 72, 9881 CrossRef CAS PubMed; (j) G. B. Kok, C. C. Pye, R. D. Singer and P. J. Scammells, J. Org. Chem., 2010, 75, 4806 CrossRef CAS PubMed; (k) G. B. Kok and P. J. Scammells, Bioorg. Med. Chem. Lett., 2010, 20, 4499 CrossRef CAS PubMed; (l) L. Werner, A. Machara, D. R. Adams, D. P. Cox and T. Hudlicky, J. Org. Chem., 2011, 76, 4628 CrossRef CAS.
- For semi-synthesis of naltrexone via oxymorphone or oripavine: (a) A. Machara, D. P. Cox and T. Hudlicky, Adv. Synth. Catal., 2012, 354, 2713 CrossRef CAS; (b) M. A. Endoma-Arias, D. P. Cox and T. Hudlicky, Adv. Synth. Catal., 2013, 355, 1869 CrossRef CAS; (c) A. Machara, L. Werner, H. Leisch, R. J. Carroll, D. R. Adams, D. Mohammad Haque, D. P. Cox and T. Hudlicky, Synlett, 2015, 2101 CAS.
- For examples of the conversion of thebaine to opioids containing the C-14 hydroxyl group: (a) M. Freund and E. Speyer, J. Prakt. Chem., 1916, 94, 135 CrossRef CAS; (b) F. H. Hauser, T.-K. Chen and F. I. Carroll, J. Med. Chem., 1974, 17, 1117 CrossRef CAS PubMed; (c) I. Iijima, K. C. Rice and A. Brossi, Helv. Chim. Acta, 1977, 60, 2135 CrossRef CAS PubMed; (d) R. Krassnig, C. Hederer and H. Schmidhammer, Arch. Pharm., 1996, 329, 325 CrossRef CAS; (e) D. Lopez, E. Quinoa and R. Riguera, Tetrahedron Lett., 1994, 35, 5727 CrossRef CAS; (f) G. B. Kok and P. J. Scammells, RSC Adv., 2012, 2, 11318 RSC.
- (a) I. Iijima, J. Minamikawa, A. E. Jacobson, A. Brossi and K. C. Rice, J. Med. Chem., 1978, 21, 398 CrossRef CAS PubMed; (b) B. R. Selfridge, X. Wang, Y. Zhang, H. Yin, P. M. Grace, L. R. Watkins, A. E. Jacobson and K. C. Rice, J. Med. Chem., 2015, 58, 5038 CrossRef CAS PubMed.
- United Nations, World Drug Report 2018, Analysis of Drug Markets, Opiates, Cocaine, Cannabis, Synthetic Drugs, United Nations Publication, 2018, booklet 3 Search PubMed.
- The synthesis of (−)-oxycodone, a semi-synthetic opioid agonist, from simple commercially available precursors has been reported: A. Kimishima, H. Umihara, A. Mizoguchi, S. Yokoshima and T. Fukuyama, Org. Lett., 2014, 16, 6244 CrossRef CAS PubMed.
- E. M. Phillips, T. Mesganaw, A. Patel, S. Duttwyler, B. Q. Mercado, K. N. Houk and J. A. Ellman, Angew. Chem., Int. Ed., 2015, 54, 12044 CrossRef CAS PubMed.
- D. D. Weller and H. Rapoport, J. Med. Chem., 1976, 19, 1171 CrossRef CAS PubMed.
- I. Iijima and K. C. Rice, Heterocycles, 1977, 6, 1157 CrossRef CAS.
- (a) M. R. Hutchinson, Y. Zhang, K. Brown, B. D. Coats, M. Shridhar, P. W. Sholar, S. J. Patel, N. Y. Crysdale, J. A. Harrison, S. F. Maier, K. C. Rice and L. R. Watkins, Eur. J. Neurosci., 2008, 28, 20 CrossRef PubMed; (b) X. Wang, Y. Zhang, Y. Peng, M. R. Hutchinson, K. C. Rice, H. Yin and L. R. Watkins, Br. J. Pharmacol., 2016, 173, 856 CrossRef CAS PubMed.
- J. M. Takacs, M. R. Jaber, F. Clement and C. Walters, J. Org. Chem., 1998, 63, 6757 CrossRef CAS.
- M. Node, S. Kodama, Y. Hamashima, T. Katoh, K. Nishide and T. Kajimoto, Chem. Pharm. Bull., 2006, 54, 1662 CrossRef CAS PubMed.
- C. H. Larsen, K. W. Anderson, R. E. Tundel and S. L. Buchwald, Synlett, 2006, 2941 CAS.
- For examples of introducing unsaturation with basic opioids via ketal protection: (a) D. Passarella, A. Consonni, A. Giardini, G. Lesma and A. Silvani, Bioorg. Med. Chem. Lett., 2002, 12, 1981 CrossRef CAS PubMed; (b) See ref. 13.
- K. C. Nicolaou, T. Montagnon and P. S. Baran, Angew. Chem., Int. Ed., 2002, 41, 993 CrossRef CAS PubMed.
- D. Huang, Y. Zhao and T. R. Newhouse, Org. Lett., 2018, 20, 684 CrossRef CAS PubMed.
- T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566 CrossRef CAS PubMed.
- B. Gutmann, P. Elsner, D. P. Cox, U. Weigl, D. M. Roberge and C. O. Kappe, ACS Sustainable Chem. Eng., 2016, 4, 6048 CrossRef CAS.
- A. Coop and K. C. Rice, Tetrahedron, 1999, 55, 11429 CrossRef CAS.
- A. Ninan and M. Sainsbury, Tetrahedron, 1992, 48, 6709 CrossRef CAS.
- Q. Zhang, J. O. Rich, I. C. Cotterill, D. P. Pantaleone and P. C. Michels, J. Am. Chem. Soc., 2005, 127, 7286 CrossRef CAS PubMed.
- Intermediate 31 is much more hydrophobic and less soluble in aqueous solution than codeinone.
- A. P. Kozikowski, W. Tuckmantel and C. George, J. Org. Chem., 2000, 65, 5371 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03748e |
| This journal is © The Royal Society of Chemistry 2019 |