
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of the structure and geometry of N-heterocyclic carbenes on Au(111) using high-resolution spectroscopy†
Giacomo
Lovat
a,
Evan A.
Doud
 b,
Deyu
Lu
c,
Gregor
Kladnik
de,
Michael S.
Inkpen
a,
Michael L.
Steigerwald
b,
Dean
Cvetko
def,
Mark S.
Hybertsen
c,
Alberto
Morgante
*dg,
Xavier
Roy
*b and
Latha
Venkataraman
*ab
b,
Deyu
Lu
c,
Gregor
Kladnik
de,
Michael S.
Inkpen
a,
Michael L.
Steigerwald
b,
Dean
Cvetko
def,
Mark S.
Hybertsen
c,
Alberto
Morgante
*dg,
Xavier
Roy
*b and
Latha
Venkataraman
*ab
aDepartment of Applied Physics and Applied Mathematics, Columbia University, New York, New York 10027, USA. E-mail: lv2117@columbia.edu
bDepartment of Chemistry, Columbia University, New York, New York 10027, USA. E-mail: xr2114@columbia.edu
cCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, New York, USA
dCNR-IOM Laboratorio Nazionale TASC, Basovizza SS-14, km 163.5, 34012 Trieste, Italy. E-mail: morgante@iom.cnr.it
eFaculty of Mathematics and Physics, University of Ljubljana, Jadranska 19, Ljubljana, Slovenia
fJ. Stefan Institute, Jamova 39, Ljubljana, SI-1000, Slovenia
gDepartment of Physics, University of Trieste, via A. Valerio 2, 34127, Trieste, Italy
First published on 5th November 2018
Abstract
N-heterocyclic carbenes (NHCs) bind very strongly to transition metals due to their unique electronic structure featuring a divalent carbon atom with a lone pair in a highly directional sp2-hybridized orbital. As such, they can be assembled into monolayers on metal surfaces that have enhanced stability compared to their thiol-based counterparts. The utility of NHCs to form such robust self-assembled monolayers (SAMs) was only recently recognized and many fundamental questions remain. Here we investigate the structure and geometry of a series of NHCs on Au(111) using high-resolution X-ray photoelectron spectroscopy and density functional theory calculations. We find that the N-substituents on the NHC ring strongly affect the molecule–metal interaction and steer the orientation of molecules in the surface layer. In contrast to previous reports, our experimental and theoretical results provide unequivocal evidence that NHCs with N-methyl substituents bind to undercoordinated adatoms to form flat-lying complexes. In these SAMs, the donor–acceptor interaction between the NHC lone pair and the undercoordinated Au adatom is primarily responsible for the strong bonding of the molecules to the surface. NHCs with bulkier N-substituents prevent the formation of such complexes by forcing the molecules into an upright orientation. Our work provides unique insights into the bonding and geometry of NHC monolayers; more generally, it charts a clear path to manipulating the interaction between NHCs and metal surfaces using traditional coordination chemistry synthetic strategies.
Introduction
N-heterocyclic carbenes (NHCs) are exceptionally strong σ-donor ligands capable of binding to virtually any transition metal. They are receiving increasing interest for their ability to form functional self-assembled monolayers (SAMs) on metal surfaces.1–8 Johnson, Crudden and their respective coworkers have demonstrated that NHC-based SAMs exhibit remarkable thermal and chemical stability that go well beyond thiol-based SAMs on Au,1,7 opening the door to novel applications in selective heterogeneous catalysis,9,10 nanotechnology11 and sensing.2 Moreover, the strength and directionality of NHC–metal bonds, by now well-established in coordination chemistry, offer exciting new possibilities for passivating and/or manipulating the work function of metal surfaces.12 While a substantial body of work has been devoted to NHCs since their discovery,13–21 this research has thus far mainly focused on the design of homogeneous catalysts. By contrast, many fundamental questions regarding the structure of NHC SAMs and their electronic coupling with metal surfaces remain unanswered.1,7,8,22 Recently, some unique insights have been provided through the application of NHCs as linker groups in molecular-scale electronics.23,24,29,32To date, most models of NHC-bound SAMs on metal surfaces postulate that the molecules adopt an upright geometry, with the heterocyclic system perpendicular to the surface.7,8,22,25–28 In this orientation, a donor–acceptor interaction from the carbene lone pair to a surface atom is the primary contribution to the NHC–metal bond. In contrast, Baddeley, Papageorgiou, and their respective co-workers recently reported that some NHCs can form flat-lying mononuclear complexes (NHC)2M (M = Cu, Ag, Au) on surfaces, where the coordinated metal site is pulled out of the surface plane.23,30 Such adatoms are key structural components in SAMs of thiol molecules on Au(111),31 and provide a clear experimental signature of surface reorganization induced by strong molecule–surface interactions. With the exception of a single report utilizing high resolution electron energy loss spectroscopy (HREELS),29 the conflicting conclusions regarding the structure of NHC SAMs may be attributed to the use of scanning tunneling microscope (STM) imaging or low resolution X-ray photoelectron spectroscopy (LR-XPS) to probe these systems.32 Such methods do not provide the chemical sensitivity required to determine the precise orientation of NHCs in SAMs, the nature of their bonding to the surface, or the unambiguous detection of any associated Au adatoms. As a result, the role of N-substituents in influencing NHC SAM structure remains ill-defined.
In this work, we use synchrotron radiation to perform high-resolution X-ray photoelectron spectroscopy (HR-XPS) and near-edge X-ray absorption fine-structure spectroscopy (NEXAFS), and combine these measurements with density functional theory (DFT) calculations to establish a detailed picture of the geometry and bonding of a series of NHCs assembled on Au(111) in ultra-high vacuum (UHV). HR-XPS is highly sensitive to the chemical composition of the NHC layer and detects small changes to core-level electron binding energies that result from NHC–Au interactions, providing unique measurements of surface coverage and surface adatom density. NEXAFS allows us to unequivocally determine the orientation of the molecules relative to the surface by probing their unoccupied electronic states. Such information is key to understanding the relationship between the NHC molecular structure and their adsorption geometry. It can be obtained clearly through spectroscopic measurements. In contrast, surface imaging techniques only focus on small areas of the surface and they cannot resolve precisely the orientation of molecules bound to the surface. By rationalizing our experimental results using DFT calculations, we quantify the impact of the NHC structure and conformation on the strength of the molecule–Au interaction. Importantly, we show that through changes in the substituents on the N atoms, we can alter the steric environment around the carbenic C atom, which strongly affects the surface tilt angle and adsorption behavior of the molecules. This study complements and extends previous important efforts to characterize these systems. It further shows how thermal annealing in combination with careful selection of N-substituents can modulate the structure of NHCs on Au(111) surfaces.
Results and discussion
Three NHCs with different steric properties are investigated: 1,3-dimethylimidazol-2-ylidene (NHCMe), 1,3-diisopropylbenzimidazol-2-ylidene (BNHCiPr), and 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (NHCdipp). Details for the syntheses of the NHC precursors are found in the ESI.† Monolayers were prepared in UHV via thermal decomposition/sublimation of NHC–CO2 precursors,22 as illustrated in Fig. 1 and described in the ESI.† Briefly here, the precursors are placed in a Pyrex cell and connected to the pre-chamber through a leak valve. This cell is evacuated, heated to ∼70 °C, and the carbene is introduced as a vapor into a pre-chamber containing a clean Au(111) crystal kept between −20 and −30 °C for ∼5 min while maintaining a partial molecular pressure of 10−7 mbar. After deposition we confirm that the molecules deposited on the substrate have lost their CO2 moieties, ostensibly through thermal decomposition, by measuring the O 1s spectrum of the layer using XPS.22 | ||
| Fig. 1 Schematic showing the molecular structure of the NHC precursors, the thermal decomposition/sublimation deposition approach to create the NHC monolayers, and the NEXAFS dichroism measurement. Free NHC molecules are generated in the gas phase upon heating. | ||
To determine the orientation of the NHC relative to the Au (111) surface normal (defined as the tilt angle θ in Fig. 1), we first present NEXAFS linear dichroism results for each molecule.33Fig. 2a shows the NEXAFS spectra collected at the N K-edge with the electric field of the incident photons perpendicular (p-polarization) and parallel (s-polarization) to the surface for NHCMe, BNHCiPr, NHCdipp monolayers. The key result is that the dependence of the NEXAFS spectra on the photon polarization (dichroism) varies with the carbene N-substituents.33 The lowest energy NEXAFS resonance at ∼401 eV arises from the N 1s → π*-LUMO (lowest unoccupied molecular orbital) transition. It is strongly enhanced with p-polarized photons for NHCMe, moderately enhanced for BNHCiPr, and virtually absent for NHCdipp; the opposite trend is observed for s-polarized photons. Since the π*-LUMO is delocalized over the whole imidazole ring for all three molecules, we can use the relative intensities of the ∼401 eV NEXAFS p-polarized and s-polarized peaks to determine the average tilt angle θ for each carbene monolayer.33 We find that NHCMe is almost flat (θ ∼ 72°), BNHCiPr has an intermediate tilt angle (θ ∼ 40°), and NHCdipp is almost standing up (θ ∼ 13°). The small tilt angle for NHCdipp can be attributed to the steric bulk introduced by the side groups forcing the molecule to stand and preventing the NHC-ring from interacting directly with the surface. This is corroborated by the NHCdipp NEXAFS spectra collected at the C K-edge (Fig. S1†).
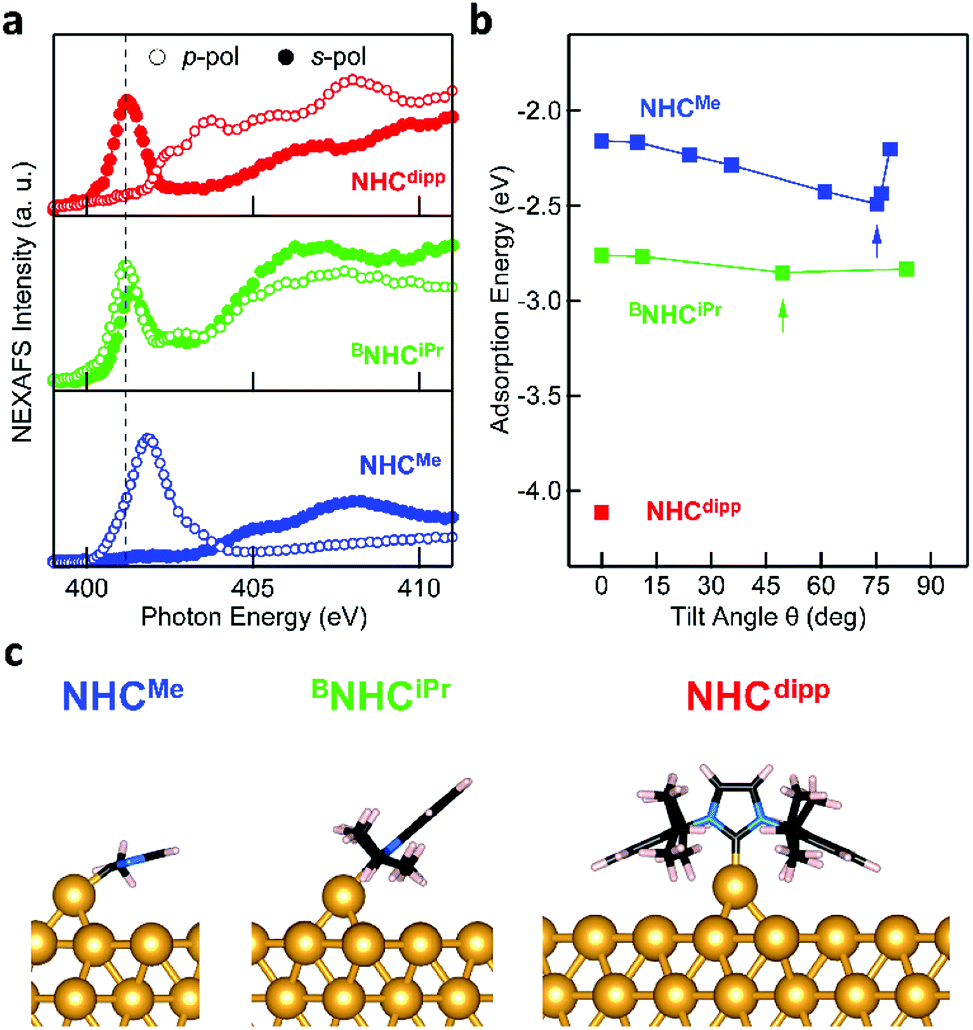 | ||
| Fig. 2 (a) NEXAFS spectra collected a the N K-edge for NHCMe (blue, bottom panel), BNHCiPr (green), and NHCdipp (red) monolayers on Au(111). Each spectrum is measured using X-ray photons with incident electric field in a plane perpendicular to the surface (p-pol, empty circles) or in a plane parallel to the surface (s-pol, filled circles). The N 1s → π*-LUMO resonance (∼401 eV, dashed black line) is significantly enhanced in p-pol for NHCMe, and in s-pol for NHCdipp, indicating a tilt angle θ ∼ 72° and ∼13° respectively. For BNHCiPr, both s- and p-pol spectra show the π*-LUMO resonance, yielding θ ∼ 40°. (b) Calculated adsorption energy of NHCMe (blue) and BNHCiPr (green) on an Au adatom as a function of θ. The adsorption energy of NHCdipp (red) is calculated only for θ = 0°. The arrows indicate the lowest energy structure tilt angles, in good agreement with experimental observations. (c) DFT-optimized energy minimum structure of a single NHCMe, BNHCiPr and NHCdipp adsorbed on an Au adatom sitting on a hollow site of an Au(111) slab. These structures are consistent with the experimentally observed tilt angles. | ||
It is remarkable that these variations in the tilt angle do not result in significant differences in the NHC monolayer stability as a function of temperature. Indeed, HR-XPS measurements show that NHCMe, BNHCiPr and NHCdipp all come off the surface around ∼300 °C either through desorption or decomposition (Fig. S2†). This implies that all three NHCs, despite their varied orientations, form strong donor–acceptor bonds to Au. The van der Waals interactions alone are unlikely to result in high desorption temperatures for such small cyclic compounds.34 The nature of the NHC–Au bond in these monolayers, and/or their macroscopic structure, is likely more complex than readily determined from the NEXAFS data alone. We therefore turn to DFT calculations to provide further insights.
We first consider two simple adsorption models for all three carbenes and use DFT calculations to evaluate their structure and bonding energy. We consider a single NHC molecule adsorbed either on a pristine Au(111) surface or an Au adatom sitting on a hollow site of the Au(111) surface. We will show later that this is not the structure that is consistent with our data for NHCMe. Total energy and geometry optimization calculations, detailed in the ESI,† are performed using Quantum ESPRESSO35 with an exchange and correlation functional that accounts for van der Waals interactions.36,37 A 4-layer Au(111) slab comprising 3 × 3, 4 × 3, and 5 × 5 surface unit cells is used to model surface-adsorbed NHCMe, BNHCiPr and NHCdipp, respectively. The adsorption energy is defined as the difference between the energy of the combined system and the sum of the energies for each component separately. It is negative for bound systems.
The adsorption energy of NHCMe in a constrained flat-lying geometry on an Au(111) slab is small (−0.91 eV). The carbene lone pair lies parallel to the Au slab and does not form a strong σ-bond to an Au atom (Fig. S3†). Removing the constraint on the tilt angle in this model results in NHCMe adopting a binding geometry nearly normal to the Au(111) surface (θ ∼ 15°). This geometrical change is accompanied by a 0.58 eV increase of the adsorption energy of the molecule (−1.49 eV; Fig. S4†). However, these results are at odds with the NEXAFS data indicating that θ ∼ 72° for NHCMe.
A significantly different outcome is obtained when the NHCs are relaxed on top of an Au adatom, as shown in Fig. 2b. The adsorption energy for the NHCs at different tilt angles is calculated by constraining the geometry of the NHC ring relative to the surface normal. In its most stable conformation (Fig. 2b and c), NHCMe has a tilt angle θ ∼ 75°, in excellent agreement with the NEXAFS data. The corresponding adsorption energy of −2.49 eV is 1 eV larger than when NHCMe is bound to a flat Au(111) surface. The maximum adsorption energy for BNHCiPr is −2.85 eV, corresponding to an optimized tilt angle θ ∼ 50°. The adsorption energy for BNHCiPr, however, is only weakly dependent on θ, a consequence of the competition between steric repulsion imparted by the bulkier side substituents and the van der Waals interaction of the benzene ring towards the Au surface. The weak dichroism in the NEXAFS spectrum of BNHCiPr (Fig. 2a) agrees well with the theoretical results. The shallow energy minimum implies that there is no strong driving force to orient the molecule in a preferential geometry. Moreover, the computed lowest energy θ for BNHCiPr is close to the theoretical angle at which no dichroism is expected (θ ∼ 55°). Due to its bulkier N-substituents, NHCdipp can only bind vertically, irrespective of the presence of an adatom (Fig. S5†). The adsorption energies for NHCdipp bound to the Au(111) surface and to an Au adatom are −2.69 eV and −4.11 eV, respectively.
For all three NHCs, the adsorption energy is larger when the molecule binds to an Au adatom. This is the result of the metal s- and d-orbitals being more accessible in the adatom resulting in a stronger donor–acceptor bond. This is reflected in the shorter NHC–Au bond length when the molecule is modeled on an Au adatom (see Table S1†). However, among the three NHCs, NHCdipp stands out. The calculated adsorption energy for the adatom-bound model is significantly larger than the corresponding values for the other two NHCs. We attribute this difference to the van der Waals interactions of the dipp groups with the Au surface. The difference in adsorption energy between the pristine surface-bound and adatom-bound models is also significantly larger for the NHCdipp system than for the other two NHCs. This can be explained by steric effects between the bulky dipp substituents and the surface, which significantly lengthen the NHC–Au bond (2.15 Å) and distort the molecule when NHCdipp is bound to a pristine Au(111) surface. By contrast, when NHCdipp is bound to an Au adatom, the dipp groups are further away from the surface, the carbenic carbon can get closer to the Au adatom (NHC–Auad bond length = 2.02 Å), and the molecule can relax to a less strained conformation. This is most easily seen looking at the orientation of the dipp groups. They are almost parallel to the surface in the perfect slab model (Fig. S5†) and close to the unstrained geometry in the adatom model (i.e. the CNHC–NNHC–Cdipp angle is ∼124°; Fig. 2c).
The DFT calculations presented above suggest that NHCs bind significantly more strongly to an Au adatom than to a perfect Au(111) surface. XPS measurements can detect the presence of such adatoms on the surface. Fig. 3a presents the N 1s core level XPS spectra of NHCMe, BNHCiPr and NHCdipp monolayers. A single N 1s peak is observed in the NHCMe and NHCdipp spectra, consistent with only one type of N-containing species on the surface. The spectrum of BNHCiPr shows two peaks. The first and more intense main peak is very close in energy to that of NHCdipp. It is attributed to surface-bound BNHCiPr. The second, lower-energy peak is attributed to incipient second-layer growth and/or a small amount of undissociated BNHCiPr–CO2 adducts on the surface. Note that the NHCMe N 1s peak (401.2 eV) is at a significantly higher binding energy than the corresponding peaks for the BNHCiPr and NHCdipp monolayers (∼400.5 eV). The correlation between this shift and the positions of the NEXAFS N 1s → π*-LUMO resonance (Fig. 2a) for these monolayers indicates that the latter is related to the N 1s core binding energy (an initial state effect). Such a shift to higher binding energy results from charge depletion at the N in the NHCMe monolayer due to interaction of its π-electron system with the Au surface. This is consistent with the orientation of the molecule as determined by NEXAFS.
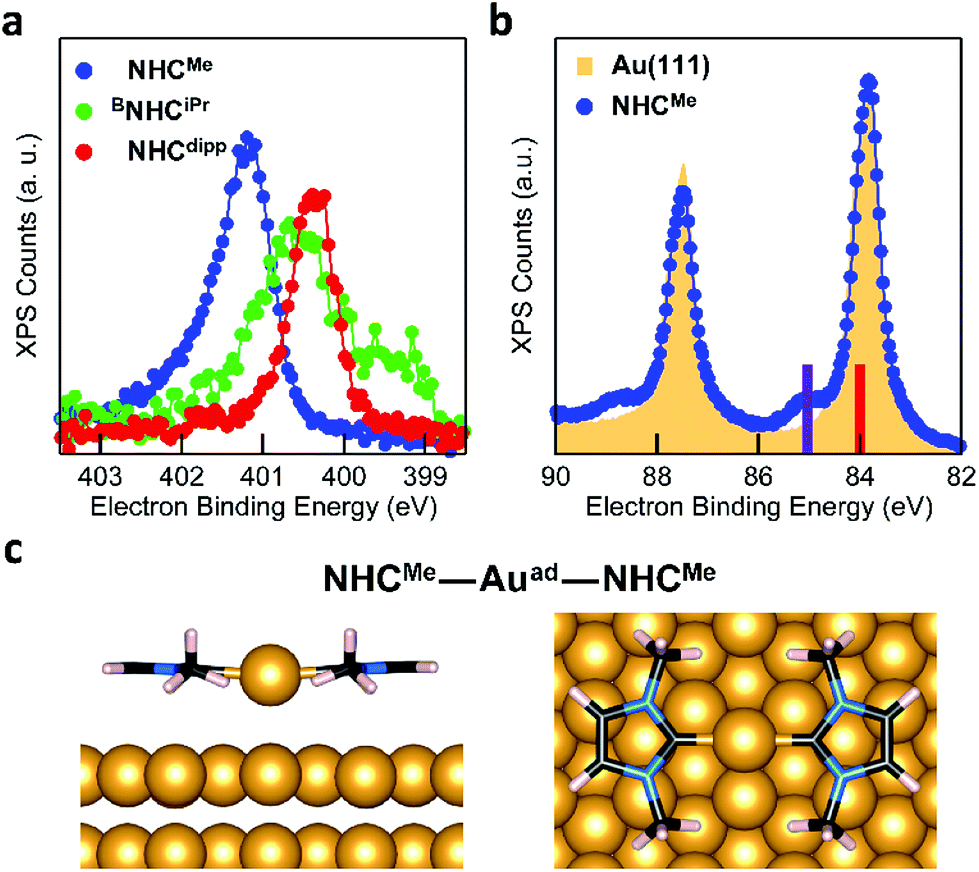 | ||
| Fig. 3 (a) XPS N 1s spectra of NHCMe (blue), BNHCiPr (green), and NHCdipp (red) monolayers on Au(111). The NHCMe N 1s peak is shifted to higher binding energy relative to both NHCdipp and BNHCiPr. (b) XPS Au 4f5/2,7/2 spectra of a clean Au(111) surface (yellow filled area) and the NHCMe monolayer (blue) on the same Au(111) surface. The satellite peaks at ∼1 eV higher binding energy are attributed to the presence of a high density of Au adatoms. Solid bars on the binding energy axis are the calculated XPS peak positions for bulk Au (red, 84.00 eV) and the Au adatom in the NHCMe–Auad–NHCMe complex (purple, 85.03 eV) adsorbed on the Au(111) slab. (c) DFT-optimized energy minimum structure of the NHCMe–Auad–NHCMe complex adsorbed on a 4-layer Au(111) slab (only the upper two layers are shown). The NHCMe rings are nearly coplanar to the surface and the adatom is on a hollow site. | ||
Fig. 3b compares the XPS spectra of the Au core level 4f spin–orbit doublet for a clean Au(111) surface and an NHCMe monolayer grown on the same Au(111) surface.38 The NHCMe monolayer spectrum features a doublet of satellite peaks at higher binding energies. Fits of the Au 4f7/2 peak indicate that the new set of peaks is shifted by +1.1 eV with respect to the Au bulk component (Fig. S6a†). Two contributions can explain the difference in the binding energy of the Au 4f peak: (1) a chemical shift; or (2) a screening shift that both result for an Au atom that is lifted significantly from the surface. Comparing the area under each peak, we find that 1/6 of the surface is covered by adatoms (Fig. S6a†).
To explore the origin of these satellite peaks, we consider two possible adsorption scenarios for NHCMe: (1) the adsorption of a single NHCMe on an Au adatom (NHCMe–Auad), and (2) the formation of a flat-lying bis(NHC) complex with an Au adatom (NHCMe–Auad–NHCMe). We calculate the binding energy shift for the Au adatom 4f core level relative to the bulk (ΔEBE) using the transition state model39,40 of the excited system and the projector augmented-wave method implemented in the Vienna Ab initio Simulation Package (VASP).41,42Fig. 3c presents the computed structure for the fully relaxed NHCMe–Auad–NHCMe complex adsorbed on a 5 × 4 unit cell. For the complex, ΔEBE = 0.91 eV, in good agreement with the experimental value (ΔEBE = 1.1 eV). By contrast, the computed binding energy shift for NHCMe–Auad (ΔEBE = 0.16 eV) is much smaller, suggesting that the satellite peaks in Fig. 3b come from NHCMe–Auad–NHCMe complexes on the surface. The formation of this bis(NHC) complex pulls the Au adatom away from the surface by more than 1 Å (Auad–Ausurf distance is 2.06 and 3.09 Å for NHCMe–Auad and NHCMe–Auad–NHCMe, respectively). The change in the electron binding energy of the Au adatom, due to chemical shift and screening effects, is similar to what has been observed in thiol-based monolayers.38
As a comparison, the Au 4f XPS spectrum of the NHCdipp monolayer shows no satellite peaks at higher binding energy. The bulky dipp groups prevent the formation of such bis(NHC) complexes (Fig. S6b†). A careful analysis of the STM images presented in a recent study22 of NHCMe monolayers on Au(111) further corroborate our conclusion that the molecules indeed form flat-lying NHCMe–Auad–NHCMe complexes as opposed to NHCMe–Auad species oriented normal to the surface, as originally proposed. To illustrate this point, Fig. S7† compares an STM image simulated from our optimized structure and an experimental STM image reproduced from Wang et al.22
The behavior of BNHCiPr differs from that of NHCMe and NHCdipp. This suggests a thermally activated process for the formation of the flat-lying bis(NHC) complexes. Fig. 4a compares the N 1s XPS spectrum of a BNHCiPr monolayer deposited at −20 °C with that of the monolayer annealed to 90 °C. The low-temperature monolayer shows a broad peak at ∼400.5 eV, indicating the formation of a mostly disordered surface structure (Fig. 2a). Remarkably, the XPS peak shape and energy change significantly upon thermal annealing. The high temperature N 1s XPS peak is sharp and shifted to a higher binding energy (∼401.5 eV). This spectrum is similar to that observed for the NHCMe–Auad–NHCMe complexes (Fig. 3a). The striking reorganization of the molecular layer is also captured in the NEXAFS dichroism collected at the N K-edge (Fig. 4b). The N 1s → π*-LUMO resonance is greatly enhanced in p-polarized light, pointing to a nearly flat geometry for the annealed monolayer.
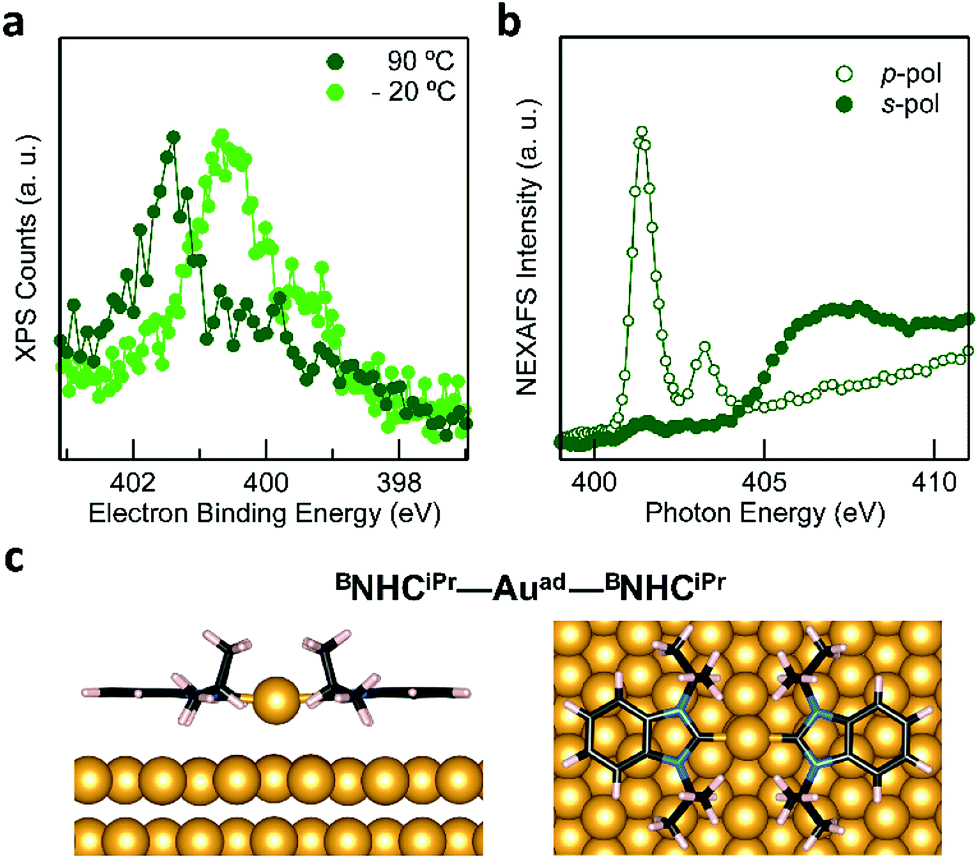 | ||
| Fig. 4 (a) XPS N 1s spectra of a BNHCiPr monolayer deposited on a cold substrate at −20 °C (light green), and then annealed to 90 °C (dark green). The broad peak in the low-temperature spectrum comprises different components likely due to multiple molecular adsorption sites and/or of second-layer molecules. Thermal annealing generates a single sharp N 1s peak shifted to higher binding energy by ∼1 eV. (b) NEXAFS spectrum collected at the N K-edge for the BNHCiPr monolayer annealed to 90 °C: a strong dichroism is clearly visible. The N 1s → π*-LUMO resonance is strongly enhanced in p-pol indicating that the molecules lie nearly flat on the surface. (c) DFT-optimized energy minimum structure of a BNHCiPr–Auad–BNHCiPr complex adsorbed on an Au(111) 5 × 7 slab. Note that the adatom is above a hollow site on the Au(111) surface. | ||
These spectroscopic changes make the adsorption behavior of BNHCiPr at high temperature similar to that of NHCMe. This prompted us to investigate theoretically the possibility that BNHCiPr forms flat-lying bis(NHC) complexes, provided sufficient energy is available. Similar to its NHCMe analogue, the modeled structure of the BNHCiPr–Auad–BNHCiPr complex on an Au(111) slab (Fig. 4c) shows that the Au adatom is pulled away from the surface by ∼1 Å (Table S1†). While this should in principle also alter the Au 4f XPS spectra, we do not observe Au satellite peaks in Fig. S6b.† The lower surface density of BNHCiPr accounts for this result. Using the intensity of the N 1s XPS peak, we estimate that the BNHCiPr surface density at 90 °C is ∼10 times lower than that of NHCMe at −20 °C. At this implied density of Au adatoms, the shifted Au 4f XPS satellite would be undetectable. Two effects combine to explain the lower surface density: (1) the footprint of the flat-lying benzannulated BNHCiPr is larger than that of NHCMe; and (2) the elevated temperature for the measurement leads to a further decrease in the packing density.
Conclusions
The strong carbene–Au interaction offers exciting opportunities as an alternative to the traditional thiol–Au bond, which suffers from limited chemical, electrochemical and thermal stability. By combining high-resolution X-ray photoelectron spectroscopy and computational modeling, this work reveals how adsorption energy, molecular orientation and metal surface structure are closely interconnected in a series of NHC monolayers. We find that NHCs bind significantly more strongly to an Au adatom than to a flat Au(111) surface. We show that with sufficient time and thermal energy, the molecule is capable of reorganizing the underlying gold surface structure. The orientation of the NHC on the surface is determined by the N-substituents. The smallest group – methyl – favors a planar geometry in which the NHC ring is parallel to the surface and organized into NHCMe–Auad–NHCMe complexes. Bulkier groups force the molecule into a more vertical orientation and prevent the formation of such flat-lying complexes. By thermally annealing the monolayer, we show it is possible to modulate the structure of surface-bound NHCs. Specifically, conversion from a single, vertically oriented NHC moiety to bis(NHC) complexes has been observed for BNHCiPr. While the initial orientation of NHCs on the surface appears to have little effect on the high thermal stability of NHCs SAMs, we expect that the geometry and electronic structure of the surface-bound carbenes will ultimately prove critical in controlling their (electro)chemical stability, reactivity and functionality, through steric hindrance of reactive sites as well as the extent of intermolecular and molecule–surface interactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Albano Cossaro for assisting with the experiments. The experimental and theoretical work was supported primarily by the Center for Precision Assembly of Superstratic and Superatomic Solids at Columbia University, an NSF MRSEC (award number DMR-1420634) and by NSF CHE-1807654. E. A. D and X. R. thank the Donors of the American Chemical Society Petroleum Research Fund for support (ACS PRF# 57062-DNI10). X. R. also acknowledges the Air Force Office of Scientific Research under AFOSR Award No. FA9550-18-1-0020. M. S. I. was supported by a Marie Skłodowska Curie Global Fellowship (MOLCLICK: 657247) within the Horizon 2020 Programme. D. C. and G. K. acknowledge partial financial support from the Slovenian Research Agency (program No. P1-0112). G. K. acknowledges financial support from the SIR grant SUNDYN [Nr. RBSI14G7TL, CUP B82I15000910001] of the Italian Ministry of Education, Universities and Research MIUR. Our research used resources of Center for Functional Nano-materials, which is a U.S. DOE Office of Science Facility, and the Scientific Data and Computing Center, a component of the Computational Science Initiative, at Brookhaven National Laboratory, under Contract No. DE-SC0012704.Notes and references
- C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H.-B. Kraatz, N. J. Mosey, T. Seki, E. C. Keske, J. D. Leake, A. Rousina-Webb and G. Wu, Nat. Chem., 2014, 1–6 Search PubMed
.
- C. M. Crudden, J. H. Horton, M. R. Narouz, Z. Li, C. A. Smith, K. Munro, C. J. Baddeley, C. R. Larrea, B. Drevniok, B. Thanabalasingam, A. B. McLean, O. V. Zenkina, I. I. Ebralidze, Z. She, H.-B. Kraatz, N. J. Mosey, L. N. Saunders and A. Yagi, Nat. Commun., 2016, 7, 12654 CrossRef CAS PubMed
- X. Ling, S. Roland and M. P. Pileni, Chem. Mater., 2015, 27, 414–423 CrossRef CAS
- X. Ling, N. Schaeffer, S. Roland and M. P. Pileni, Langmuir, 2015, 31, 12873–12882 CrossRef CAS PubMed
- M. J. MacLeod and J. A. Johnson, J. Am. Chem. Soc., 2015, 137, 7974–7977 CrossRef CAS PubMed
- S. Roland, X. Ling and M. P. Pileni, Langmuir, 2016, 32, 7683–7696 CrossRef CAS PubMed
- A. V. Zhukhovitskiy, M. G. Mavros, T. Van Voorhis and J. A. Johnson, J. Am. Chem. Soc., 2013, 135, 7418–7421 CrossRef CAS PubMed
- T. Weidner, J. E. Baio, A. Mundstock, C. Grosse, S. Karthauser, C. Bruhn and U. Siemeling, Aust. J. Chem., 2011, 64, 1177–1179 CrossRef CAS PubMed
- J. A. Mata, M. Poyatos and E. Peris, Coord. Chem. Rev., 2007, 251, 841–859 CrossRef CAS
- K. V. S. Ranganath, S. Onitsuka, A. K. Kumar and J. Inanaga, Catal. Sci. Technol., 2013, 3, 2161–2181 RSC
- R. W. Y. Man, C. H. Li, M. W. A. MacLean, O. V. Zenkina, M. T. Zamora, L. N. Saunders, A. Rousina-Webb, M. Narnbo and C. M. Crudden, J. Am. Chem. Soc., 2018, 140, 1576–1579 CrossRef CAS PubMed
- H. K. Kim, A. S. Hyla, P. Winget, H. Li, C. M. Wyss, A. J. Jordan, F. A. Larrain, J. P. Sadighi, C. Fuentes-Hernandez, B. Kippelen, J.-L. Brédas, S. Barlow and S. R. Marder, Chem. Mater., 2017, 29, 3403–3411 CrossRef CAS
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS
- S. Diez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef CAS PubMed
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS PubMed
- W. A. Herrmann, M. Elison, J. Fischer, C. Kocher and G. R. J. Artus, Angew. Chem., Int. Ed., 1995, 34, 2371–2374 CrossRef CAS
- W. A. Herrmann, M. Elison, J. Fischer, C. Kocher and G. R. J. Artus, Chem.–Eur. J., 1996, 2, 772–780 CrossRef CAS
- W. A. Herrmann and C. Kocher, Angew. Chem., Int. Ed., 1997, 36, 2162–2187 CrossRef CAS
- N. Marion, S. Diez-Gonzalez and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS PubMed
- N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776–1782 RSC
- G. Wang, A. Ruhling, S. Amirjalayer, M. Knor, J. B. Ernst, C. Richter, H. J. Gao, A. Timmer, H. Y. Gao, N. L. Doltsinis, F. Glorius and H. Fuchs, Nat. Chem., 2017, 9, 152–156 CrossRef CAS PubMed
- E. A. Doud, M. S. Inkpen, G. Lovat, E. Montes, D. W. Paley, M. L. Steigerwald, H. Vázquez, L. Venkataraman and X. Roy, J. Am. Chem. Soc., 2018, 140, 8944–8949 CrossRef CAS PubMed
- G. Foti and H. Vazquez, Nanotechnology, 2016, 27, 125702 CrossRef PubMed
- E. C. Hurst, K. Wilson, I. J. S. Fairlamb and V. Chechik, New J. Chem., 2009, 33, 1837–1840 RSC
- C. Richter, K. Schaepe, F. Glorius and B. J. Ravoo, Chem. Commun., 2014, 50, 3204–3207 RSC
- Q. Tang and D. E. Jiang, Chem. Mater., 2017, 29, 6908–6915 CrossRef CAS
- A. V. Zhukhovitskiy, M. J. MacLeod and J. A. Johnson, Chem. Rev., 2015, 115, 11503–11532 CrossRef CAS PubMed
- C. R. Larrea, C. J. Baddeley, M. R. Narouz, N. J. Mosey, J. H. Horton and C. M. Crudden, ChemPhysChem, 2017, 18, 3536–3539 CrossRef CAS PubMed
- L. Jiang, B. Zhang, G. Medard, A. P. Seitsonen, F. Haag, F. Allegretti, J. Reichert, B. Kuster, J. V. Barth and A. C. Papageorgiou, Chem. Sci., 2017, 8, 8301 RSC
- A. Cossaro, R. Mazzarello, R. Rousseau, L. Casalis, A. Verdini, A. Kohlmeyer, L. Floreano, S. Scandolo, A. Morgante, M. L. Klein and G. Scoles, Science, 2008, 321, 943–946 CrossRef CAS PubMed
- A. Bakker, A. Timmer, E. Kolodzeiski, M. Freitag, H. Y. Gao, H. Monig, S. Amirjalayer, F. Glorius and H. Fuchs, J. Am. Chem. Soc., 2018, 140, 11889–11892 CrossRef CAS PubMed
-
J. Stohr, NEXAFS Spectroscopy, Heidelberg, 1992 Search PubMed
- S. M. Wetterer, D. J. Lavrich, T. Cummings, S. L. Bernasek and G. Scoles, J. Phys. Chem. B, 1998, 102, 9266–9275 CrossRef CAS
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, J. Phys.: Condens. Matter, 2009, 21, 084203 CrossRef PubMed
- K. Berland, V. R. Cooper, K. Lee, E. Schröder, T. Thonhauser, P. Hyldgaard and B. I. Lundqvist, Rep. Prog. Phys., 2015, 78, 119601 CrossRef PubMed
- D. C. Langreth, B. I. Lundqvist, S. D. Chakarova-Kack, V. R. Cooper, M. Dion, P. Hyldgaard, A. Kelkkanen, J. Kleis, L. Z. Kong, S. Li, P. G. Moses, E. Murray, A. Puzder, H. Rydberg, E. Schroder and T. Thonhauser, J. Phys.: Condens. Matter, 2009, 21, 134203 CrossRef PubMed
- A. Cossaro, L. Floreano, A. Verdini, L. Casalis and A. Morgante, Phys. Rev. Lett., 2009, 103, 119601 CrossRef PubMed
- C. Goransson, W. Olovsson and I. A. Abrikosov, Phys. Rev. B, 2005, 72, 134203 CrossRef
- J. F. Janak, Phys. Rev. B, 1978, 18, 7165–7168 CrossRef CAS
- G. Kresse and J. Furthmuller, Phys. Rev. B, 1996, 54, 11169 CrossRef CAS
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details for the NHC precursors. Additional details regarding the preparation of the samples, high-resolution XPS measurements, NEXAFS measurements, theoretical calculations, as well as any additional experimental and theoretical data. See DOI: 10.1039/c8sc03502d |
| This journal is © The Royal Society of Chemistry 2019 |