DOI:
10.1039/C8SC03178A
(Edge Article)
Chem. Sci., 2019,
10, 614-619
Topological self-template directed synthesis of multi-shelled intermetallic Ni3Ga hollow microspheres for the selective hydrogenation of alkyne†
Received
18th July 2018
, Accepted 18th October 2018
First published on 19th October 2018
Abstract
Multi-shelled hollow structured materials featuring large void volumes and high specific surface areas are very promising for a variety of applications. However, controllable synthesis of multi-shelled hollow structured intermetallic compounds remains a formidable challenge due to the high annealing temperature commonly required for the formation of intermetallic phases. Here, a topological self-template strategy was developed to solve this problem. Using this strategy, we prepared well-defined multi-shelled intermetallic Ni3Ga hollow microspheres (Ni3Ga-MIHMs) as disclosed by the HAADF-STEM, HRTEM, and EDS characterizations, and the BET specific surface areas of them measured as much as 153.4 m2 g−1. XRD and EXAFS spectral characterizations revealed the atomically ordered intermetallic phase nature of the Ni3Ga-MIHMs. The selective hydrogenation of acetylene catalytic evaluation results further demonstrated excellent catalytic properties of the Ni3Ga-MIHMs, which results from the more energetically facile reaction pathway for acetylene hydrogenation and ethylene desorption over it as revealed by DFT calculations. Besides, this strategy is also extendable to synthesize other multi-shelled intermetallic Ni3Sn4 hollow microspheres, and is expected to open up new opportunities for rational design and preparation of novel structured and highly efficient intermetallics.
Introduction
Atomically ordered intermetallic compounds have gained great research interest in a variety of fields such as heterogeneous catalysis,1–6 energy conversion,7–10 and environmental engineering,11 by virtue of their intriguing electronic and structural properties. However, the formation of intermetallic phases commonly demands a high annealing temperature, making it a formidable challenge to fabricate intermetallic materials in a manipulatable manner.12,13 For example, a conventional high-temperature melting route can only make bulk intermetallic compounds.14 Wet chemistry methods provide more opportunities to prepare nano-sized intermetallics by employing capping ligand-assisted protocols, but unavoidable particle aggregation during the high-temperature annealing treatment or removal of capping ligands process generally occurs.15,16 As a consequence, it remains a major challenge to synthesize well-defined intermetallic compounds with novel morphologies such as multi-shelled hollow structures featuring large void volumes and high specific surface areas.17–19 Actually, multi-shelled hollow structured materials have been widely employed in many promising fields due to their more abundant surface-exposed active sites and more facile mass transfer pathways.20–23 Great research efforts have been directed to the fabrication of multi-shelled hollow structured metal oxides, metal sulfides, and metal phosphides, etc.24 However, it is quite surprising that little attention is given to the preparation of multi-shelled intermetallics, which may arise from the great difficulty in keeping the multi-shelled hollow structures under high annealing temperatures. Herein, we propose a topological self-template strategy that involves a simultaneous annealing-reduction process of an initial densely stacked template to create well-defined multi-shelled intermetallics. Using this method, we successfully fabricated multi-shelled intermetallic Ni3Ga hollow microspheres (Ni3Ga-MIHMs) featuring large void volumes and high specific surface areas (as much as 153.4 m2 g−1). The prepared Ni3Ga-MIHMs exhibit excellent catalytic properties toward selective hydrogenation of acetylene, an industrially important process,25,26 benefiting from their novel structures and energetically more favorable pathway for acetylene hydrogenation and ethylene desorption over it as revealed by DFT calculations. Besides, this strategy is also extendable to prepare other multi-shelled intermetallic Ni3Sn4 hollow microspheres, showing excellent generality of this approach.
Results and discussion
Synthesis and characterization of Ni3Ga-MIHMs
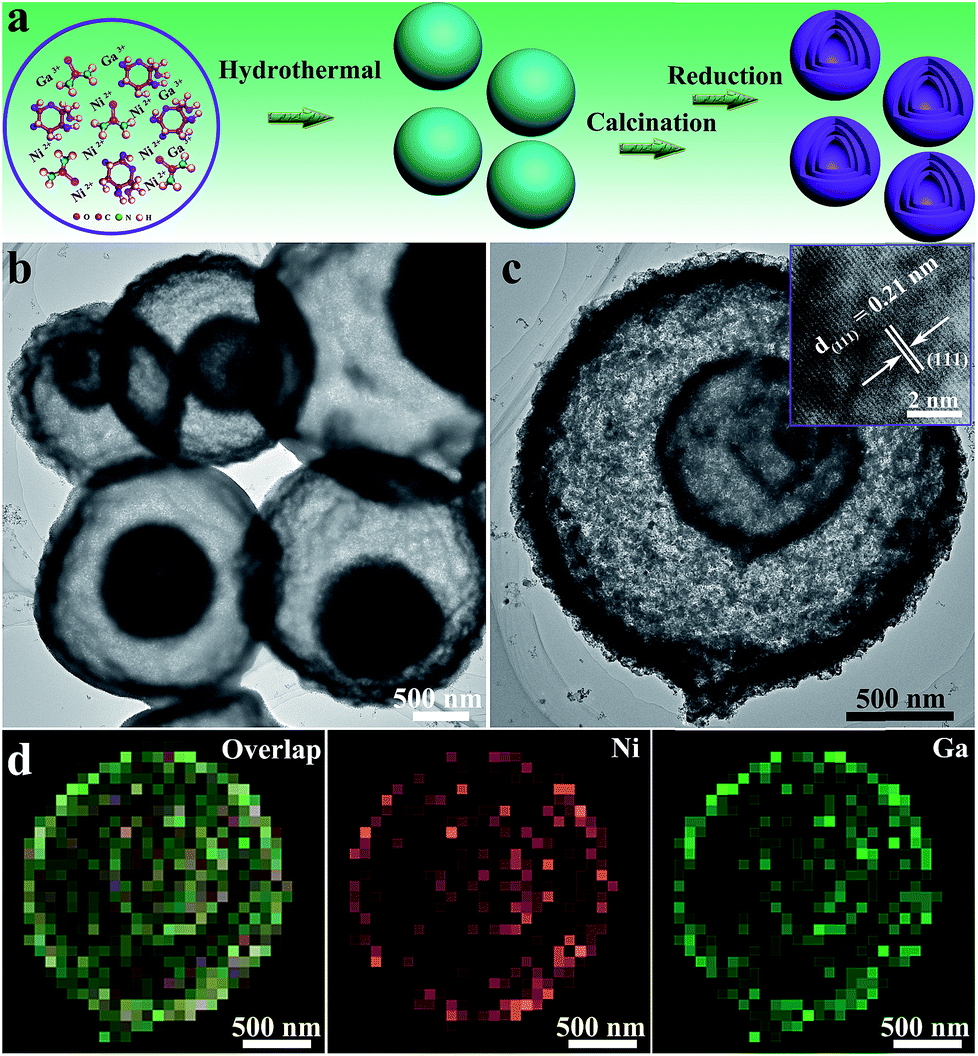
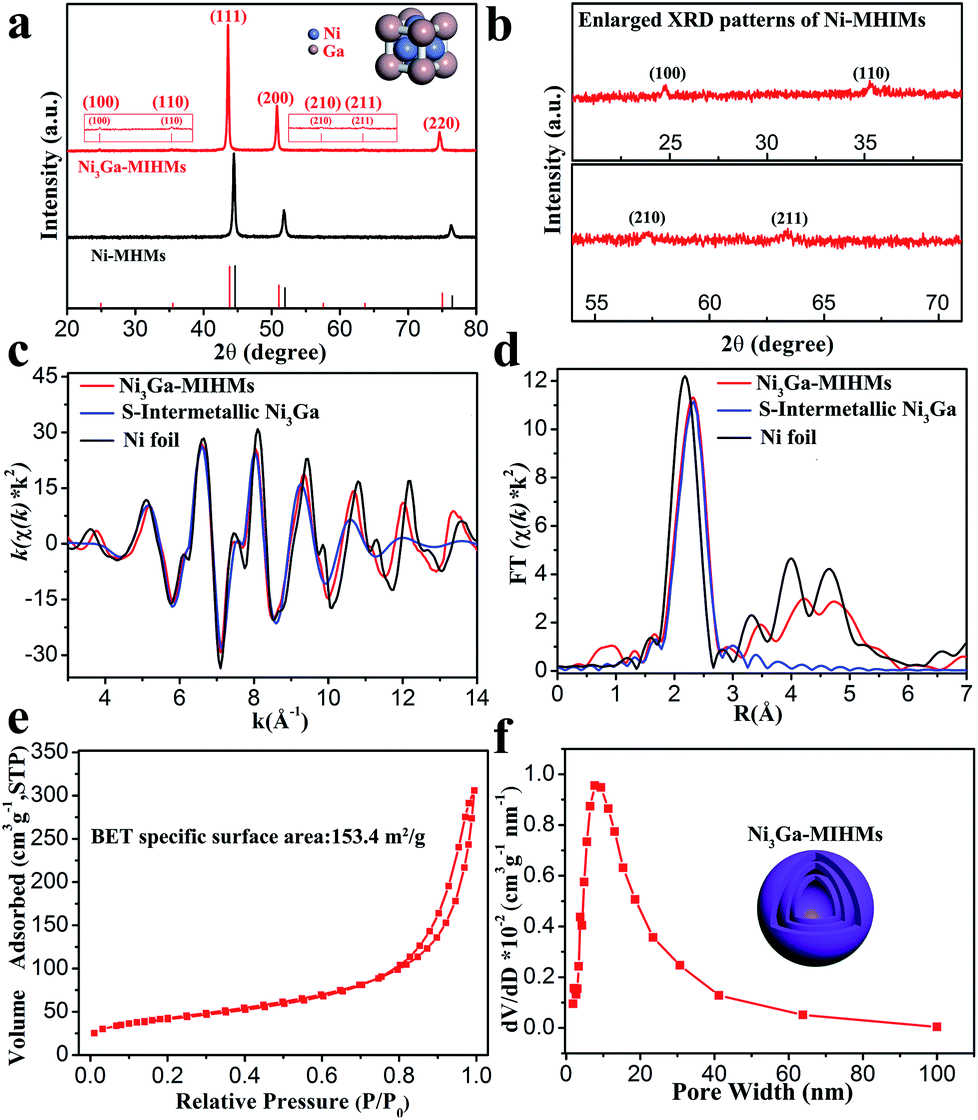
The schematic preparation process of Ni3Ga-MIHMs is displayed in Fig. 1a. By a facile topological self-template strategy, well-structured multi-shelled intermetallic Ni3Ga hollow microspheres were successfully prepared. Fig. S1† shows that after a hydrothermal process, composite solid spheres composed of evenly dispersed metal elements (Ni and Ga), carbon, and oxygen are first obtained. XRD analysis in Fig. S2† further reveals the amorphous nature of these composite solid spheres. To explore the appropriate calcination temperature, these composite solid spheres were then subjected to thermogravimetric analysis. Based on the TGA results in Fig. S3,† their calcination temperature was determined as 600 °C. Fig. S4† is the TEM image of the composite solid spheres after 600 °C calcination treatment in air. It was found that after calcination the morphology of these spheres was readily transformed from solid to hollow, consisting of densely stacked nanoparticles. Corresponding XRD analysis in Fig. S5† further indicates that these amorphous composite solid spheres have also been turned into a mixed-phase of NiO (JCPDS 47-1049) and Ga2O3 (JCPDS 20-0426) after 600 °C calcination. Taking advantage of a further reduction treatment, the above mixed-phase hollow spheres become more loosely piled, generating well-defined multi-shelled hollow microspheres as displayed by the TEM images in Fig. 1b and c and SEM images in Fig. S6.† The high-resolution TEM (HRTEM) image in the inset of Fig. 1c shows that the lattice fringe of 0.21 nm is well assigned to that of the (111) plane of cubic intermetallic Ni3Ga (JCPDS 65-0141), showing the atomically ordered intermetallic phase nature of these multi-shelled hollow microspheres, which is well in accord with the following XRD and EXAFS characterizations. The well-defined multi-shelled hollow structures of these Ni3Ga-MIHMs are further confirmed by the corresponding EDS elemental mapping characterizations as displayed in Fig. 1d. The homogeneously distributed Ni and Ga elements show the well-defined ensemble structures of Ni3Ga-MIHMs. Additionally, the Ni/Ga atom ratio of these Ni3Ga-MIHMs is measured as 2.96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by ICP-OES, which is in excellent agreement with the stoichiometric composition of Ni3Ga. XRD analysis in Fig. 2a and b reveal the intermetallic phase nature of these multi-shelled Ni3Ga hollow microspheres and the diffraction peaks observed at 2θ values of 24.7°, 35.3°, 43.6°, 50.8°, 57.4°, 63.5°, and 74.8° are well indexed to the (100), (110), (111), (200), (210), (211), and (220) planes of the cubic intermetallic Ni3Ga (JCPDS 65-0141), agreeing well with the above HRTEM analysis in Fig. 1. The intermetallic phase nature of the obtained Ni3Ga-MIHMs was further confirmed by EXAFS characterizations. As displayed in Fig. 2c, the Fourier transforms of the Ni K-edge EXAFS oscillations of the Ni3Ga-MIHMs in the k space are in excellent agreement with the simulated standard peaks of intermetallic Ni3Ga, as is also observed in the R space shown in Fig. 2d. Data fitting results of the EXAFS spectra are shown in Table S1.† It was found that the first shell neighbour of Ni was Ga coordination with a Ni–Ga bond length of about 2.5 Å. The second shell neighbour of Ni was Ni coordination and the Ni–Ni bond length of it was about 2.6 Å. The coordination numbers of Ni–Ga and Ni–Ni were calculated to be 7.9 and 4.0, respectively. These results are well in accord with previous reports for intermetallic Ni3Ga, which undoubtedly reveals the intermetallic Ni3Ga phase nature of the above prepared Ni3Ga-MIHMs.27 XPS analysis was further employed to characterize the surface structures of the prepared Ni3Ga-MIHMs. Fig. S7a† is the Ga 2p XPS spectra of the prepared Ni3Ga-MIHMs. It was found that binding energy positions of the Ni3Ga-MIHMs centered at 1116.6 eV and 1118.3 eV were well assigned to the Ga 2p3/2 peaks of Ga0 and Ga+, respectively.28 Ni 2p XPS spectra of the prepared Ni3Ga-MIHMs in Fig. S7b† further demonstrates that binding energy positions centered at 852.2 eV and 855.2 eV are well in line with the Ni 2p3/2 peaks of Ni0 and Ni2+.5,29,30 The binding energy values of the Ga 2p XPS peaks and Ni 2p XPS peaks of Ni3Ga-MIHMs are in excellent agreement with a previous report of intermetallic Ni3Ga.5 In the current case, the emerged high valence oxidation species in Ni3Ga-MIHMs are most likely due to surface oxidation when exposed to ambient atmosphere as has been frequently revealed by reported findings.31,32 The Brunauer–Emmett–Teller (BET) specific surface area and pore volume of these multi-shelled Ni3Ga hollow microspheres were measured by nitrogen adsorption–desorption characterizations to be as much as 153.4 m2 g−1 and 0.461 cm3 g−1 with an average pore size of 14 nm as displayed in Fig. 2e and f, which is derived from the multi-shelled hollow sphere structures with large void volume.
:1 by ICP-OES, which is in excellent agreement with the stoichiometric composition of Ni3Ga. XRD analysis in Fig. 2a and b reveal the intermetallic phase nature of these multi-shelled Ni3Ga hollow microspheres and the diffraction peaks observed at 2θ values of 24.7°, 35.3°, 43.6°, 50.8°, 57.4°, 63.5°, and 74.8° are well indexed to the (100), (110), (111), (200), (210), (211), and (220) planes of the cubic intermetallic Ni3Ga (JCPDS 65-0141), agreeing well with the above HRTEM analysis in Fig. 1. The intermetallic phase nature of the obtained Ni3Ga-MIHMs was further confirmed by EXAFS characterizations. As displayed in Fig. 2c, the Fourier transforms of the Ni K-edge EXAFS oscillations of the Ni3Ga-MIHMs in the k space are in excellent agreement with the simulated standard peaks of intermetallic Ni3Ga, as is also observed in the R space shown in Fig. 2d. Data fitting results of the EXAFS spectra are shown in Table S1.† It was found that the first shell neighbour of Ni was Ga coordination with a Ni–Ga bond length of about 2.5 Å. The second shell neighbour of Ni was Ni coordination and the Ni–Ni bond length of it was about 2.6 Å. The coordination numbers of Ni–Ga and Ni–Ni were calculated to be 7.9 and 4.0, respectively. These results are well in accord with previous reports for intermetallic Ni3Ga, which undoubtedly reveals the intermetallic Ni3Ga phase nature of the above prepared Ni3Ga-MIHMs.27 XPS analysis was further employed to characterize the surface structures of the prepared Ni3Ga-MIHMs. Fig. S7a† is the Ga 2p XPS spectra of the prepared Ni3Ga-MIHMs. It was found that binding energy positions of the Ni3Ga-MIHMs centered at 1116.6 eV and 1118.3 eV were well assigned to the Ga 2p3/2 peaks of Ga0 and Ga+, respectively.28 Ni 2p XPS spectra of the prepared Ni3Ga-MIHMs in Fig. S7b† further demonstrates that binding energy positions centered at 852.2 eV and 855.2 eV are well in line with the Ni 2p3/2 peaks of Ni0 and Ni2+.5,29,30 The binding energy values of the Ga 2p XPS peaks and Ni 2p XPS peaks of Ni3Ga-MIHMs are in excellent agreement with a previous report of intermetallic Ni3Ga.5 In the current case, the emerged high valence oxidation species in Ni3Ga-MIHMs are most likely due to surface oxidation when exposed to ambient atmosphere as has been frequently revealed by reported findings.31,32 The Brunauer–Emmett–Teller (BET) specific surface area and pore volume of these multi-shelled Ni3Ga hollow microspheres were measured by nitrogen adsorption–desorption characterizations to be as much as 153.4 m2 g−1 and 0.461 cm3 g−1 with an average pore size of 14 nm as displayed in Fig. 2e and f, which is derived from the multi-shelled hollow sphere structures with large void volume.
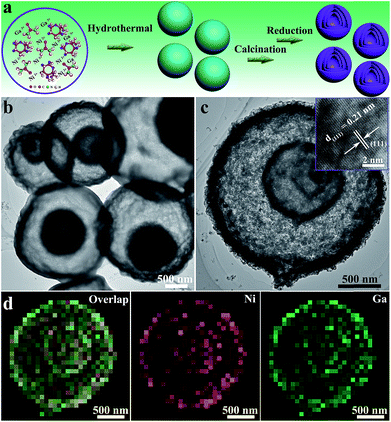 |
| | Fig. 1 (a) Schematic preparation process of the multi-shelled intermetallic Ni3Ga hollow microspheres (Ni3Ga-MIHMs) using a topological self-template strategy. (b) Overall TEM images of the prepared Ni3Ga-MIHMs. (c) TEM image of a single multi-shelled intermetallic Ni3Ga hollow microsphere (Ni3Ga-MIHM). The inset of (c) is a corresponding HRTEM image of the Ni3Ga-MIHMs. (d) EDS elementary mapping images of the Ni3Ga-MIHM. | |
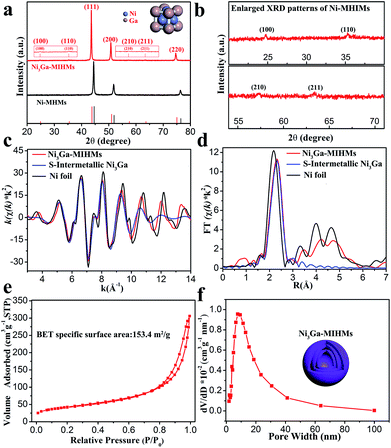 |
| | Fig. 2 (a) XRD patterns of the prepared Ni3Ga-MIHMs and Ni-MHMs. The red vertical lines represent the standard peaks of Ni3Ga (JCPDS 65-0141) and the black vertical lines represent the standard peaks of Ni (JCPDS 04-0850). (b) Enlarged XRD patterns of the Ni3Ga-MIHMs. Fourier transforms of the Ni K-edge EXAFS oscillations in the (c) k space and in the (d) R space of the prepared Ni3Ga-MIHMs and Ni foil. S-Intermetallic Ni3Ga represents the simulated standard Ni3Ga oscillation mode. (e) Nitrogen adsorption–desorption isotherm and (f) corresponding pore size distribution of the prepared Ni3Ga-MIHMs. | |
Selective hydrogenation of alkyne evaluation over Ni3Ga-MIHMs
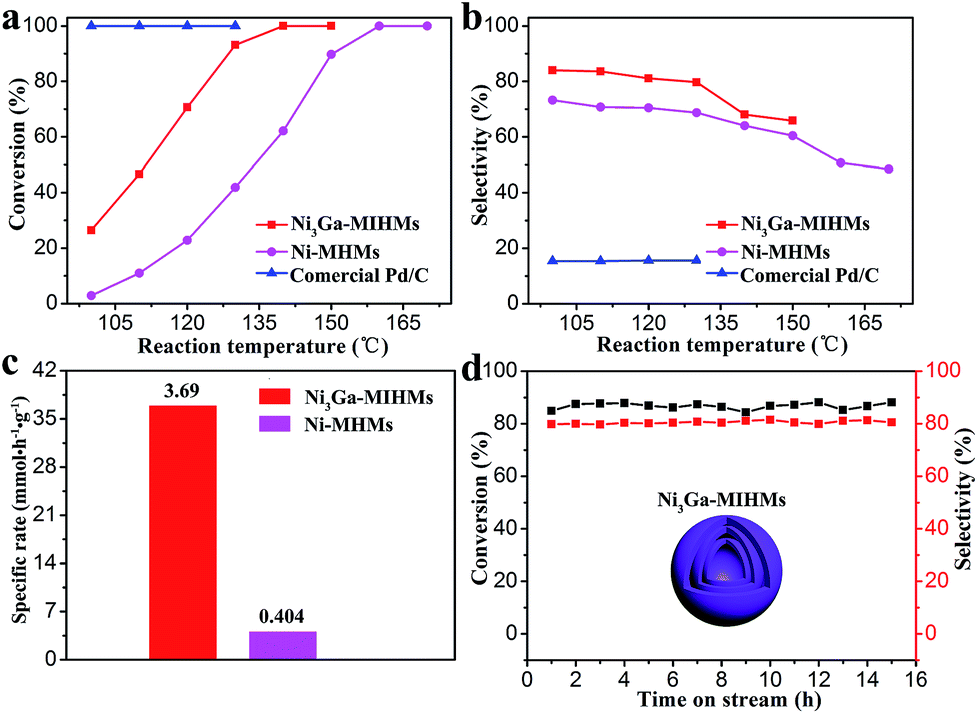
The well-defined Ni3Ga intermetallic phase nature, multi-shelled hollow sphere structures and large specific surface areas impart these Ni3Ga-MIHMs with great potentials for selective hydrogenation of acetylene, an industrially significant reaction.33–38 As shown in Fig. 3a and b, commercial Pd/C catalysts demonstrate full conversion of acetylene in the testing temperatures ranging from 100 °C to 130 °C, but the corresponding ethylene selectivity is quite poor and below 20%. Strikingly, the prepared Ni3Ga-MIHMs exhibit excellent catalytic selectivity toward ethylene (around 80%) in a wide range of reaction temperatures (100–130 °C). Only when acetylene content was near to total conversion, did the corresponding ethylene selectivity demonstrate a slight decrease. This was a common phenomenon in the acetylene hydrogenation reaction and could be ascribed to the different adsorption strengths of acetylene and ethylene as has already well been disclosed by previous reports.39 Acetylene is more thermodynamically favorable to realize adsorption and activation than ethylene. However, when acetylene was near to full conversion, ethylene could competitively adsorb on the active sites, which led to the decrease of ethylene selectivity. Moreover, the full conversion of acetylene over Ni3Ga-MIHMs was obtained as low as 140 °C.
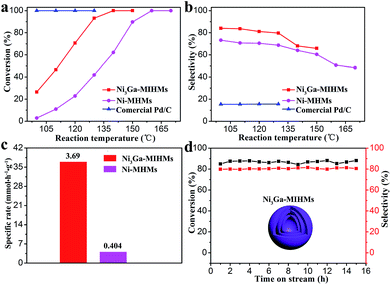 |
| | Fig. 3 (a) Acetylene conversion and (b) ethylene selectivity as a function of temperature over the Ni3Ga-MIHMs, Ni-MHMs, and commercial Pd/C catalysts. (c) The specific rate of the prepared Ni3Ga-MIHMs and Ni-MHMs at 100 °C. (d) Stability test over the Ni3Ga-MIHMs as a function of time at 125 °C. | |
The multi-shelled Ni hollow microspheres (Ni-MHMs) are also fabricated as a contrast catalyst as shown in Fig. 2 and S8.† It was found that the fabricated Ni-MHMs demonstrated poor catalytic activity toward acetylene hydrogenation and the total conversion temperature of acetylene reached as high as 160 °C, showing an obvious increase in comparison with that of the Ni3Ga-MIHMs. Besides, corresponding ethylene selectivity over the Ni-MHMs also shows an evident decrease as compared with the Ni3Ga-MIHMs displayed in Fig. 3b. Fig. 3c is the specific rate of the Ni3Ga-MIHMs and Ni-MHMs. It shows that the specific rate of the Ni3Ga-MIHMs (3.69 mmol h−1 g−1) is more than 9-fold than that of the Ni-MHMs (0.404 mmol h−1 g−1).
DFT analysis of corresponding reaction mechanism over Ni3Ga-MIHMs
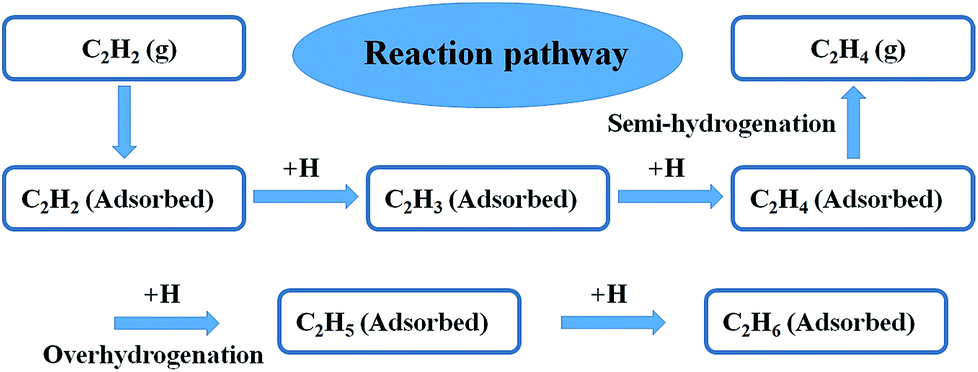
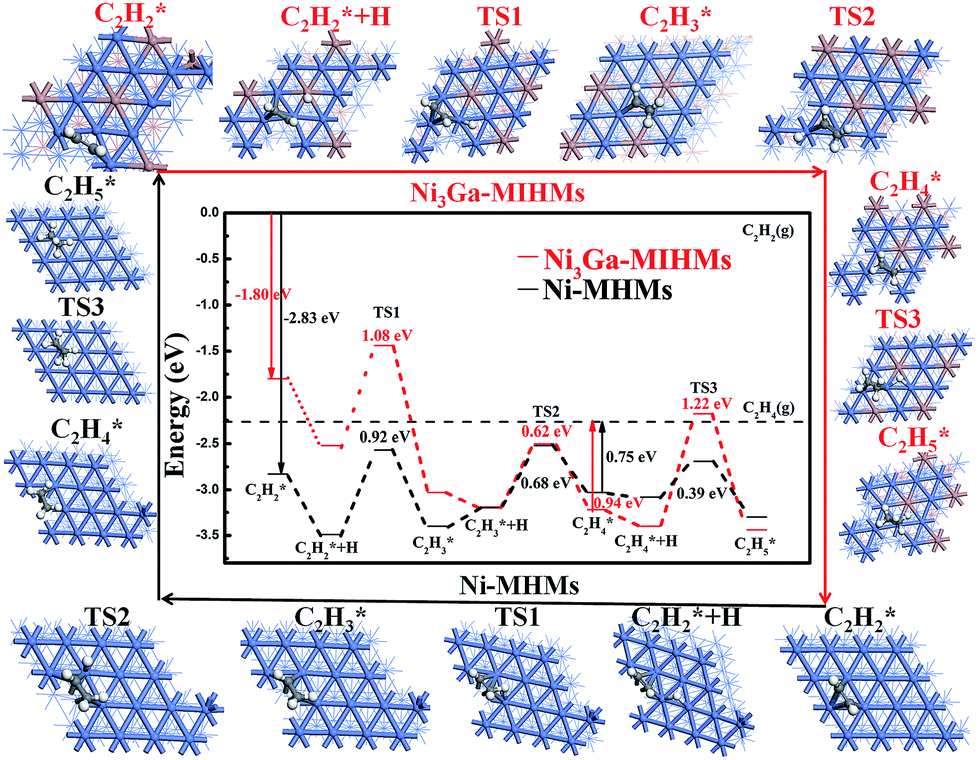
To better understand the underlying reason of the astonishing catalytic properties difference between the Ni3Ga-MIHMs and Ni-MHMs, DFT calculations were employed as shown in Fig. 4, 5, and S8–S12.† We investigated the whole catalytic process from the following three aspects: adsorption of acetylene hydrogenation species, transition states of acetylene hydrogenation, and selectivity of acetylene hydrogenation to ethylene. To make a more generalized model system, we further employed a model unit cell of 1:1 intermetallic cubic with a space group of Pm3m as a model system to simulate the intermetallic Ni3Ga structures. Periodic slab models of Ni3Ga (111) and Ni (111) were used to simulate hydrogenation reactivity of Ni3Ga and Ni based on thermodynamics. To further demonstrate the reaction pathway of selective hydrogenation of alkyne over the built model Ni3Ga (111) and Ni (111) systems, we have also drawn a reaction pathway map as displayed in Fig. 4. Initially, acetylene (C2H2) gas would adsorb on the surface of the catalyst. Then, the adsorbed C2H2 was successively hydrogenated to generate C2H3(ad) and C2H4(ad). In the following step, the generated C2H4(ad) was further over hydrogenated to produce C2H5(ad) or selectively semi-hydrogenated to form target product (C2H4 gas). This step is of great importance in determining corresponding C2H4 selectivity.
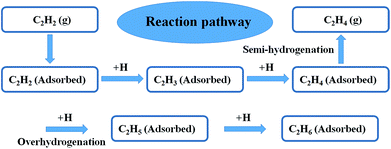 |
| | Fig. 4 The reaction pathways of selective hydrogenation of alkyne. | |
Adsorption of acetylene hydrogenation species
The adsorption structures and corresponding energies of C2H2 and C2H4 were calculated and are shown in Fig. S9–S13 and Tables S1 and S2.† The adsorption energies of C2H2 on the Ni3Ga (111) and Ni (111) are −1.80 and −2.83 eV with a C–H bond length of 1.94 Å and 1.89 Å, respectively, showing effective reactivity of Ni sites of the model Ni3Ga (111) and Ni (111). Moreover, the adsorption energy of C2H4 on Ni3Ga (111) and Ni (111) are −0.94 and −0.75 eV, with C–H bond lengths of 2.15 Å and 2.03 Å, respectively, which implies that C2H4 can be further hydrogenated to C2H6. On the Ni (111) surface, C2H2 tends to bind to Ni atoms with a multi-σ bonding mode and a μ-bridge configuration.5,40 On the Ni3Ga (111) surface, all the adsorbents are bonded to Ni atoms with a similar adsorption mode as on the Ni (111) surface rather than Ga atoms because Ga is not the prime active component.5 Besides the adsorption and activation of C2H2 molecules, H2 adsorption and activation are also of great importance in determining corresponding reactivity. According to the Horiuti–Polanyi mechanism,41 H2 adsorption energies on Ni3Ga (111) and Ni (111) are calculated as 0.127 eV and −0.285 eV, respectively, as shown in Fig. S13.† To be noted, H2 dissociation over the Ni3Ga (111) is almost barrierless with a barrier energy of only 0.0086 eV, but H2 molecule dissociation is more challenging upon the Ni (111) with the barrier energy increased over 4-fold than that over the Ni3Ga (111). Besides, the H2 dissociation exothermic energy value of Ni3Ga (111) (1.51 eV) is also much larger than that of Ni (111) (0.98 eV), further revealing the Ni3Ga (111) is more reactive for hydrogen disassociation and activation, which is of great significance to corresponding reaction activity.
Transition states of acetylene hydrogenation
Energy profiles (energy vs. reaction coordinates) of the whole acetylene hydrogenation pathways are depicted in Fig. 5. The alkyne hydrogenation barriers over the Ni (111) are 0.92 eV (TS1), 0.68 eV (TS2), and 0.39 eV (TS3), respectively. The rate-determining step is the first hydrogenation step (from C2H2 to C2H3) because the barriers of this step are the largest among the TS1, TS2, and TS3. By comparison, the hydrogenation barriers of alkyne over the Ni3Ga (111) are 1.08 eV (TS1), 0.61 eV (TS2), and 1.22 eV (TS3) with an exothermic energy of 0.61 eV, 0.25 eV, and 0.04 eV, respectively as demonstrated in Table S2.† The rate-determining step of it is the third step owing to largest barrier energy, which is different from the Ni (111) reaction pathway in which the first hydrogenation step is the rate-determining step. This result also implies that C2H4 is more difficult to hydrogenate to C2H5(ad) over the Ni3Ga (111), which is very favorable to realize high C2H4 selectivity.
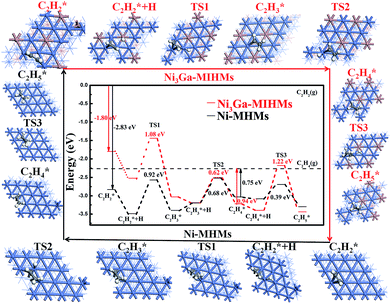 |
| | Fig. 5 Potential energy diagram by DFT calculations for the C2H2 hydrogenation process over the Ni3Ga-MIHMs and Ni-MHMs. | |
Selectivity of acetylene hydrogenation to ethylene
Since the adsorbed ethylene still has an unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, it can be over hydrogenated to form ethane, which decreases corresponding ethylene selectivity. To avoid this effect, we should first reveal the underlying reason that determines ethylene selectivity and develop more effective measures to improve ethylene selectivity. As indicated in Fig. 5, the ethylene gas desorption energy value upon the Ni3Ga (111) is 0.94 eV, which is much lower than that of the corresponding ethylene hydrogenation barrier (TS3, 1.22 eV), disclosing that ethylene is more prone to desorb in the form of gas rather than to be hydrogenated to ethane. However, as for the Ni (111), the ethylene desorption energy value over it is 0.75 eV, which is much higher than that of the corresponding ethylene hydrogenation barrier (TS3, 0.39 eV), which reveals that ethylene prefers to further hydrogenate to ethane rather than desorption, leading to the low ethylene selectivity of the it. What's more, the selectivity of ethylene formation could be estimated by the difference (ΔEa) between the hydrogenation barrier and the desorption barrier of ethylene, and a more positive ΔEa value indicates better selectivity to ethylene.42 The ΔEa of Ni3Ga (111) and Ni (111) are calculated to be 0.28 eV and −0.36 eV, respectively, illustrating that Ni3Ga (111) is more effective in obtaining high selectivity of ethylene. The generally simulated reaction pathway nicely explains the experimental results as displayed in Fig. 3–5, and is also in good agreement with previous reports.5,40 We believe that the DFT results are not limited to the current system and are also extendable to other systems because of the built models Ni3Ga (111) and Ni (111).
C bond, it can be over hydrogenated to form ethane, which decreases corresponding ethylene selectivity. To avoid this effect, we should first reveal the underlying reason that determines ethylene selectivity and develop more effective measures to improve ethylene selectivity. As indicated in Fig. 5, the ethylene gas desorption energy value upon the Ni3Ga (111) is 0.94 eV, which is much lower than that of the corresponding ethylene hydrogenation barrier (TS3, 1.22 eV), disclosing that ethylene is more prone to desorb in the form of gas rather than to be hydrogenated to ethane. However, as for the Ni (111), the ethylene desorption energy value over it is 0.75 eV, which is much higher than that of the corresponding ethylene hydrogenation barrier (TS3, 0.39 eV), which reveals that ethylene prefers to further hydrogenate to ethane rather than desorption, leading to the low ethylene selectivity of the it. What's more, the selectivity of ethylene formation could be estimated by the difference (ΔEa) between the hydrogenation barrier and the desorption barrier of ethylene, and a more positive ΔEa value indicates better selectivity to ethylene.42 The ΔEa of Ni3Ga (111) and Ni (111) are calculated to be 0.28 eV and −0.36 eV, respectively, illustrating that Ni3Ga (111) is more effective in obtaining high selectivity of ethylene. The generally simulated reaction pathway nicely explains the experimental results as displayed in Fig. 3–5, and is also in good agreement with previous reports.5,40 We believe that the DFT results are not limited to the current system and are also extendable to other systems because of the built models Ni3Ga (111) and Ni (111).
According to DFT calculations in Table S4,† the crystal formation energy of intermetallic Ni3Ga and Ni3Sn4 are −16.35 eV and −59.46 eV, respectively, which demonstrates a thermodynamically favorable path for the formation of multi-shelled intermetallic inter-metallic Ni3Ga and Ni3Sn4 phases. Taking a similar approach, well-defined multi-shelled intermetallic Ni3Sn4 hollow microspheres were further successfully synthesized as evidenced by the detailed XRD, HAADF-STEM, and EDS elemental mapping characterizations as demonstrated in Fig. S14–16,† showing effective generality of the topological self-template protocol. To be noted, the BET specific surface area of these multi-shelled Ni3Sn4 hollow microspheres amounts to 69.1 m2 g−1, as shown in Fig. S16b.†
Conclusions
In summary, we have prepared well-defined multi-shelled intermetallic Ni3Ga hollow microspheres (Ni3Ga-MIHMs) via a topological self-template strategy. Detailed characterization revealed the novel structures and atomically ordered intermetallic Ni3Ga phase nature of the fabricated Ni3Ga-MIHMs. Selective hydrogenation of the acetylene catalytic evaluation results further showed the excellent catalytic properties of the multi-shelled intermetallic Ni3Ga hollow microspheres, due to the more energetically favorable reaction pathway for acetylene hydrogenation and ethylene desorption over it as revealed by the corresponding DFT calculations. This strategy is also extendable to the preparation of other multi-shelled intermetallic Ni3Sn4 hollow microspheres, opening up new opportunities for the rational design and synthesis of novel structured and highly efficient intermetallic compound materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
M. Z. H., W. J. Y., and S. Z. L. contributed equally to this work. This work was supported by the National Natural Science Foundation of China (21872076, 21573119, 21866032, 21590792, 21473199, 51651201), the National Key R&D Program of China (2017YFA0700101, 2016YFA0202801) and Beijing Municipal Science & Technology Commission and Chinese Academy of Sciences. China Postdoctoral Science Foundation (2018M631486). We thank Tsinghua National Laboratory for Information Science and Technology for providing grid resources.
References
- Q. Feng, S. Zhao, Y. Wang, J. Dong, W. Chen, D. He, D. Wang, J. Yang, Y. Zhu, H. Zhu, L. Gu, Z. Li, Y. Liu, R. Yu, J. Li and Y. Li, J. Am. Chem. Soc., 2017, 139, 7294 CrossRef PubMed.
- E. W. Zhao, R. Maligal-Ganesh, C. Xiao, T.-W. Goh, Z. Qi, Y. Pei, H. E. Hagelin-Weaver, W. Huang and C. R. Bowers, Angew. Chem., 2017, 129, 3983 CrossRef.
- N. Taccardi, M. Grabau, J. Debuschewitz, M. Distaso, M. Brandl, R. Hock, F. Maier, C. Papp, J. Erhard, C. Neiss, W. Peukert, A. Görling, H. P. Steinrück and P. Wasserscheid, Nat. Chem., 2017, 9, 862 CrossRef PubMed.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Science, 2008, 320, 1320 CrossRef PubMed.
- Y. Liu, X. Liu, Q. Feng, D. He, L. Zhang, C. Lian, R. Shen, G. Zhao, Y. Ji, D. Wang, G. Zhou and Y. Li, Adv. Mater., 2016, 28, 4747 CrossRef PubMed.
- Y. Bai, H. Huang, C. Wang, R. Long and Y. Xiong, Mater. Chem. Front., 2017, 1, 1951 RSC.
- K. Jiang, P. Wang, S. Guo, X. Zhang, X. Shen, G. Lu, D. Su and X. Huang, Angew. Chem., Int. Ed., 2016, 55, 9030 CrossRef PubMed.
- Z. Qi, C. Xiao, C. Liu, T. W. Goh, L. Zhou, R. Maligal-Ganesh, Y. Pei, X. Li, L. A. Curtiss and W. Huang, J. Am. Chem. Soc., 2017, 139, 4762 CrossRef PubMed.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Nørskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329 CrossRef PubMed.
- S. Thota, Y. Wang and J. Zhao, Mater. Chem. Front., 2018, 2, 1074 RSC.
- M. Vandichel, A. Moscu and H. Grönbeck, ACS Catal., 2017, 7, 7431 CrossRef.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2012, 12, 81 CrossRef PubMed.
- G. Jiang, H. Zhu, X. Zhang, B. Shen, L. Wu, S. Zhang, G. Lu, Z. Wu and S. Sun, ACS Nano, 2015, 9, 11014 CrossRef PubMed.
- C. Suryanarayana, E. Ivanov and V. V. Boldyrev, Mater. Sci. Eng., A, 2001, 304–306, 151 CrossRef.
- D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044 CrossRef PubMed.
- M. Luo, Y. Sun, L. Wang and S. Guo, Adv. Energy Mater., 2016, 7, 1602073 CrossRef.
- Z. Dong, X. Lai, J. E. Halpert, N. Yang, L. Yi, J. Zhai, D. Wang, Z. Tang and L. Jiang, Adv. Mater., 2012, 24, 1046 CrossRef PubMed.
- Z. Dong, H. Ren, C. M. Hessel, J. Wang, R. Yu, Q. Jin, M. Yang, Z. Hu, Y. Chen, Z. Tang, H. Zhao and D. Wang, Adv. Mater., 2014, 26, 905 CrossRef PubMed.
- J. Wang, N. Yang, H. Tang, Z. Dong, Q. Jin, M. Yang, D. Kisailus, H. Zhao, Z. Tang and D. Wang, Angew. Chem., 2013, 125, 6545 CrossRef.
- L. Yu, X. Y. Yu and X. W. Lou, Adv. Mater., 2018, 1800939, DOI:10.1002/adma.201800939.
- X. Zhao, R. Yu, H. Tang, D. Mao, J. Qi, B. Wang, Y. Zhang, H. Zhao, W. Hu and D. Wang, Adv. Mater., 2017, 29, 1700550 CrossRef PubMed.
- D. Li, X. Zhao, R. Yu, B. Wang, H. Wang and D. Wang, Inorg. Chem. Front., 2018, 5, 535 RSC.
- J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu, G. Ma, Z. Su, H. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749 RSC.
- J. Wang, H. Tang, H. Wang, R. Yu and D. Wang, Mater. Chem. Front., 2017, 1, 414 RSC.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209 CrossRef PubMed.
- A. Borodziński and G. C. Bond, Catal. Rev., 2008, 50, 379 CrossRef.
- L.-S. Hsu, Y. K. Wang, Y. L. Tai and J. F. Lee, J. Alloys Compd., 2006, 413, 11 CrossRef CAS.
- K. Schutte, A. Doddi, C. Kroll, H. Meyer, C. Wiktor, C. Gemel, G. van Tendeloo, R. A. Fischer and C. Janiak, Nanoscale, 2014, 6, 5532 RSC.
- L. S. Hsu, G. H. Gweon and J. W. Allen, J. Phys. Chem. Solids, 1999, 60, 1627 CrossRef CAS.
- A. P. Tsai, S. Kameoka, K. Nozawa, M. Shimoda and Y. Ishii, Acc. Chem. Res., 2017, 50, 2879 CrossRef CAS PubMed.
- H. Zhou, X. Yang, L. Li, X. Liu, Y. Huang, X. Pan, A. Wang, J. Li and T. Zhang, ACS Catal., 2016, 6, 1054 CrossRef CAS.
- D. A. Torelli, S. A. Francis, J. C. Crompton, A. Javier, J. R. Thompson, B. S. Brunschwig, M. P. Soriaga and N. S. Lewis, ACS Catal., 2016, 6, 2100 CrossRef CAS.
- M. B. Boucher, B. Zugic, G. Cladaras, J. Kammert, M. D. Marcinkowski, T. J. Lawton, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Phys. Chem. Chem. Phys., 2013, 15, 12187 RSC.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, Science, 2008, 320, 86 CrossRef CAS PubMed.
- H. Fei, L. Hongyang and S. Dangsheng, Sci. China Mater., 2017, 60, 1149 Search PubMed.
- L. Qiang, J. Shufang, L. Mufan and D. Xiangfeng, Sci. China Mater., 2018 DOI:10.1007/s40843-018-9282-1.
- C. Wu, Y. Chen, R. Shen, W. Zhu, Y. Gong, L. Gu, Q. Peng, H. Guo and W. He, Nano Res., 2018, 11, 4883 CrossRef CAS.
- Y. Wang, Z. Chen, R. Shen, X. Cao, Y. Chen, C. Chen, D. Wang, Q. Peng and Y. Li, Nano Res., 2016, 9, 1209 CrossRef CAS.
- A. J. McCue and J. A. Anderson, Front. Chem. Sci. Eng., 2015, 9, 142 CrossRef CAS.
- D.-M. Rao, S.-T. Zhang, C.-M. Li, Y.-D. Chen, M. Pu, H. Yan and M. Wei, Dalton Trans., 2018, 47, 4198 RSC.
- B. Yang, R. Burch, C. Hardacre, P. Hu and P. Hughes, Catal. Sci. Technol., 2017, 7, 1508 RSC.
- B. Yang, R. Burch, C. Hardacre, G. Headdock and P. Hu, ACS Catal., 2012, 2, 1027 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and DFT calculation information. See DOI: 10.1039/c8sc03178a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Qing
Peng
a,
Chen
Chen
*b,
Qing
Peng
a,
Chen
Chen