
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFolding of unstructured peptoids and formation of hetero-bimetallic peptoid complexes upon side-chain-to-metal coordination†
Maria
Baskin
a,
Hui
Zhu
b,
Zheng-Wang
Qu
b,
Jordan H.
Chill
c,
Stefan
Grimme
 b and
Galia
Maayan
*a
b and
Galia
Maayan
*a
aSchulich Faculty of Chemistry, Technion – Israel Institute of Technology, Technion City, Hailfa 32000, Israel. E-mail: gm92@technion.ac.il
bMulliken Center for Theoretical Chemistry, Rheinische Friedrich-Wilhelms-Universität Bonn, Beringstrasse 4, 53115 Bonn, Germany
cDepartment of Chemistry, Bar-Ilan University, Ramat Gan 52900, Israel
First published on 17th October 2018
Abstract
Helices are key structural features in biopolymers, enabling a variety of biological functions. Mimicking these secondary structure motifs has wide potential in the development of biomimetic materials. Peptoids, N-substituted glycine oligomers, are an important class of peptide mimics that can adopt polyproline type helices if the majority of their sequence consists of chiral bulky pendent groups. Such side-chains are structure inducers but they have no functional value. We present here the inclusion of several metal-binding groups in one peptoid oligomer as a new platform towards the development of functional helical peptoids. Thus, we describe the coordination of two metal ions to unstructured peptoids incorporating four 8-hydroxyquinoline (HQ) ligands at fixed positions as two (HQ, HQ) metal binding sites, and a mixture of chiral benzyl and alkyl substituents in varied positions along the peptoid backbone. For the first time, we demonstrate by circular dichroism spectroscopy, solution NMR techniques and high-level DFT calculations that some of these unstructured peptoids can fold upon metal binding to form helical structures. Replacing one HQ ligand with a terpyridine (Terpy) ligand resulted in unique sequences that can selectively coordinate Cu2+ to the (Terpy, HQ) and Zn2+ (or Co2+) to the (HQ, HQ) sites from a solution mixture containing Cu2+ and Zn2+ (or Co2+) ions. Interestingly, the binding of Cu2+ to the (Terpy, HQ) site in one of these peptoids can initiate a conformational change that in turn facilitates the coordination of Zn2+ (or Co2+) ions to the (HQ, HQ) site, demonstrating a unique example of positive allosteric cooperativity in peptide mimics.
Introduction
The remarkable efficiency and selectivity of biopolymers in carrying out valuable functions such as molecular recognition, information storage and catalysis, stem from the ability of their polypeptide backbone to adopt well-defined three-dimensional structures. The folding of biopolymers can be controlled by a variety of interactions including hydrogen bonding, metal–ligand interactions, and solvophobic effects.1 Among those, metal–ligand interactions are particularly interesting because metal ions are key elements not only in the structure but also in the function of biopolymers, being employed in tasks spanning from regulation to catalysis. In addition, there is a direct relationship between folding and metal coordination; while some biopolymers fold upon metal binding, others can only bind metal ions in their folded state.1 As a consequence, biopolymers exhibit high affinity and especially high selectivity towards specific metal ions that are required for their utility. Notably, many metalloproteins found in nature have multiple coordination sites suitable for the binding of at least two different metal ions, each at a distinct site, thus enabling cooperative and challenging catalytic tasks. Therefore, an important goal in the design of metal-binding proteins, peptides and peptide mimics is to generate structures that selectively bind different metals in distinct sites.2 The coordination of the metal ions Cu and Zn is especially intriguing because these metals, being the two most abundant trace elements in biological and ecological systems, play central roles in the structure and function of proteins and are both essential components in many enzymes, e.g. the zinc–copper superoxide dismutase.3 Mimicking such hetero bimetallic motifs using synthetic oligomers that can also fold upon binding and exhibit selective recognition of each metal ion might lead to unique functional compounds in which each metal ion plays a distinct role in their overall activity.In recent years, chemists have been starting to explore possibilities to imitate these unique recognition properties, mostly by developing biomimetic foldamers4 capable of binding metal ions.5–7 Peptoids, N-substituted glycine oligomers, are an important class of peptide mimics capable of forming well-defined secondary structures,8 with demonstrated biological and chemical applications including drug development,9 catalysis10 and selective recognition of metal ions.11 Peptoids can be synthesized efficiently using the solid phase sub-monomer method, which utilizes primary amines as synthons.12 This method enables the facile incorporation of a wide variety of side chains, resulting in highly versatile sequences, with the potential to include various functional groups at specified N-positions along the peptoid spine.13 Thus, several metal-binding ligands were incorporated within the peptoid sequences, leading to the formation of metallopeptoids.14 In the absence of hydrogen bonding, the formation of peptoid helices requires that at least two thirds of the peptoid side chains are chiral and bulky. Such sequences form stable folded structures, which resemble polyproline type helices (PP-I and PP-II).15 Although this approach is well established in peptoid chemistry, it only enables the production of very specific structures, dramatically limiting the sequence and functional diversity of peptoid helices, which currently, can only be generated from a few distinct side chains out of the numerous ones that exist. Moreover, these helix-inducing side chains only serve a structural purpose, and have no functional contribution to the peptoids. Therefore, it will be beneficial to develop alternative platforms for the creation of peptoid helices that increase their diversity on one hand, while providing some added functional value on the other hand. This may be performed by metal coordination, which proved efficient in the case of stabilization of peptide helices.16 Although metal binding was shown to increase the conformational order of helical peptoids,14b,17 the folding of unstructured peptoids upon metal coordination was not yet achieved.14j Moreover, there is currently one example of a peptoid dodacamer that forms homobimetallic metallopeptoids,14j but selective recognition of two different metal ions by one oligomer to form heterobimetallic metallopeptoid (or any other metallo-peptidomimetic) complexes was not demonstrated.
To accomplish these challenging tasks, we describe here the design, synthesis and characterization of four unstructured peptoid dodecamers containing a combination of chiral-bulky and chiral-small substituents and two metal binding sites based on four 8-hydroxyquinoline (HQ) side-chain ligands. We demonstrate for the first time that the binding of Cu2+, Zn2+ and Co2+ ions to un-structured peptoids can induce the formation of peptoid helices as evidenced by circular dichroism (CD) and NMR spectra, and using high-level DFT calculations. Due to the large size and high complexity of such metallopeptoids, both their experimental characterization and DFT analysis in solution and in the solid-state are highly challenging. We further show that two of these peptoids, modified by replacing one HQ with a terpyridine (Terpy) ligand, can selectively bind Cu2+ ions to the (Terpy, HQ) site and Zn2+ (or Co2+) to the other (HQ, HQ) site and form unique heterobimetallic metallopeptoids. Finally, we discovered that one of these peptoids can already fold upon the first and selective coordination of Cu2+ to the (Terpy, HQ) site, and that this event facilitates the binding of Zn2+ (or Co2+) ions to the second (HQ, HQ) site, presenting the first example of positive allosteric cooperativity in metallopeptoids.
Results and discussion
Rational design of unstructured peptoids with two (HQ, HQ) metal binding sites
It was previously suggested that both the overall number and the position of the chiral (S)-(−)-1-phenylethylamine (Nspe) side-chains, especially their presence at the N- and C-termini as well as next to the N-terminus, are important for deriving peptoid folding.17,18 Thus, we initially designed a peptoid dodecamer that has two Nspe groups at the N- and C-termini, four HQ ligands at the 2nd and 5th (site 1) as well as 7th and 10th (site 2) side-chain positions (i and i + 3 for each HQ pair as potential metal binding sites) and six S-methoxy-propylamine (Nsmp) groups at the other side-chain positions along the peptoid sequence (12P1, Fig. 1). This design relied on our previous observation that (a) sequences mainly composed of Nsmp side-chain groups lead to unstructured petoids14j and (b) the cooperative binding of metal ions to two ligands is much stronger at the i and i + 3 positions (such as sites 1 and 2) than at the i and i + 2 positions (here 5th and 7th, site 3).11a We therefore anticipated that this peptoid will be unstructured but the coordination of the two metal ions (one at each site) might lead to a significant increase in its conformational order and result in a helical structure. Moreover, we decided to synthesize peptoid dodecamers 12P2 and 12P3, in which one Nspe group is located at the position next to the N-terminus and the other at the C- or N-terminus, respectively (Fig. 1). The three peptoids were synthesized on a solid support, cleaved at the end of the synthesis and purified by high performance liquid chromatography (HPLC, >95%). Their molecular weights measured by electrospray mass spectrometry (ESI-MS) were consistent with the expected masses (see ESI†).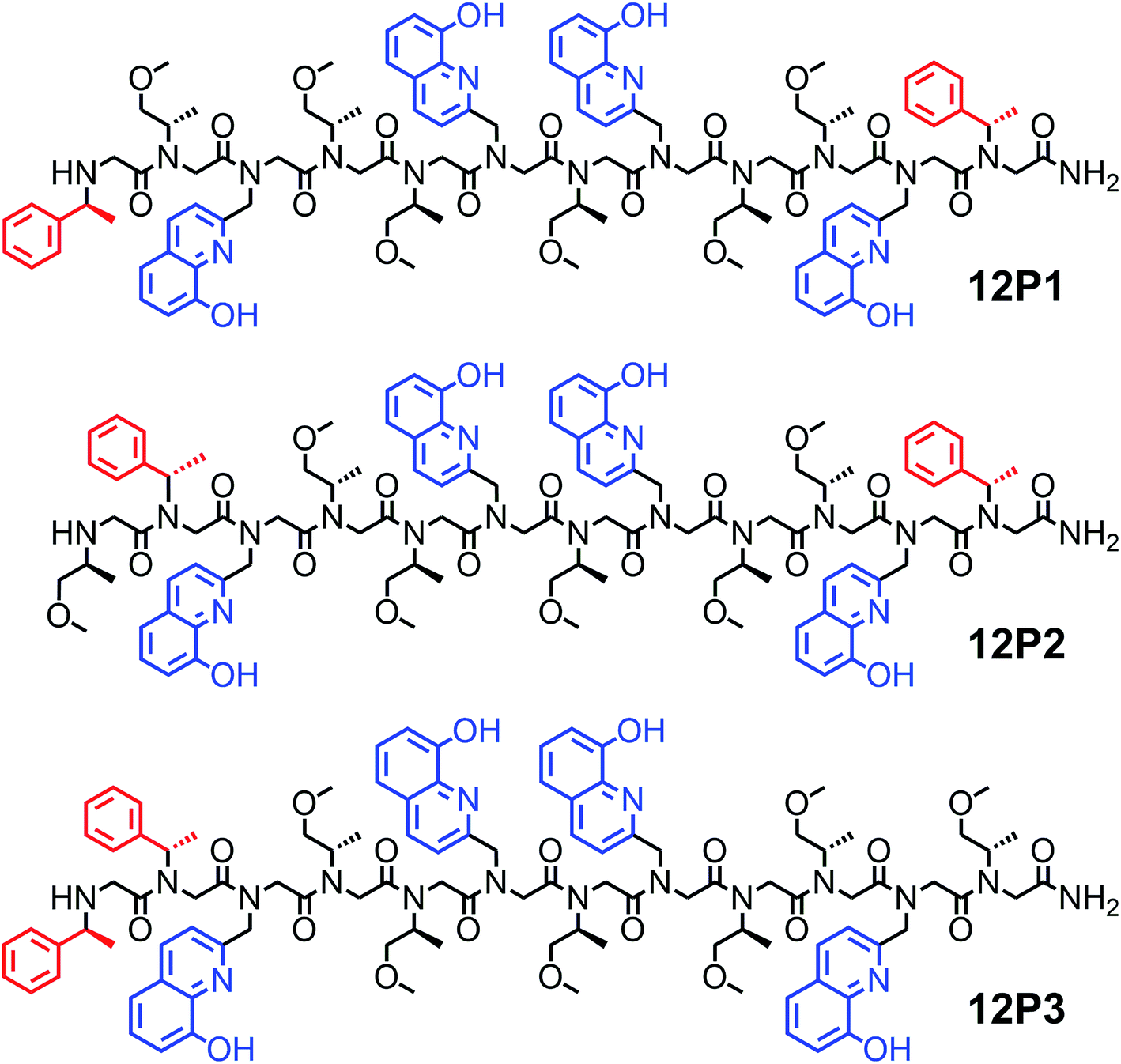 | ||
| Fig. 1 Chemical sequences of oligomers 12P1–12P3. | ||
Characterization of the metallopeptoids formed upon metal coordination
Metal-free peptoid 12P1 exhibits absorption bands near λ = 245 nm in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH:H2O solution. Titration with Cu2+ acetate led to the appearance of a new absorption band at λ = 264 nm, which corresponds to the formation of a peptoid–copper complex (Fig. 2a). In order to determine the stoichiometry of this complex we conducted a Job plot experiment by keeping the total molar concentrations of Cu2+ and of 12P1 constant at 7 μM and varying their relative proportions (mole fraction). The absorbance that is proportional to complex formation was plotted against the mole fraction and from the intersection point the peptoid-to-metal stoichiometry ratio was determined to be 2.06,19 demonstrating the formation of the 1:2 peptoid:metal complex (Fig. 2a, inset). UV-Vis titration experiments of 12P1 were further conducted with Zn2+ and with Co2+ ions and from the peptoid-to-metal ratio plot a stoichiometry of 1:2 was confirmed. The formation of intramolecular Cu2+, Zn2+ and Co2+ complexes was further corroborated by electrospray mass spectrometry (ESI-MS). Notably, no evidence for the formation of higher order complexes (e.g. 2:4 complexes) was detected. The MS-MS techniques further supported our assumption that metal binding occurs in sites 1 and 2 (see ESI†). Similar UV-Vis titration spectra were obtained upon the addition of the metal ions Cu2+, Zn2+ and Co2+ to 12P2 and 12P3 (see ESI†). Based on the peptoid-to-metal ratio plots, the stoichiometry of these metallopeptoids was shown to also be 1:2, as was further verified by their ESI-MS analysis in solution (see ESI†). Overall, the spectroscopic data demonstrate the formation of homo-bimetallic metallopeptoids, in which the metal ions are bound in sites 1 and 2 via intramolecular chelation.
:1 MeOH:H2O solution. Titration with Cu2+ acetate led to the appearance of a new absorption band at λ = 264 nm, which corresponds to the formation of a peptoid–copper complex (Fig. 2a). In order to determine the stoichiometry of this complex we conducted a Job plot experiment by keeping the total molar concentrations of Cu2+ and of 12P1 constant at 7 μM and varying their relative proportions (mole fraction). The absorbance that is proportional to complex formation was plotted against the mole fraction and from the intersection point the peptoid-to-metal stoichiometry ratio was determined to be 2.06,19 demonstrating the formation of the 1:2 peptoid:metal complex (Fig. 2a, inset). UV-Vis titration experiments of 12P1 were further conducted with Zn2+ and with Co2+ ions and from the peptoid-to-metal ratio plot a stoichiometry of 1:2 was confirmed. The formation of intramolecular Cu2+, Zn2+ and Co2+ complexes was further corroborated by electrospray mass spectrometry (ESI-MS). Notably, no evidence for the formation of higher order complexes (e.g. 2:4 complexes) was detected. The MS-MS techniques further supported our assumption that metal binding occurs in sites 1 and 2 (see ESI†). Similar UV-Vis titration spectra were obtained upon the addition of the metal ions Cu2+, Zn2+ and Co2+ to 12P2 and 12P3 (see ESI†). Based on the peptoid-to-metal ratio plots, the stoichiometry of these metallopeptoids was shown to also be 1:2, as was further verified by their ESI-MS analysis in solution (see ESI†). Overall, the spectroscopic data demonstrate the formation of homo-bimetallic metallopeptoids, in which the metal ions are bound in sites 1 and 2 via intramolecular chelation.
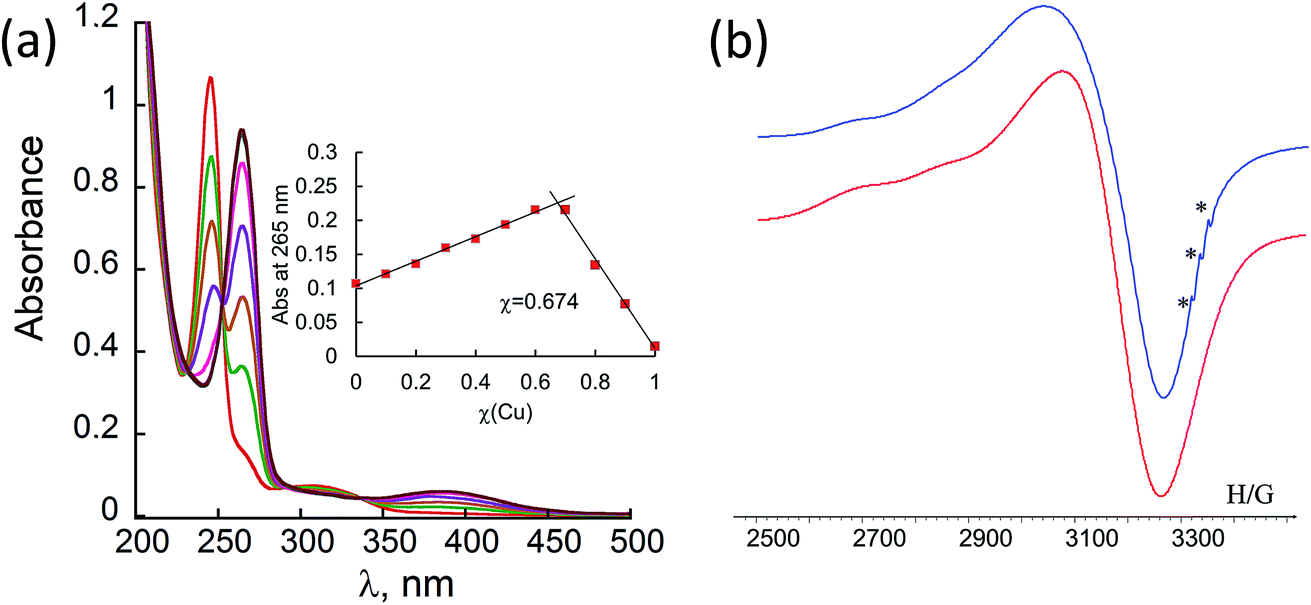 | ||
| Fig. 2 (a) UV-Vis titration (8.5 μM) and a Job plot of 12P1 (7 μM) with Cu2+ in 4:1 MeOH:H2O. (b) Room temperature X-band EPR spectrum of the copper–peptoid copper complex (12P1)Cu2 (blue line) in solid state and its simulated spectrum (red line). Reference – TEMPO (marked by *), g = 2.0058. | ||
In order to gain some more information about the structure of the peptoid–copper complexes, the X-band EPR spectra of the powdered samples were measured at room temperature (Fig. S2†). In all cases, the EPR signals clearly indicated the presence of Cu2+ ions, and the Hamiltonian parameters obtained from the spectra are shown in Table S3.† Interestingly, all the spectra exhibit a broadening of the EPR signals, which is due to an exchange interaction between two Cu2+ ions located within a distance of less than 1.5 nm.20 A representative EPR spectrum of (12P1)Cu2 is depicted in Fig. 2b. Based on calculated molecular models and EPR studies, it was previously suggested that the trans-planar configuration of Cu(hydroxyquinolate)2 is not possible in the intramolecular Cu2+(HQ2 peptoid) complexes due to steric hindrance from the peptoid backbone that should result in a pseudotetrahedral geometry.14b,17 The values of the EPR parameters of (12P1)Cu2, (12P2)Cu2 and (12P3)Cu2 (g‖ = 2.276, 2.279 and 2.275, g⊥ = 2.082, 2.084 and 2.080 and A‖ = 149, 148 and 147 G, respectively) are similar to those measured for other Cu2+(HQ2 peptoid) complexes, implying also here pseudotetrahedral coordination geometries.
Secondary structure of free and metal-coordinated peptoids: CD spectroscopy
Peptoids having only Nspe or (R)-N-(1-phenylethyl)glycine (Nrpe) side-chains (alpha-methyl-benzyl peptoids), were shown to generally adopt PP-I helices with three residues per turn,15 with a minor population of terminal trans-amide bonds.15c Typical CD spectra of helical alpha-methyl-benzyl peptoids measured in organic solvents exhibit double minima near 200 and 220 nm due to the mixed population of the cis-and trans-amide bonds.15c The peptoid 12P1 exhibits only an intense minimum near 195 nm (without a minimum near 220 nm) similar to the CD spectra of a random coil polypeptide, indicating that the peptoid is unstructured in solution (Fig. 3a and b, blue line). The coordination of the Cu2+, Zn2+ or Co2+ ions to the free peptoid 12P1 led to an increase in the absorption band near 220 nm as well as a slight decrease in the band near 195 nm, resulting in a double minima typical for helical alpha-methyl-benzyl peptoids (Fig. 3a and b). We also tested the thermal stability of the (12P1)Cu2 complex by recording the CD spectra between 20 °C and 70 °C in 4:1 MeOH:H2O solution. Almost no change in the intensity of the absorbance bands was observed upon heating, indicating that the metallopeptoid structure is quite stable in solution (Fig. S28†). In addition to the spectral changes observed in the 190–230 nm region, new CD bands also appeared between 240 and 300 nm due to the HQ π–π* transitions. The CD spectra in this region exhibited exciton couplets caused by the dipole–dipole interaction between two HQ chromophores. These exciton couplets indicate that chiral induction from the peptoid side-chains to the achiral HQ ligand eventually results in chiral metal complexes.14b,j,k,17,21 To evaluate whether both metal binding sites are crucial for the folding of 12P1 upon the binding of (two) metal ions, we have synthesized two control dodecamer peptoids 12P1a and 12P1b bearing only one (HQ, HQ) binding site at sites 1 and 2, respectively. The CD spectra of 12P1a and 12P1b displayed only one intense minimum near 195 nm, which was not changed upon metal binding to both peptoids (see ESI†), suggesting that both metal binding sites are essential for secondary structure induction upon metal coordination.
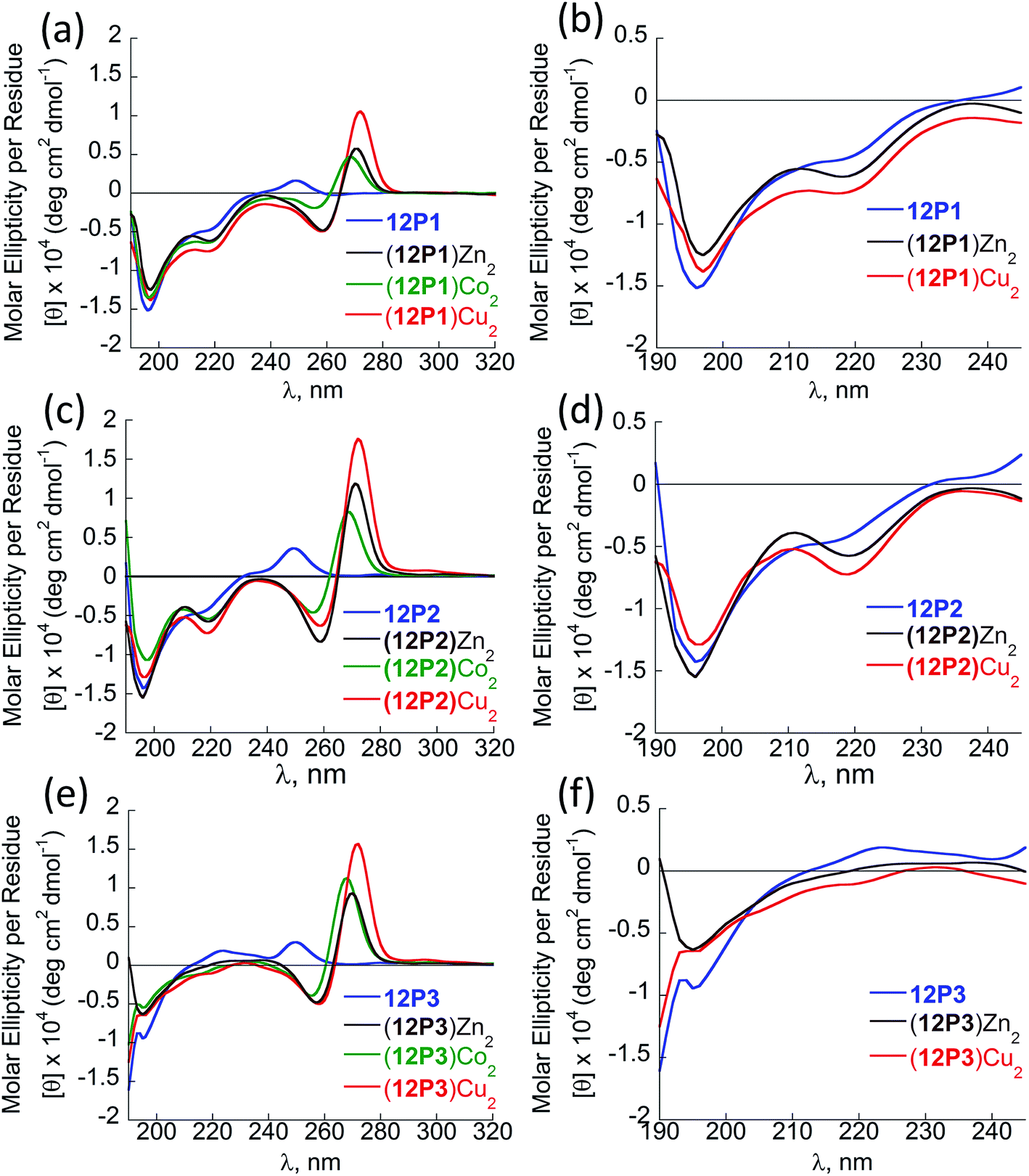 | ||
| Fig. 3 CD spectra of the peptoid olygomers 12P1 (a and b) 12P2 (c and d) and 12P3 (e and f) and their metal complexes measured at the concentration of 100 μM in 4:1 MeOH:H2O solution. | ||
The CD spectra of free 12P2 and its metal complexes exhibited trends similar to those of 12P1, indicating again metal-coordination induced peptoid folding into helical structures (Fig. 3c and d). The CD spectrum of the free peptoid 12P3 also displayed an intense minimum near 195 nm (Fig. 3e and f, blue line). However, upon the addition of Cu2+, Zn2+ or Co2+ ions, no new CD bands were detected in the region between 190 and 240 nm (Fig. 3e and f), suggesting that the peptoid 12P3, which lacks the Nspe group at the C-terminus, does not fold into helical structures upon metal binding. These results demonstrate that the Nspe group at the C-terminus of the alpha-methyl-benzyl peptoids plays a crucial role in the stabilization of the helical secondary structure.15b Following these results we synthesized another dodecamer peptoid 12P4 that contains only one Nspe group at the C-terminus (Fig. 4a). The new peptoid 12P4 was synthesized, purified and analyzed by UV-Vis titrations with Cu2+, Zn2+ and Co2+ to verify 2:1 coordination stoichiometry, which was further confirmed by ESI-MS (see ESI†). CD measurements of 12P4 before and after the addition of metal ions revealed the formation of double minima lower in intensity but similar to the ones formed upon metal binding to 12P1 and 12P2 (Fig. 4b). This result is remarkable because it confirms that a folding process can be initiated by metal coordination to unstructured peptoids bearing only one chiral bulky group at the C-terminus.
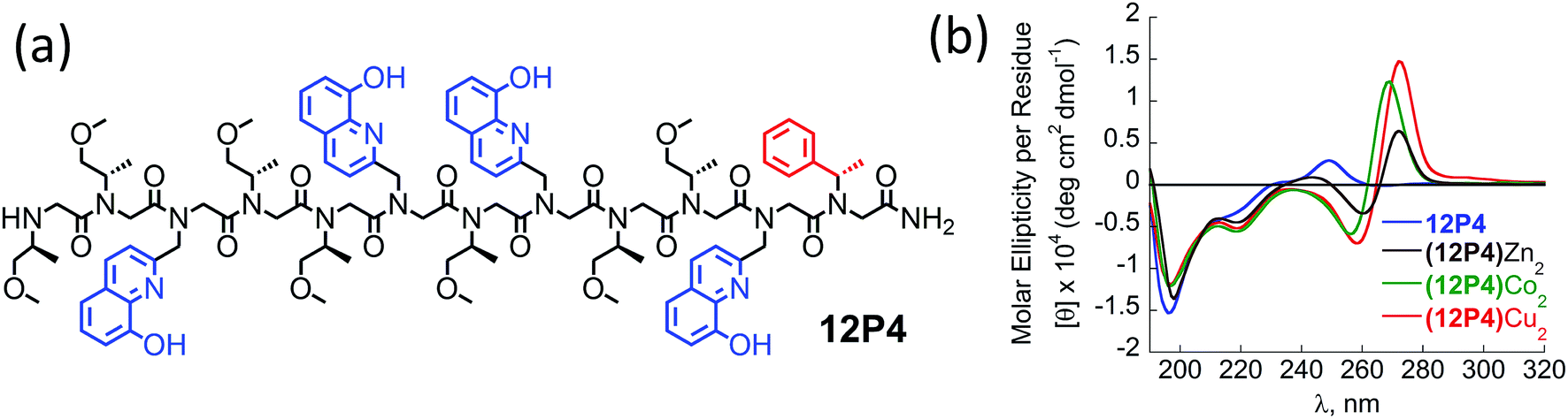 | ||
| Fig. 4 (a) Chemical sequence of 12P4 and (b) CD spectra of 12P4 and its metal complexes measured at the concentration of 100 μM in 4:1 MeOH:H2O solution. | ||
DFT calculations and assignment for the CD spectra of free and metal-coordinated peptoids
Overall, our experimental CD results suggest that the unstructured peptoids 12P1 and 12P2 could fold into helical structures while 12P3 remained unstructured upon metal binding. However, obtaining single crystals of the metal-coordinated peptoids 12P1 and 12P2 suitable for X-ray analysis was still not possible. Therefore we sought to perform some high-level DFT calculations to support our experimental findings. Previous molecular mechanics calculations showed that the homo-octamer (Nspe)8 should form a right-handed PP-I helix with the cis amide bonds and with backbone dihedrals (ω, ϕ, ψ) around (0°, −75°, 170°),22 leading to the typical double-minima near 200 and 220 nm observed in the experimental CD spectrum. No theoretical calculations have been done before for Nsmp-containing peptoids and other higher peptoid oligomers. Currently, no X-ray structure of any Nsmp-containing peptoids has been reported. Thus, detailed DFT calculations were performed to support our experimental results. Both right-handed cis PP-I and left-handed trans PP-II helices with the respective backbone (ω, ϕ, ψ) dihedrals around (0°, −75°, 170°) and (170°, −75°, 180°) of 12P1, (12P1)Zn2, 12P3 and (12P3)Zn2 are constructed for further conformational searching of the lowest energy conformations in our DFT calculations. The complexes of the closed-shell Zn2+ ions are chosen in our DFT study to avoid more computational efforts required for the open-shell Cu2+ and Co2+ ions while similar peptoid conformations can be expected for all these ions. The DFT-optimized helical structures were used to calculate the UV-Vis and CD spectra for comparison with our experimental spectra to provide useful insights into the electronic assignment of spectral bands as well as the actual secondary structures observed in the experiment.The recently developed GFN-xTB tight-binding method23 as implemented in the xtb program24 was used for efficient initial conformational searching in methanol solution employing the GBSA solvation model, followed by further DFT-refinement. The TURBOMOLE V7.0 suite of programs25 was used for all DFT calculations. The prescreened peptoid structures were fully optimized at the TPSS-D3/def2-TZVP + COSMO (methanol) level of theory, which combines the TPSS meta-GGA density functional26 with the BJ-damped DFT-D3 dispersion correction27 and the large def2-TZVP AO basis set,28 together with the COSMO (for methanol solvent: dielectric constant εr = 32.6, Rsol = 2.53 Å) solvation model.29 The density-fitting RI-J approach30 was used to accelerate the geometry optimization and harmonic frequency calculations. Vibrational frequency analysis was used to identify the nature of the located stationary points and to provide thermal and free-energy corrections according to the modified ideal gas-rigid rotor-harmonic oscillator model.31 The structures were characterized as true minima (with no imaginary frequency). The helical cis PP-I-like structures turned out to be at least 10 kcal mol−1 more stable than the corresponding trans PP-II-like structures.
Fig. 5 shows the DFT-optimized cis PP-I-like structure of (12P1)Zn2 and the structure of (12P3)Zn2 for comparison (optimized structures of cis PP-I-like 12P1 and 12P3 as well as other trans PP-II-like structures can be found in the ESI†). The optimized (12P1)Zn2 complex adopts a quasi-linear helical backbone with a rise of ∼6 Å (roughly 3.3 residues) per turn with two right-handed side-chain Zn(HQ)2 complexes, both found in a similar tetrahedral geometry between one central Zn2+ ion and two deprotonated HQ ligands with typical ∠O–Zn–O, ∠N–Zn–N and ∠N–Zn–O bond angles of 129 ± 5°, 130 ± 3° and 84.9 ± 0.3°, respectively. In contrast, the optimized structure of (12P3)Zn2 adopts a much less ordered backbone, with the respective side-chain Zn(HQ)2 complex near the N- and C-terminus being more planar (∠N–Zn–N = 150°) and more pyramidal (∠N–Zn–N = 120°), respectively, consistent with the modified sites of one bulky Nspe-substituents. In contrast, the corresponding trans PPII-like structures of both complexes, with a usual rise of ∼9 Å (3 residues) per turn, adopt much more bent backbones that may be further deformed by the possible coordination of the amide carbonyl groups to side-chain Zn(HQ)2 complexes (see ESI Fig. S80†). Without the support from side-chain Zn(HQ)2 complexes, the free peptoids 12P1 and 12P3 are structurally much more flexible and can be found in many possible H-bonded (HB) conformations with the acidic OH groups of the side-chain HQ ligands as HB donors and the main-chain amide carbonyl and side-chain Nsmp groups as HB acceptors.
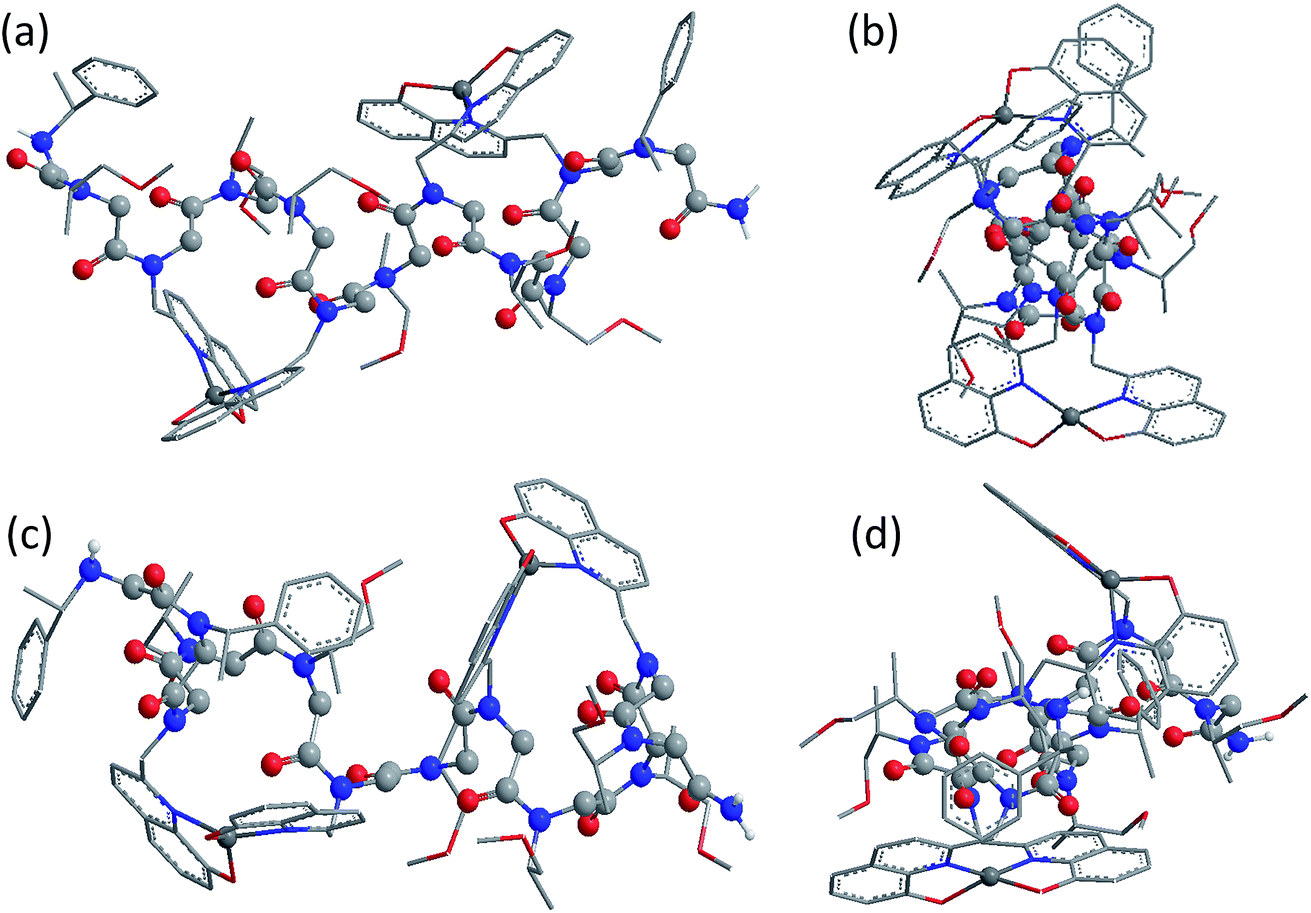 | ||
| Fig. 5 Comparison of the side and top views of (12P1)Zn2 (a and b) and (12P3)Zn2 (c and d) optimized at the TPSS-D3/def2-TZVP + COSMO (methanol) level, both assuming the right-handed cis PP-I helical structure. The main-chain C, N, O atoms and acidic H atoms (HQ hydroxyl and amide NH groups) are highlighted as grey, blue, red and white balls, respectively. | ||
The sTDA program package32 was used to efficiently compute the UV and CD spectra up to the excitation energies of 7.0 eV (∼180 nm) according to the simplified time-dependent density functional theory (sTDDFT)33 at the BHLYP/def2-TZVP + COSMO (methanol) level. About 400 excited electronic states are involved in the spectral range of up to 180 nm, making direct assignment almost impossible especially for high-energy bands below 230 nm. Clear electronic assignment was made using smaller model systems including the free HQ, Zn(HQ)2 complex, tetramer peptoid with and without single Zn(HQ)2 (p4c and ZnQ2c, respectively) truncated from the best cis PP-I structure of (12P1)Zn2 (See ESI†). The experimental UV-vis spectra could be well reproduced by our DFT-calculations, which are insensitive to the detailed peptoid secondary structures. A more detailed analysis shows that the main-chain amide groups contribute only to the high-energy bands below 230 nm while the side-chain HQ (or Zn(HQ)2) groups dominate the lower-energy bands.
Fig. 6 shows the comparison between the experimental and calculated CD spectra of 12P1, (12P1)Zn2, 12P3 and (12P3)Zn2. In general, the CD signal near 380 nm is rather low and thus is not shown. Taking (12P1)Zn2 as an example, the experimental positive and negative bands near 270 nm are well reproduced using our DFT calculations, which are due to the exciton couplet π → π* transitions of the side-chain Zn(HQ)2 groups, thus confirming the correct right-handed configuration of the Zn(HQ)2 groups predicted in our DFT calculations (reversed sign expected for left-handed configuration). Such CD bands in 12P1 are blue-shifted to about 250 nm having much reduced intensities, with residual signals of the side-chains HQ induced by the local chiral peptoid structures. Similarly, the experimental negative double minima bands near 220 and 200 nm are reasonably reproduced using our DFT calculations, which are assigned to the n → π* and π → π* transitions of the amide groups of the right-handed PP-I secondary structure with some contributions from the π → π* transitions of the side-chain Zn(HQ)2 groups. In this region, DFT calculations tend to overestimate the n → π* and π → π* transition energies of the amide groups by about 10 nm (or 0.17 eV) than the π → π* transitions of the side-chain ZnQ2 groups. For the (12P3)Zn2 complex, the well reproduced CD bands around 270 nm again suggest the formation of right-handed side-chain Zn(HQ)2 complexes; however, the absence of negative bands around 220 nm (characteristic for right-handed cis PP-I) in the experiment strongly suggests that the regular PP-I secondary structure is not formed upon Zn2+ coordination. Collectively, for the first time, clear electronic assignment to the experimental UV-Vis and CD spectra is postulated by high-level DFT calculations, providing strong evidence for both the detailed side-chain metal complexes and peptoid backbone secondary structures of large metallopeptoid complexes such as (12P1)Zn2 in solution.
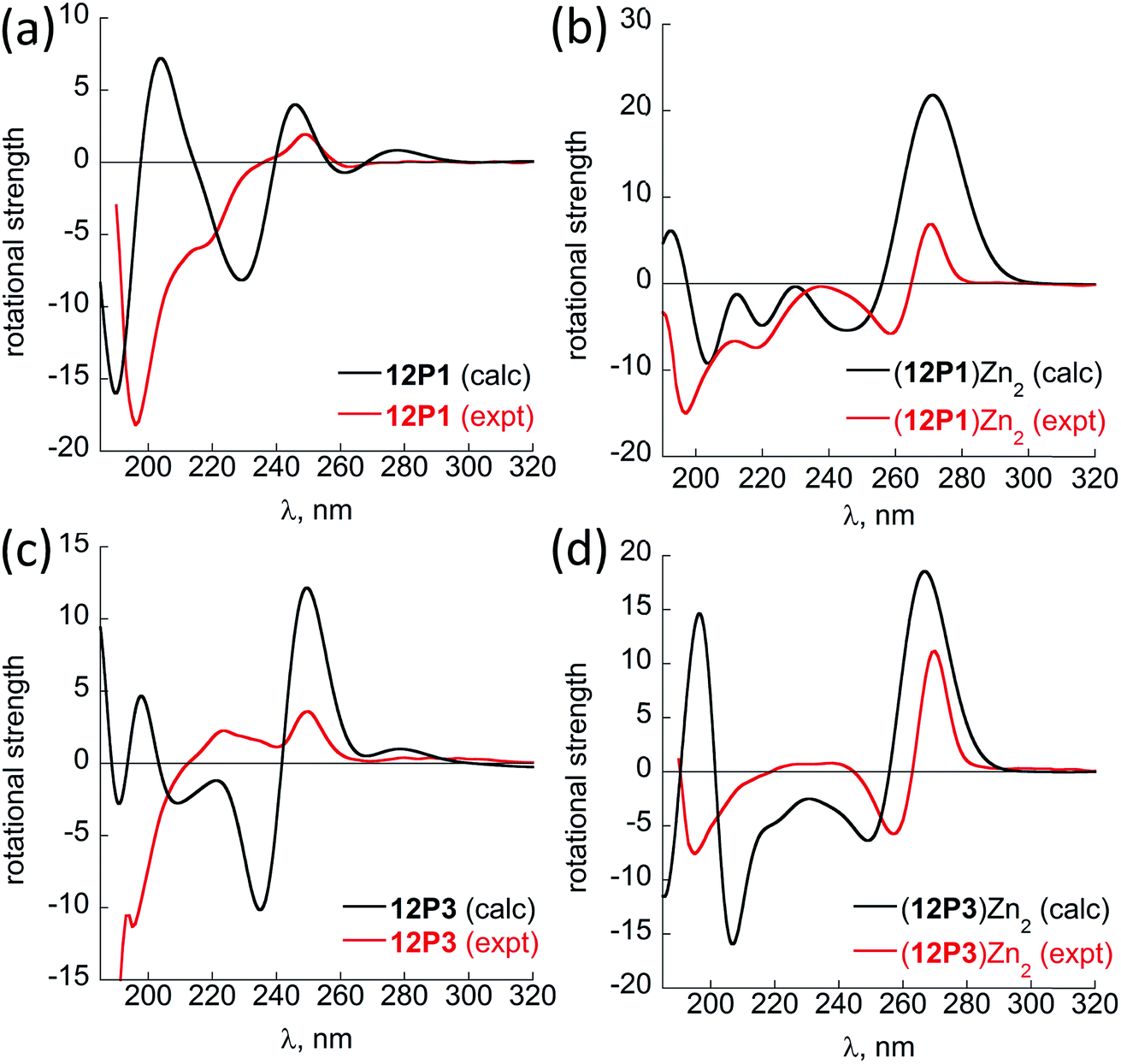 | ||
| Fig. 6 A comparison between the sTD-BHLYP/def2-TZVP + COSMO computed (black lines) and experimental (red lines) CD spectra of (a) 12P1, (b) (12P1)Zn2, (c) 12P3 and (d) (12P3)Zn2. The assignment of the bands to excited electronic states is based on smaller model systems of free HQ, Zn(HQ)2, p4c and truncated ZnQ2c molecules (see ESI†). | ||
NMR analysis of peptoid folding induced by Zn2+ coordination
The free 12P1, 12P2, 12P3 and 12P4 peptoids as well as their Zn2+ complexes were further investigated by the 1H–1H COSY 2D-NMR technique, which among the other 2D-NMR methods is most suitable for peptoid characterization in solution.34 When compared to peptides, the peptoids characteristically exhibit a multitude of distinct and slowly interchanging conformations in solution caused by the cis–trans interconversions at the backbone amides. As a result, rather than a single set of resonances the NMR spectrum of the peptoids will exhibit ‘clusters’ of cross-peaks in characteristic spectral regions, and these can be used to follow structural changes in peptoid conformation. This inherent conformational flexibility is also expected to dually frustrate attempts of structure determination using standard homonuclear TOCSY and NOESY spectra, due to the inefficient dipole–dipole magnetization transfer resulting in poor NOESY spectra, as well as the absence of any dominant structural species. However, the ensemble of the peptoid structures could still be characterized by following the typical shifts of the signal clusters. Thus, COSY and TOCSY remain as the preferred experiments; of these, the less complex COSY experiment (correlating only adjacent protons connected by 3J coupling constants) has given us the best results in monitoring the conformational changes occurring in our peptoids.Fig. 7 displays the COSY correlation map for 12P1 and 12P3. Two notable regions are seen between the prochiral geminal protons in the glycine-like backbone (near the diagonal, between 3.5 and 4.7 ppm, Fig. 7a and b) and correlations between the methyl groups and various vicinal protons (1.0–1.7 ppm for the methyls, Fig. 7c and d, 4.0–6.0 ppm for the neighboring protons). Correlations of the latter region are generally divided into Nsmp (1.0–1.2 ppm) and Nspe (1.5–1.7 ppm) methyls, in agreement with the aliphatic or aromatic nature of the side chains, respectively. Previous studies33 show that in both cases the downfield cluster corresponds to the cis confirmation of the peptoid bond, and the upfield cluster corresponds to the trans conformation. The third cluster in the Nspe region represents the terminal ‘edge’ Nspe group present in the peptoids, which, as expected for the secondary amine, resonates further upfield. Similar maps are shown for the peptoids 12P2 and 12P4 in the ESI.† The geminal proton region of 12P1 exhibits many well-resolved off-diagonal cross-peaks, which is an indication of a relatively rigid and structured backbone. However, the solution-phase structure of 12P1 could not be obtained by simulated annealing using distance restraints derived from key inter-residue NOEs, as previously done for several other peptoids,8f,34 probably because of a mixture of differently populated conformations that would confound the NMR analysis. The geminal proton region for unstructured 12P3 exhibits less well-resolved off-diagonal cross-peaks and more diffuse signals, indicating a relatively flexible backbone, supporting the observations from the CD analysis.
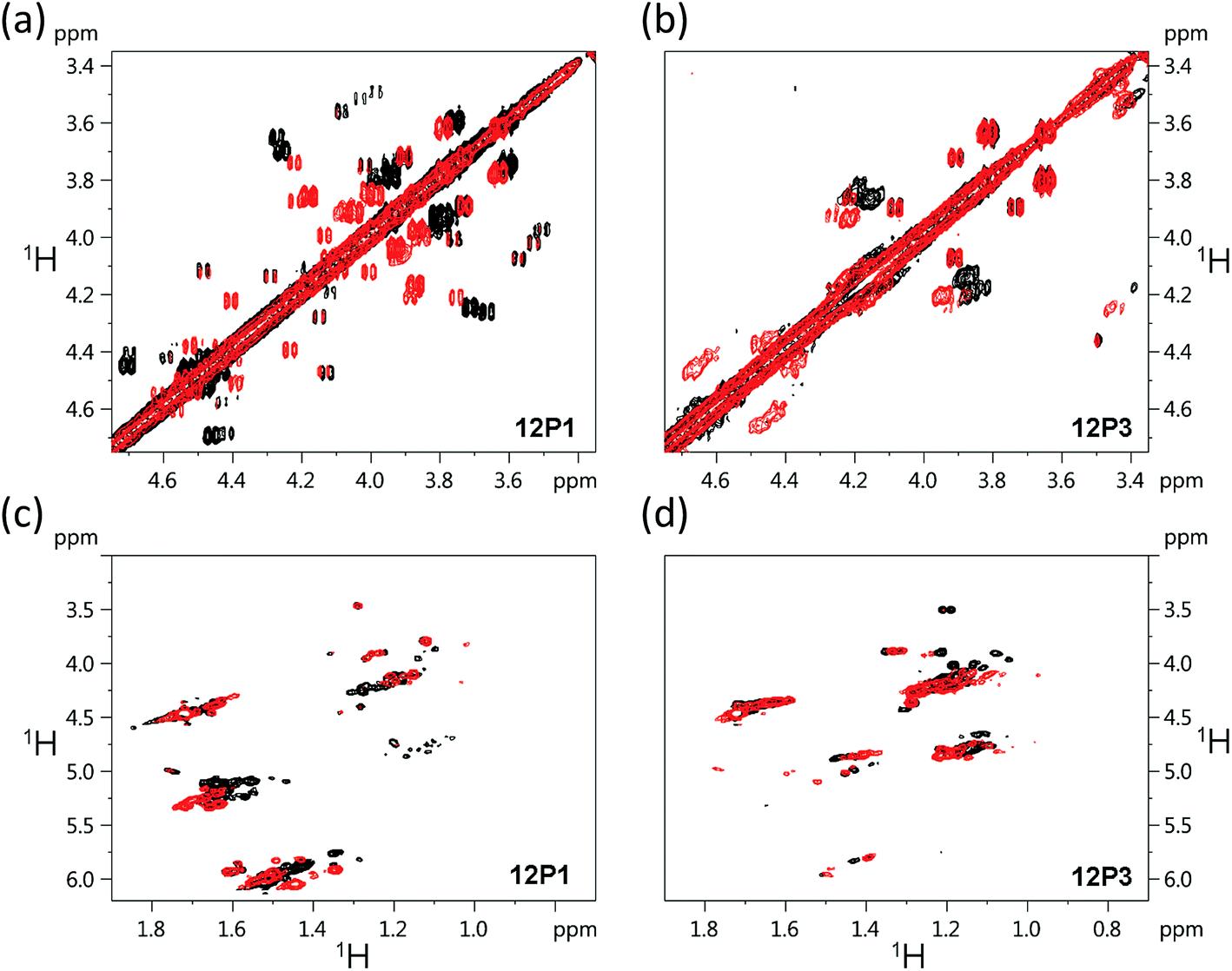 | ||
| Fig. 7 1H–1H COSY 2D-NMR spectra (700 MHz) of the free peptoids 12P1 and 12P3 (black) and their corresponding (12P1)Zn2 and (12P3)Zn2 complexes (red) measured at 283 K at 4 mM concentration in CD3OD. | ||
Such correlation maps were then used to evaluate the structural changes induced by the present of Zn2+. Since Zn2+ coordination occurs via the side-chain HQ ligands, the effects observed upon the geminal protons reflect an overall structural influence rather than a local electronic influence. Upon addition of Zn2+ to 12P1 several of the geminal proton signals in the correlation map are shifted, indicating a conformational change in the peptoid backbone. In addition, the number of cross-peaks is reduced, suggesting a decrease in the conformational heterogeneity of the backbone. Also, the Nspe methyl region exhibits an increase in spectral dispersion as well as resolution, once again indicating that 12P1 undergoes a rigidification in side-chain conformations (Fig. 7). In contrast, addition of Zn2+ to 12P3 fails to exhibit such changes. Both the geminal proton and the methyl regions exhibit less shifts in peak positions and they remain relatively diffuse in nature, suggesting that the structural changes in the peptoid are minimal. A comparison of these results to those obtained for the other two peptoids 12P2 and 12P4 (see ESI†) shows that they are closer in behavior to 12P1, although the structural changes are less pronounced.
Taken together, the experimental as well as the computational spectroscopic results provide strong evidence for the folding of the initially unstructured 12P1, 12P2 and 12P4 upon metal binding assisted by a small amount of structure-directing side-chains (about 8–17% Nspe groups, much less than those reported at least 66%). Moreover, DFT calculations provide for the first time the detailed solution structure as well as clear assignment of UV-Vis and CD spectra of long linear peptoids and metallopeptoids.35
From structure to function: rationally designed peptoids for the selective binding of two different metal ions
At this point we wanted to explore whether peptoids folding upon the coordination of one metal ion can lead to selective recognition of a second, different metal ion, and result in heterobimetallic metallopeptoid complexes. The peptoids 12P1, 12P2, 12P3 and 12P4 include four identical HQ ligands each, pre-organized into two similar binding sites, and thus selectivity in metal binding to one site over the other is not likely to occur. We have previously found that the incorporation of one HQ and one 2,2′:6′,2′′-terpyridine (Terpy) side-chain ligand located at the i and i + 3 positions within a helical peptoid leads to a strong and selective coordination of Cu2+ ions from a mixture containing additional Zn2+ and Co2+ ions.11a,36 Replacing the HQ groups at the 2nd position of 12P1–12P4 by a Terpy group should provide access to peptoids with two distinct intramolecular metal binding sites that can selectively bind different metal ions such as Cu2+, Co2+ and Zn2+ ions. To explore this possibility we decided to focus on the peptoids 12P1, which exhibits the highest conformational change upon metal coordination, and 12P3, which remains unstructured upon metal binding. Replacing one HQ side-chain with a Terpy side-chain in each peptoid leads to peptoids 12P5 and 12P6, respectively (Fig. 8a and 9a).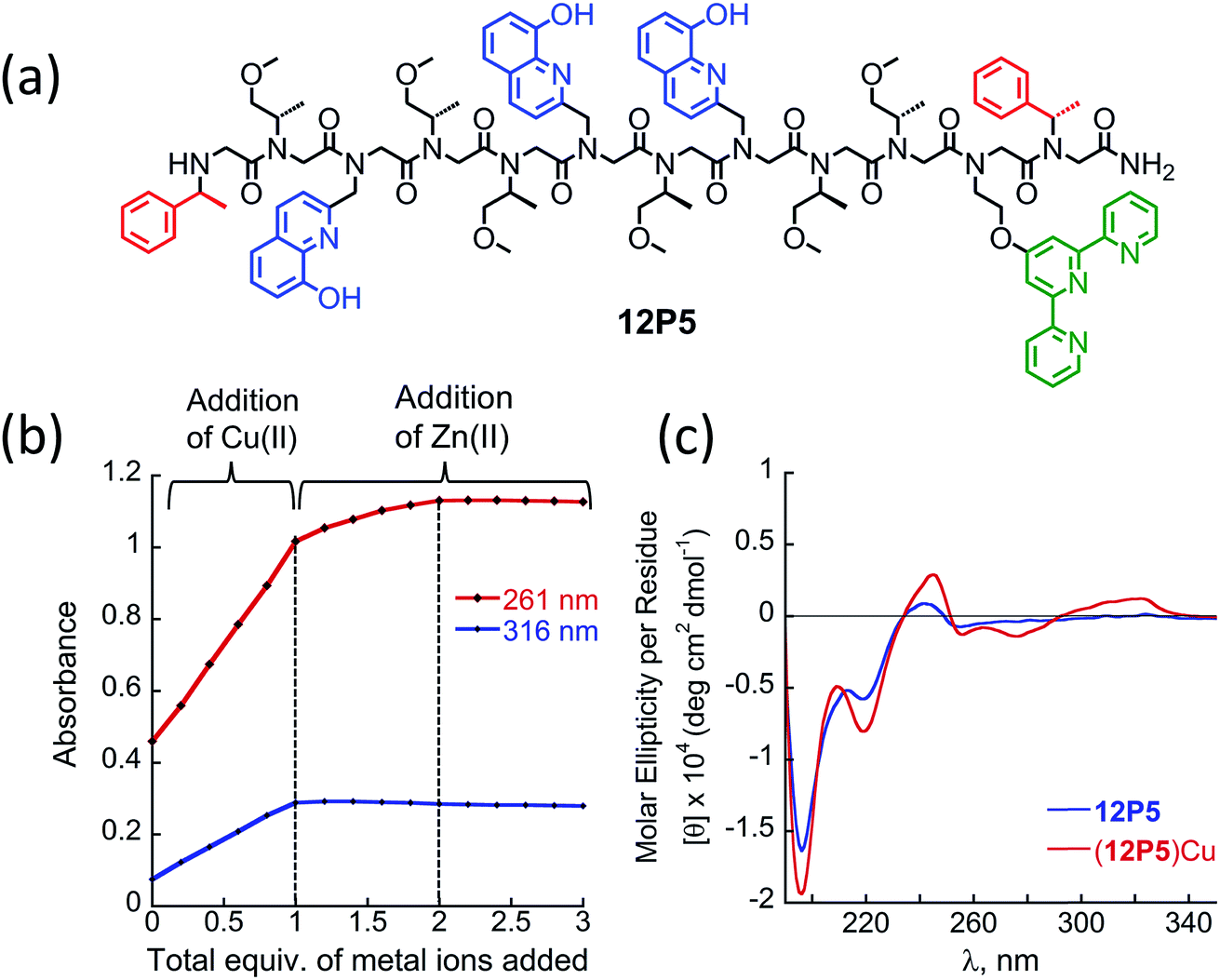 | ||
| Fig. 8 (a) Chemical sequence of the peptoid oligomer 12P5. (b) A plot representing absorbance at 261 nm and 316 nm as a function of total equivalents of metal ions added from the UV-Vis titration of 12P5 with first 1 equiv. Cu2+ followed by addition of Zn2+, at the concentration of 17 μM in 4:1 MeOH:H2O solution. (c) CD spectra of the peptoid 12P5 and (12P5)Cu complex, at the concentration of 100 μM in 4:1 MeOH:H2O solution. | ||
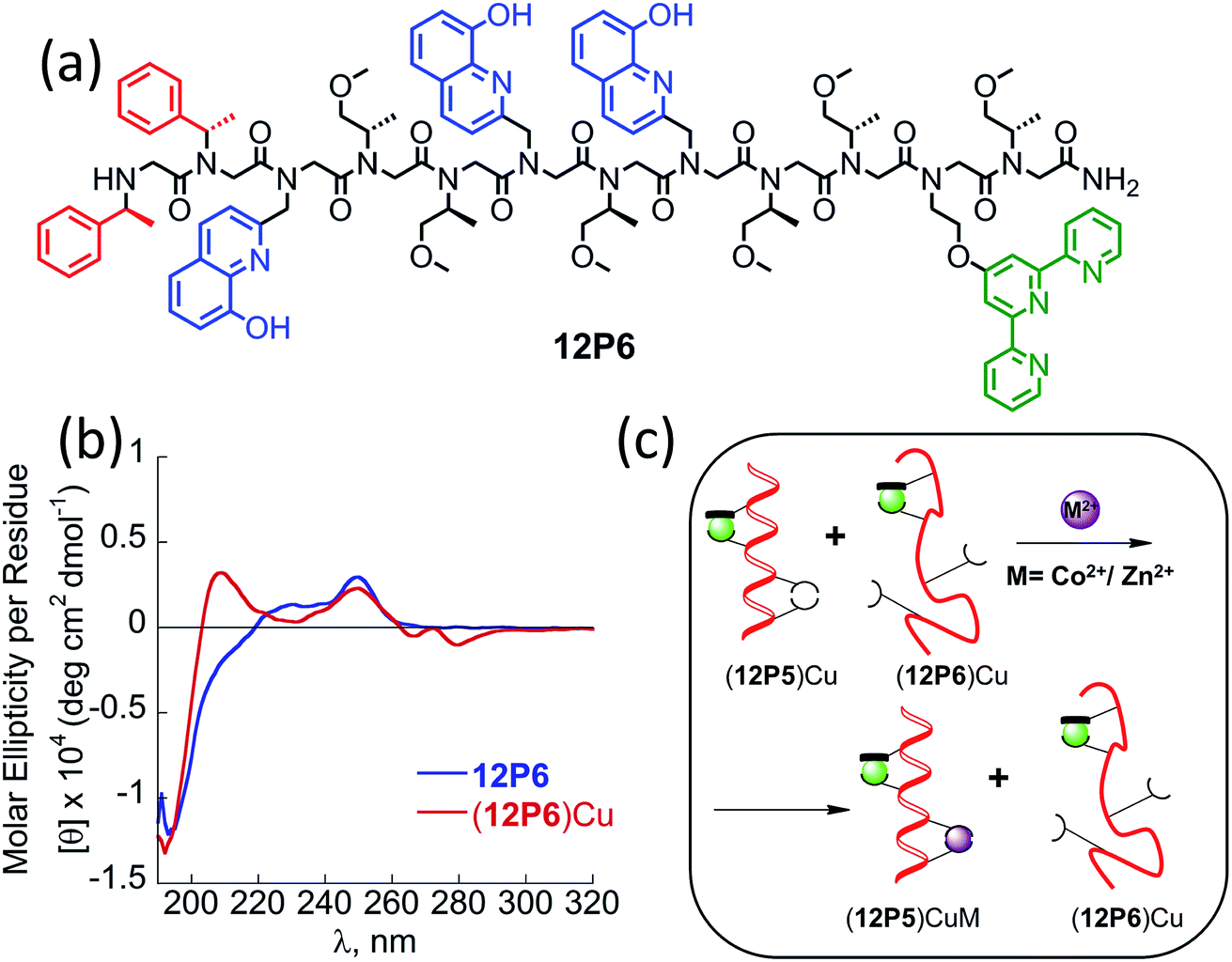 | ||
| Fig. 9 (a) Chemical sequence of the peptoid oligomers 12P6. (b) CD spectra of the peptoid 12P6 and (12P6)Cu complex, 100 μM in 4:1 MeOH:H2O. (c) A cartoon demonstrating positive allosteric cooperative binding to (12P5)Cu. | ||
Peptoids 12P5 and 12P6 were synthesized on a solid support, cleaved and purified; their identity was verified by ESI-MS. UV-Vis titration of 12P5 using a Cu2+ solution was performed to follow the intensity changes at 261 and 316 nm that correspond to the absorbance of metal-bound HQ and Terpy ligands, respectively. A linear increase in the intensity of both bands was observed (Fig. 8b) until 1 equiv. of Cu2+ ions was added; further addition of another 1 equiv. of Zn2+ (or Co2+) ions led to a gradual intensity increase only for the band at 261 nm but not for the band at 316 nm. This clearly demonstrated that Cu2+ ions are bound to the combined (Terpy, HQ) site at first, followed by Zn2+ (or Co2+) binding to the other (HQ, HQ) site. The formation of the 2:1 metal:peptoid bimetallic complexes from 12P5 was further confirmed by ESI-MS analysis (see ESI†). Moreover, the Hamiltonian parameters obtained from simulating the EPR spectra of (12P5)Cu, (12P5)CuCo and (12P5)CuZn revealed a Hamiltonian parameter A‖ of 165 in all cases, consistent with the square pyramidal coordination geometry37 expected for the combined coordination of one Terpy and one HQ ligands to Cu2+ ions.11a,36 To explore whether we can obtain the hetero-bimetallic complexes (12P5)CuZn (or (12P5)CuCo) by self-assembly, a solution of 12P5 was treated with a solution mixture containing 1 equiv. of Cu2+ and 1 equiv. Zn2+ (or Co2+). This led to the complete disappearance of the UV-Vis bands at 245 and 278 nm and the appearance of two new bands at 261 and 316 nm identical to those obtained from the sequential addition of the two metal ions, indicating selective binding of two different metal ions in distinct binding sites to form (12P5)CuZn (or (12P5)CuCo) via self-assembly from the solution mixture (Fig. S18 and S19†). Similar results were also found for 12P6.
The CD spectra of both 12P5 and 12P6 exhibited an intense minimum at 200 nm and low intensity minimum (12P5) or no minimum at all (12P6) at 220 nm. Interestingly, after the addition of 1 equiv. of Cu2+ ions to 12P5, the intensity of the peak at 220 nm was increased, implying that the conformational order of the Cu-peptoid is higher (Fig. 8c). A further addition of 1 equiv. of Zn2+ (or Co2+) did not significantly change the CD spectra, suggesting that the second metal binding has little effect on the peptoid secondary structure (Fig. S24†). The CD spectrum of 12P6, on the other hand, did not change in the region of 200–220 nm upon the addition of metal ions (see ESI† and Fig. 9b), indicating that the conformational order of this peptoid is not affected by metal coordination. Based on these results we wanted to further explore whether the folding of 12P5 upon the first coordination of Cu2+ ions to the (Terpy, HQ) binding site can facilitate the second coordination of Zn2+ (or Co2+) ions to the (HQ, HQ) binding site of (12P5)Cu, compared with the unstructured (12P6)Cu complex, thus demonstrating positive allosteric cooperativity.
To this aim, we conducted a direct competition experiment between (12P5)Cu and 12P6Cu on the binding of Zn2+ (or Co2+).38 Thus, a solution mixture of (12P5)Cu and (12P6)Cu (both 0.5 mM in MeOH:H2O 4:1) was treated with 1 equiv. of Zn2+ (or Co2+) and analyzed by ESI-MS. The (12P5)CuM and (12P6)CuM (M = Zn2+ or Co2+) show the same molecular weight but different peptoid sequence, and therefore could be analyzed by the MS-MS technique. The observed fragmentations in the mass spectra matched only those of (12P5)CuZn (or (12P5)CuCo), with no indication of those related to (12P6)CuM (Fig. S66–S69†). These results demonstrate that both Zn and Co are selectively bound to (12P5)Cu rather than to (12P6)Cu, suggesting that the conformational change that occurs only when Cu2+ binds 12P5 facilitates the second binding event compared to the same binding event that takes place by the unstructured (12P6)Cu. As such, we propose that the structural change induced by the selective metal binding of Cu2+ to 12P5 leads to its ability to perform a biomimetic function, i.e. positive allosteric cooperativity in the binding of Zn2+ (or Co2+).
Conclusions
This paper reports on two advances in peptidomimetic chemistry: (i) the ability of rationally designed unstructured peptoids to fold upon metal binding, and (ii) the ability of a peptoid bearing two different metal-binding sites to bind one metal ion in one site, change its conformational order and demonstrate positive allosteric cooperative binding of a second metal ion in the other site. Metal-binding peptide mimics are an important class of biomimetic oligomers with demonstrated functionalities of selective recognition and catalysis; however, examples of metal-coordination driven folding and allosteric cooperativity of peptidomimetics are rare. Therefore, the demonstration that peptoids can perform these biomimetic tasks is a milestone in the development of functional complex structures from peptidomimetic scaffolds.Currently, the stabilization of peptoid helices mainly relies on the incorporation of specific chiral bulky side chains. In this work, for the first time, our CD and NMR experimental data as well as DFT-predicted solution structures and spectral assignment confirmed the folding of unstructured peptoids upon metal coordination to two side-chain (HQ, HQ) metal binding sites. By replacing one HQ with a Terpy ligand we created two distinct metal binding sites within one peptoid. The selective coordination of one Cu2+ ion to the (HQ, Terpy) binding site leads to the peptoid folding that in turn facilitates the second coordination of a Zn2+ (or Co2+) ion to the (HQ, HQ) binding site. We showed that the affinity of the folded Cu-bound peptoid (12P5)Cu to Zn2+ (or Co2+) is higher than that of less structured (12P6)Cu as evidenced by mass spectra analysis, suggesting a unique example of allosteric cooperativity in peptide mimics.
We believe that our findings introduce a new platform not only in peptoid folding, but also in the design of novel complex biomimetic structures based on bimetallic and hetero-bimetallic peptidomimetic composites. The observation that a synthetic oligomer can perform a complicated biomimetic function such as positive allosteric cooperative metal binding can impact the design of novel functional foldamers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the European Union's – Seventh Framework Program (FP7/2007–2013) under grant agreement no. 333034-MC--MF STRC AND FCN. G. M. and M. B. thank Mrs Larisa Panz for her assistance with the various MS measurements and Dr Boris Tumanskii for his assistance with the EPR measurements. M. B. thanks the Schulich foundation and the Gutwirth foundation for her PhD fellowship. S. G., H. Z. and Z. W. Q. thank the DFG for financial support in the framework of the Leibniz price. J. C. acknowledges support of the Christians for Israel Chair for Medical Research.References
- Foldamers: Proteins Folding and Metal ions, Mechanisms, Biology and Disease, ed. C. M. Gomes and P. Wittung-Stafshede Huc, CRC Press, Taylor & Francis group, FL, USA, 2011 Search PubMed.
- M. L. Zastrow and V. l. Pecoraro, Coord. Chem. Rev., 2013, 257, 2565–2588 CrossRef PubMed.
- R. H. Holm, P. Kennepohl and I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef PubMed.
- (a) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef PubMed; (b) Foldamers: Structure, Properties, and Applications, ed. S. Hecht and I. Huc, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (c) S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef; (d) D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366–1375 CrossRef PubMed; (e) M. Berg and A. Lützen, in Structural aspects of helicates, Metallofoldamers. Supramolecular Architecturesfrom Helicates to Biomimetics, ed. G. Maayan and M. Albrecht, John Wiley& Sons, Ltd., 2013, ch. 4, p. 130. See also ref. 16–27 in this chapter Search PubMed.
- Reviews: (a) G. Maayan, Eur. J. Org. Chem., 2009, 33, 5699–5710 CrossRef; (b) Metallofoldamers. Supramolecular Architectures from Helicates to biomimetics, ed. G. Maayan and M. Albrecht, John Wiley& Sons, Ltd, 2013 Search PubMed.
- (a) F. Zhang, S. Bai, G. P. A. Yap, V. Tarwade and J. M. Fox, J. Am. Chem. Soc., 2005, 127, 10590–10599 CrossRef PubMed; (b) Z. Dong Jr., R. J. Karpowicz, S. Bai, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2006, 128, 14242–14243 CrossRef PubMed.
- S. Tashiro, K. Matsuoka, A. Minoda and M. Shionoya, Angew. Chem., Int. Ed., 2012, 51, 13123–13127 CrossRef PubMed.
- (a) K. T. Nam, S. A. Shelby, P. H. Choi, A. B. Marciel, R. Chen, L. Tan, T. K. Chu, R. A. Mesch, B.-C. Lee, M. D. Connolly, C. Kisielowski and R. N. Zuckermann, Nat. Mater., 2010, 9, 454–460 CrossRef PubMed; (b) J. A. Crapster, I. A. Guzei and H. E. Blackwell, Angew. Chem., Int. Ed., 2013, 52, 5079–5084 CrossRef PubMed; (c) R. V. Mannige, T. K. Haxton, C. Proulx, E. J. Robertson, A. Battigelli, G. L. Butterfoss, R. N. Zuckermann and S. Whitelam, Nature, 2015, 516, 415–420 CrossRef PubMed; (d) J. S. Laursen, P. Harris, P. Fristrup and C. A. Olsen, Nat. Commun., 2015, 6, 7013 CrossRef PubMed; (e) S. J. Laursen, J. E. Andreasen and C. A. Olsen, Acc. Chem. Res., 2015, 48, 2696–2704 CrossRef PubMed; (f) B. C. Gorske, E. M. Mumford, C. G. Gerrity and I. Ko, J. Am. Chem. Soc., 2017, 139, 8070–8073 CrossRef PubMed; (g) O. Roy, G. Dumonteil, S. Faure, L. Jouffret, A. Kriznik and C. Taillefumier, J. Am. Chem. Soc., 2017, 139, 13533–13540 CrossRef PubMed.
- (a) R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg and C. K. Marlowe, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367–9371 CrossRef PubMed; (b) J. A. Patch, K. Kirshenbaum, S. L. Seurynck, R. N. Zuckermann and A. E. Barron, in Drug Discovery, Wiley-VCH, Weinheim, Germany, 2004, pp. 1–31 Search PubMed; (c) R. N. Zuckermann and T. Kodadek, Curr. Opin. Mol. Ther., 2009, 11, 299–307 Search PubMed; (d) M. T. Dohm, R. Kapoor and A. E. Barron, Curr. Pharm. Des., 2011, 17, 2732–2747 CrossRef CAS PubMed; (e) K. H. A. Lau, Biomater. Sci., 2014, 2, 627–633 RSC; (f) A. S. Knight, E. Y. Zhou, M. B. Francis and R. N. Zuckermann, Adv. Mater., 2015, 27, 5665 CrossRef CAS PubMed.
- (a) R. Schettini, F. D. Riccardis, G. D. Sala and I. Izzo, J. Org. Chem., 2016, 81, 2494–2505 CrossRef CAS PubMed; (b) K. J. Prathap and G. Maayan, Chem. Commun., 2015, 51, 11096–11099 RSC; (c) D. ChandraMohan, A. Sadhukha and G. Maayan, J. Catal., 2017, 355, 139–144 CrossRef CAS; (d) G. Maayan, M. D. Ward and K. Kirshenbaum, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13679–13684 CrossRef CAS PubMed.
- (a) M. Baskin and G. Maayan, Chem. Sci., 2016, 7, 2809–2820 RSC; (b) A. S. Knight, E. Y. Zhou, J. G. Pelton and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17488–17493 CrossRef CAS PubMed; (c) A. S. Knight, E. Y. Zhou and M. B. Francis, Chem. Sci., 2015, 6, 4042–4048 RSC.
- R. N. Zuckermann, J. M. Kerr, S. B. W. Ken and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- A. S. Culf and R. J. Ouellette, Molecules, 2010, 15, 5282–5335 CrossRef CAS PubMed.
- (a) B. C. Lee, T. K. Chu, K. A. Dill and R. N. Zuckermann, J. Am. Chem. Soc., 2008, 130, 8847–8855 CrossRef CAS PubMed; (b) G. Maayan, M. D. Ward and K. Kirshenbaum, Chem. Commun., 2009, 56–58 RSC; (c) N. Maulucci, I. Izzo, G. Bifulco, A. Aliberti, C. De Cola, D. Comegna, C. Gaeta, A. Napolitano, C. Pizza, C. Tedesco, D. Flot and F. De Riccardis, Chem. Commun., 2008, 3927–3929 RSC; (d) C. De Cola, S. Licen, D. Comegna, E. Cafaro, G. Bifulco, I. Izzo, P. Tecilla and F. De Riccardis, Org. Biomol. Chem., 2009, 7, 2851 RSC; (e) G. Della Sala, B. Nardone, F. De Riccardis and I. Izzo, Org. Biomol. Chem., 2013, 11, 726–731 RSC; (f) I. Izzo, G. Ianniello, C. De Cola, B. Nardone, L. Erra, G. Vaughan, C. Tedesco and F. De Riccardis, Org. Lett., 2013, 15, 598–601 CrossRef CAS PubMed; (g) C. De Cola, G. Fiorillo, A. Meli, S. Aime, E. Gianolio, I. Izzo and F. De Riccardis, Org. Biomol. Chem., 2014, 12, 424 RSC; (h) C. Tedesco, L. Erra, I. Izzo and F. De Riccardis, CrystEngComm, 2014, 16, 3667–3687 RSC; (i) T. Zabrodski, M. Baskin, J. K. Prathap and G. Maayan, Synlett, 2014, 25 Search PubMed; (j) M. Baskin and G. Maayan, Biopolymers, 2015, 104, 577–584 CrossRef CAS PubMed; (k) M. Baskin, L. Panz and G. Maayan, Chem. Commun., 2016, 52, 10350–10353 RSC; (l) T. Ghosh, N. Fridman, M. Kosa and G. Maayan, Angew. Chem., Int. Ed., 2018, 57, 7703–7708 CrossRef CAS PubMed.
- (a) K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4303–4308 CrossRef CAS PubMed; (b) C. W. Wu, T. J. Sanborn, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2001, 123, 2958–2963 CrossRef CAS PubMed; (c) C. W. Wu, T. J. Sanborn, K. Huang, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2001, 123, 6778–6784 CrossRef CAS PubMed; (d) C. W. Wu, K. Kirshenbaum, T. J. Sanborn, J. A. Patch, K. Huang, K. A. Dill, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2003, 125, 13525–13530 CrossRef CAS PubMed; (e) B. C. Gorske, B. L. Bastian, G. D. Geske and H. E. Blackwell, J. Am. Chem. Soc., 2007, 129, 8928–8929 CrossRef CAS PubMed; (f) N. H. Shah, G. L. Butterfoss, K. Nguyen, B. Yoo, R. Bonneau, D. L. Rabenstein and K. Kirshenbaum, J. Am. Chem. Soc., 2008, 130, 16622–16632 CrossRef CAS PubMed; (g) B. C. Gorske, J. R. Stringer, B. L. Bastian, S. A. Fowler and H. E. Blackwell, J. Am. Chem. Soc., 2009, 131, 16555–16567 CrossRef CAS PubMed; (h) O. Roy, C. Caumes, Y. Esvan, C. Didierjean, S. Faure and C. Taillefumier, Org. Lett., 2013, 15, 2246–2249 CrossRef CAS PubMed; (i) C. Caumes, O. Roy, S. Faure and C. Taillefumier, J. Am. Chem. Soc., 2012, 134, 9553–9556 CrossRef CAS PubMed.
- (a) M. J. Kelso, H. N. Hoang, T. G. Appleton and D. P. Fairlie, J. Am. Chem. Soc., 2000, 122, 10488–10489 CrossRef CAS; (b) M. T. Ma, H. N. Hoang, C. C. G. Scully, T. G. Appleton and D. P. Fairlie, J. Am. Chem. Soc., 2009, 131, 4505–4512 CrossRef CAS PubMed; (c) M. R. Ghadiri and C. Choi, J. Am. Chem. Soc., 1990, 112, 1630–1632 CrossRef CAS; (d) M. R. Ghadiri and A. K. Fernholz, J. Am. Chem. Soc., 1990, 112, 9633–9635 CrossRef CAS; (e) F. Ruan, Y. Chen and P. B. Hopkins, J. Am. Chem. Soc., 1990, 112, 9403–9404 CrossRef CAS; (f) P. Rossi, P. Tecilla, L. Baltzer and P. Scrimin, Chem.–Eur. J., 2004, 10, 4163–4170 CrossRef CAS PubMed; (g) S. R. Gilbertson, G. Chen and M. McLoughlin, J. Am. Chem. Soc., 1994, 116, 4481–4482 CrossRef CAS; (h) S. R. Gilbertson, C. A. Klug, J. Schaefer, X. Wang and G. S. Hoge, Organometallics, 1996, 15, 4678–4680 CrossRef CAS; (i) N. Ousaka, N. Tani, R. Sekiya and R. Kuroda, Chem. Commun., 2008, 2894–2896 RSC; (j) S. Tashiro, K. Matsuoka, A. Minoda and M. Shionoya, Angew. Chem., Int. Ed., 2012, 51, 13123–13127 CrossRef CAS PubMed; (k) S. J. Smith, R. J. Radford, R. H. Subramanian, B. R. Barnett, J. S. Figueroa and F. A. Tezcan, Chem. Sci., 2016, 7, 5453–5461 RSC; (l) S. J. Smith, K. Du, R. J. Radford and F. A. Tezcan, Chem. Sci., 2013, 4, 3740–3747 RSC.
- L. Zborovsky, A. Smolyakova, M. Baskin and G. Maayan, Chem.–Eur. J., 2018, 24, 1159–1167 CrossRef CAS PubMed.
- H. M. Shin, C. M. Kang, M. H. Yoon and J. Seo, Chem. Commun., 2014, 50, 4465–4468 RSC.
- The peptoid-to-metal stoichiometry ratio (a) was determined by calculation the intersection point from the two equations:
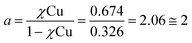 .
. - (a) J. W. Karr, L. J. Kaupp and V. A. Szalai, J. Am. Chem. Soc., 2004, 126, 13534–13538 CrossRef CAS PubMed; (b) J. E. Weder, T. W. Hambley, B. J. Kennedy, P. A. Lay, D. MacLachlan, R. Bramley, C. Delfs, D. K. Murray, S. B. Moubaraki, B. Warwick, J. R. Biffin and H. L. Regtop, Inorg. Chem., 1999, 38, 1736–1744 CrossRef CAS PubMed.
- M. Baskin and G. Maayan, Dalton Trans., 2018, 47, 10767–10774 RSC.
- P. Armand, K. Kirshenbaum, A. Falicov, R. L. Dunbrack Jr, K. A. Dill, R. N. Zuckermann and F. E. Cohen, Folding Des., 1997, 2, 369–375 CrossRef CAS PubMed.
- S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed.
- S. Grimme and C. Bannwarth, J. Chem. Phys., 2016, 145, 054103–054120 CrossRef PubMed.
- TURBOMOLE V7.0 2015, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- J. M. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- (a) A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS; (b) F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS; (c) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 2, 799–805 RSC.
- (a) K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed; (b) P. Deglmann, K. May, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2004, 384, 103–107 CrossRef CAS.
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- C. Bannwarth and S. Grimme, Comput. Theor. Chem., 2014, 1040–1041, 45–53 CrossRef CAS.
- A detailed discussion on the calculated vs. experimental UV-Vis spectra including relevant figures is described in the ESI.†.
- (a) J. R. Stringer, J. A. Crapster, I. A. Guzei and H. E. Blackwell, J. Am. Chem. Soc., 2011, 133, 15559–15567 CrossRef CAS PubMed; (b) P. Armand, K. Kirshenbaum, R. A. Goldsmith, S. Farr-Jones, A. E. Barron, K. T. V. Truong, K. A. Dill, D. F. Mierke, F. E. Cohen, R. N. Zuckermann and E. K. Bradley, Proc. Natl. Acad. Sci. U. S. A., 1998, 14, 4309–4314 CrossRef.
- G. L Butterfoss, B. Yoob, J. N. Jaworski, I. Chorny, K. A. Dill, R. N. Zuckermann, R. Bonneau, K. Kirshenbaum and V. A. Voelz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14320–14325 CrossRef PubMed.
- M. Baskin, N. Fridman, M. Kosa and G. Maayan, Dalton Trans., 2017, 46, 15330–15339 RSC.
- (a) J. Peisach and W. E. Blumberg, Arch. Biochem. Biophys., 1974, 165, 691–708 CrossRef CAS PubMed; (b) N. Wei, N. N. Murthy and K. D. Karlin, Inorg. Chem., 1994, 33, 6093–6100 CrossRef CAS; (c) B. Lucches, K. S. Humphreys, D.-H. Lee, C. D. Incarvito, R. D. Sommer, A. L. Rheingold and K. D. Karlin, Inorg. Chem., 2004, 43, 5987–5998 CrossRef PubMed.
- Numerical evaluation of the association constants of (12P5)Cu and (12P6)Cu with either Zn2+ or Co2+ was not possible. Initially, we have used UV-Vis titrations in low concentrations to estimate these K values, but the obtained numbers reflected very strong binding of the second metal ion to either (12P5)Cu or (12P6)Cu, which were at least five orders of magnitude higher than the concentration of (12P5)Cu or (12P6)Cu in these experiments and thus the obtained numbers could not be considered accurate (See ESI†). Typical competition experiments with other chelators (e.g. EGTA) were attempted, however these strong chelators could take out the bound Cu2+ ion and thus these experiments also could not be used for determining the association constants. Therefore, direct competition between the two peptoids was performed..
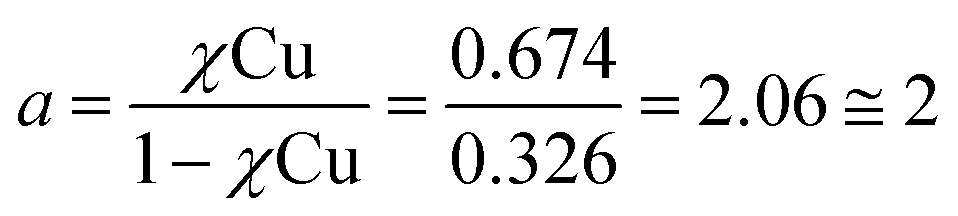
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03616k |
| This journal is © The Royal Society of Chemistry 2019 |