
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterologous biosynthesis of elsinochrome A sheds light on the formation of the photosensitive perylenequinone system†
Jinyu
Hu
 a,
Farzaneh
Sarrami
a,
Hang
Li
a,
Guozhi
Zhang
a,
Keith A.
Stubbs
a,
Ernest
Lacey
bc,
Scott G.
Stewart
a,
Amir
Karton
a,
Andrew M.
Piggott
c and
Yit-Heng
Chooi
*a
a,
Farzaneh
Sarrami
a,
Hang
Li
a,
Guozhi
Zhang
a,
Keith A.
Stubbs
a,
Ernest
Lacey
bc,
Scott G.
Stewart
a,
Amir
Karton
a,
Andrew M.
Piggott
c and
Yit-Heng
Chooi
*a
aSchool of Molecular Sciences, University of Western Australia, Perth, WA 6009, Australia. E-mail: yitheng.chooi@uwa.edu.au
bMicrobial Screening Technologies, Smithfield, NSW 2164, Australia
cDepartment of Molecular Sciences, Macquarie University, Sydney, NSW 2109, Australia
First published on 22nd November 2018
Abstract
Perylenequinones are a class of aromatic polyketides characterised by a highly conjugated pentacyclic core, which confers them with potent light-induced bioactivities and unique photophysical properties. Despite the biosynthetic gene clusters for the perylenequinones elsinochrome A (1), cercosporin (4) and hypocrellin A (6) being recently identified, key biosynthetic aspects remain elusive. Here, we first expressed the intact elc gene cluster encoding 1 from the wheat pathogen Parastagonospora nodorum heterologously in Aspergillus nidulans on a yeast-fungal artificial chromosome (YFAC). This led to the identification of a novel flavin-dependent monooxygenase, ElcH, responsible for oxidative enolate coupling of a perylenequinone intermediate to the hexacyclic dihydrobenzo(ghi)perylenequinone in 1. In the absence of ElcH, the perylenequione intermediate formed a hexacyclic cyclohepta(ghi)perylenequinone system via an intramolecular aldol reaction resulting in 6 and a novel hypocrellin 12 with opposite helicity to 1. Theoretical calculations supported that 6 and 12 resulted from atropisomerisation upon formation of the 7-membered ring. Using a bottom-up pathway reconstruction approach on a tripartite YFAC system developed in this study, we uncovered that both a berberine bridge enzyme-like oxidase ElcE and a laccase-like multicopper oxidase ElcG are involved in the double coupling of two naphthol intermediates to form the perylenequinone core. Gene swapping with the homologs from the biosynthetic pathway of 4 showed that cognate pairing of the two classes of oxidases is required for the formation of the perylenequinone core, suggesting the involvement of protein–protein interactions.
Introduction
Perylenequinones are a class of polyketide-derived photosensitisers and dark-coloured pigments that share a pentacyclic 4,9-dihydroxy-3,10-perylenequinone core (Fig. 1). The fungal perylenequinones, known as the Class B perylenequinones, are characterised by the stereogenic C7,C7′ side chains as well as the oxidative substitutions at C2,C2′ and C6,C6′.1 The substitutions on the perylenequinone core render the structures non-planar, resulting in axial chirality classified as either (M) or (P) (Fig. 1). These compounds can absorb light to generate reactive oxygen species (ROS) and possess a wide range of bioactivities, most notably their protein kinase C inhibitory activity, and they have potential application in photodynamic cancer therapy.1–3 Well-known members of this class of compounds include elsinochromes 1–3, cercosporin 4, hypocrellins (e.g.5–6) and calphostins (Fig. 1). Due to their photochemical properties, cytotoxic activities, interesting axial chirality, and their involvement in fungal virulence, perylenequinones have attracted much interest from both chemists and biologists.3,4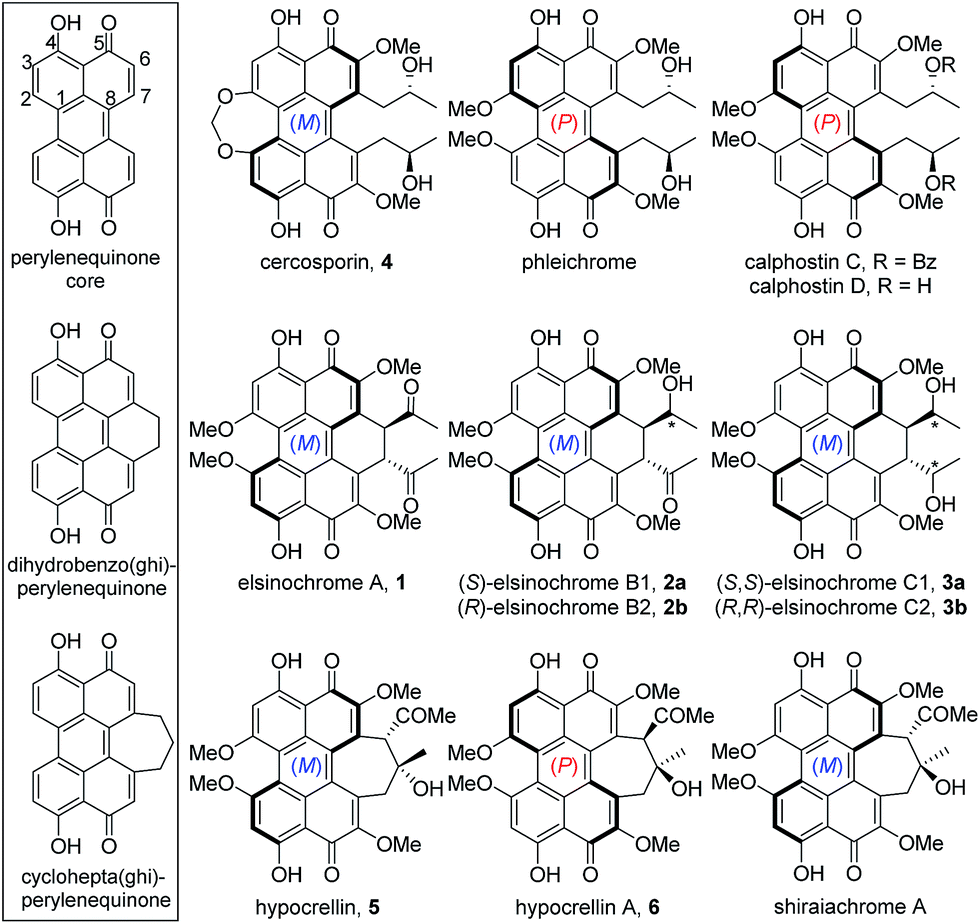 | ||
| Fig. 1 Class B perylenequinones grouped into three subclasses based on the core structures (box). | ||
To date, the biosynthetic gene clusters (BGCs) responsible for the biosynthesis of the three Class B perylenequinones, 4, 6 and 3, have been identified (Fig. 2A). Cercosporin 4 was the first perylenequinone to be isolated.5 It has a typical pentacyclic perylenequinone core, but is distinguished from other members of the class by the presence of an asymmetric 1,3-dioxepine moiety. The CTB cluster for biosynthesis of 4 from Cercospora nicotianae was also the first to be identified and several studies of the genes have been reported (Fig. 2B).6,7 Hypocrellins 5–6 and related analogues are known to be present in traditional Chinese medicine preparations derived from Hypocrella bambusae and Shiraia bambusicola parasitising on bamboo, which have been used to treat vitiligo, psoriasis, rheumatoid arthritis and other diseases.8 The hypocrellins are characterised by the hexacyclic cyclohepta(ghi)perylenequinone core (Fig. 1). The HYP cluster for 6 was identified in Shiraia sp. Slf14 based on gene expression correlation,9 and subsequently verified by targeted gene deletion.10 Elsinochromes 1–3 are characterised by the hexacyclic dihydrobenzo(ghi)perylenequinone core (Fig. 1). Our group recently identified the elc cluster from the wheat pathogen Parastagonospora nodorum for 3, which has been shown to be important for its virulence.11
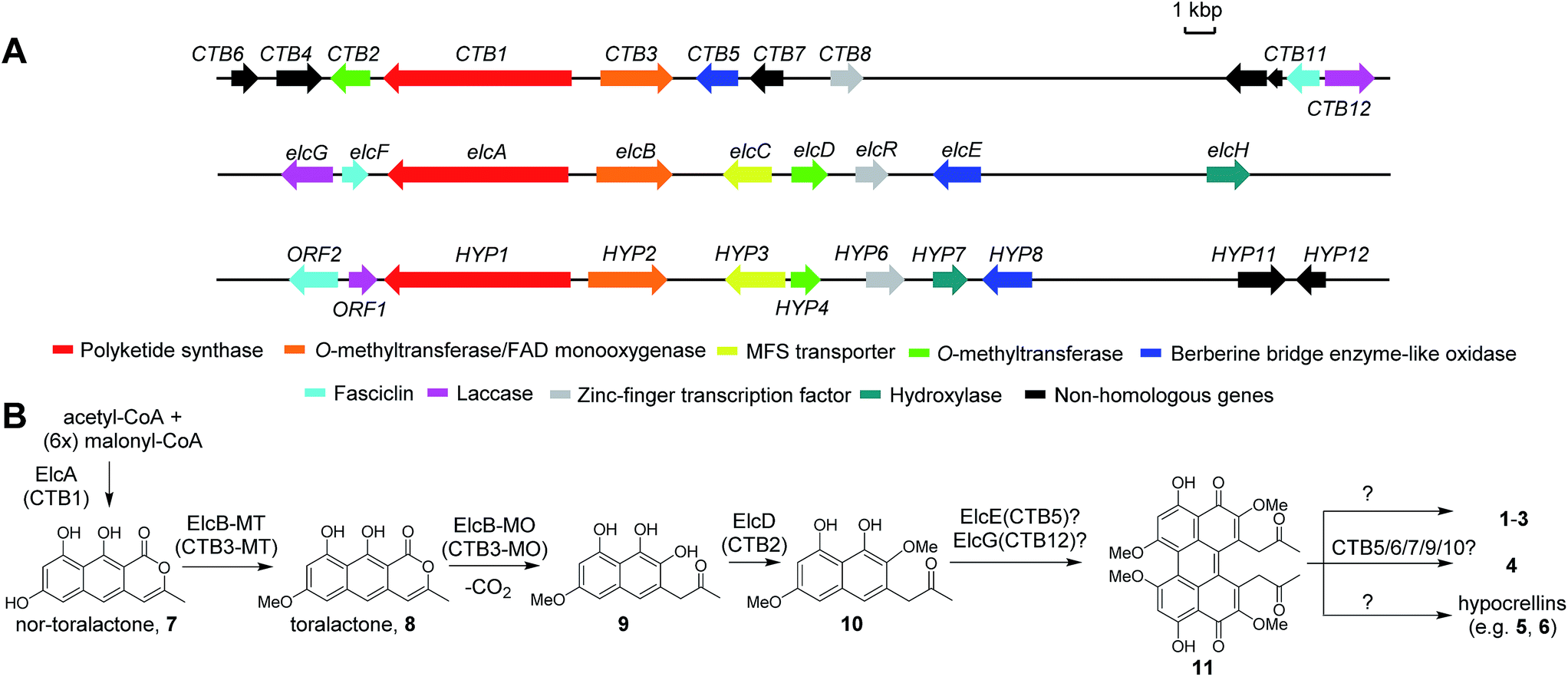 | ||
| Fig. 2 The biosynthetic gene clusters for 4 (CTB), 1 (elc) and 6 (HYP) (A) and their proposed biosynthetic pathway (B). | ||
The biosynthesis of perylenequinone has been shown to start from a common aromatic polyketide precursor, nor-toralactone (7), which is synthesised by the non-reducing polyketide synthase (PKS) CTB1/ElcA (Fig. 2B).11,12 More insights into the biosynthesis of perylenequinones have been gained recently with the characterisation of a bifunctional enzyme CTB3, which possesses fused O-methyltransferase and flavin-dependent monooxygenase domains, from the C. nicotianae CTB cluster. This enzyme is responsible for hydroxylation, ring opening, decarboxylation and O-methylation of the nor-toralactone polyketide precursor synthesised by CTB1 PKS (Fig. 2B).7 Nevertheless, the key steps in perylenequinone biosynthesis, i.e. the oxidative coupling steps to generate the pentacyclic core, the enolate coupling to yield the hexacyclic system in 1–3, and the 7-membered ring in 5–6, remained unresolved.
The elc, CTB and HYP gene clusters contain multiple shared homologs (Fig. 2A and Table S4†).11,13 The most recent study reported that the CTB cluster,13 like the elc and HYP clusters, encodes homologs of ElcF (fasciclin domain-containing protein) and ElcG (laccase-like multicopper oxidase (LMCO)), which were found in the HYP cluster but were missing in the originally defined CTB cluster (Fig. 2A).6 We hypothesised that the shared homologs in the three clusters encode common biosynthetic steps and are responsible for the pentacyclic perylenequinone core and the common substitutions (Fig. 2B). Herein, we shed light on the biosynthesis of Class B perylenequinones by reconstituting the pathway of 1 in Aspergillus nidulans. We demonstrate that formation of the three C–C bridges between the two naphthol monomers for 1 involves three distinct classes of oxidative enzymes.
Results
Heterologous expression of putative elc cluster produced two hypocrellins
To determine whether the putative elc cluster has all the genes required to synthesise 1–3, we first cloned the entire 27 kbp elc cluster onto a yeast-fungal hybrid artificial chromosome (YFAC) system, pYFAC-CH1, containing CEN/ARS and AMA1 for DNA replication in Saccharomyces cerevisiae and A. nidulans derived from pKW20088 vector backbone.14 The 27-kbp fragment containing the entire putative elc cluster (elcA-G and elcR) was cloned into pYFAC-CH1, resulting in pYFAC-CH6 (Table S3†). An engineered A. nidulans strain LO7890 with low metabolic background was used as the heterologous host.15 Expression of the elc cluster was achieved by replacing the native promoter of the pathway-specific transcription factor gene, elcR,11 with a PalcA alcohol-inducible promoter. LC-DAD-MS analysis of the acetone mycelium extract from a four-day post-induction culture showed that A. nidulans harbouring pYFAC-CH6 produced two new compounds, both of which had a mass ion of m/z 547 [M + H]+ (2 Da less in mass compared to 3) and near-identical UV-vis spectra to elsinochromes 1–3 (Fig. 3A). LC-DAD-MS comparison with standards of 1–3 showed that their retention times (tR) were different from 2 but close to 1 (Fig. S2†). The two compounds were isolated from scaled-up A. nidulans liquid culture (see ESI†). Extensive 1D and 2D NMR analysis of the compound 12 eluting earlier at 7.8 min (Fig. 3A) confirmed its structure to have the same atom connectivity as 5/6 with a cyclohepta(ghi)perylenequinone core (Table S5, Fig. S14–S18†). This prompted us to compare the two compounds with a standard of hypocrellin A 6. LC-DAD-MS analysis showed that the compound eluting later at 7.95 min had the same tR as 6 (Fig. 3A). The 1H NMR data along with the ECD spectrum supported that the compound is identical to 6, which has the (P) helicity (Table S7, Fig. S3, S20 and S21†).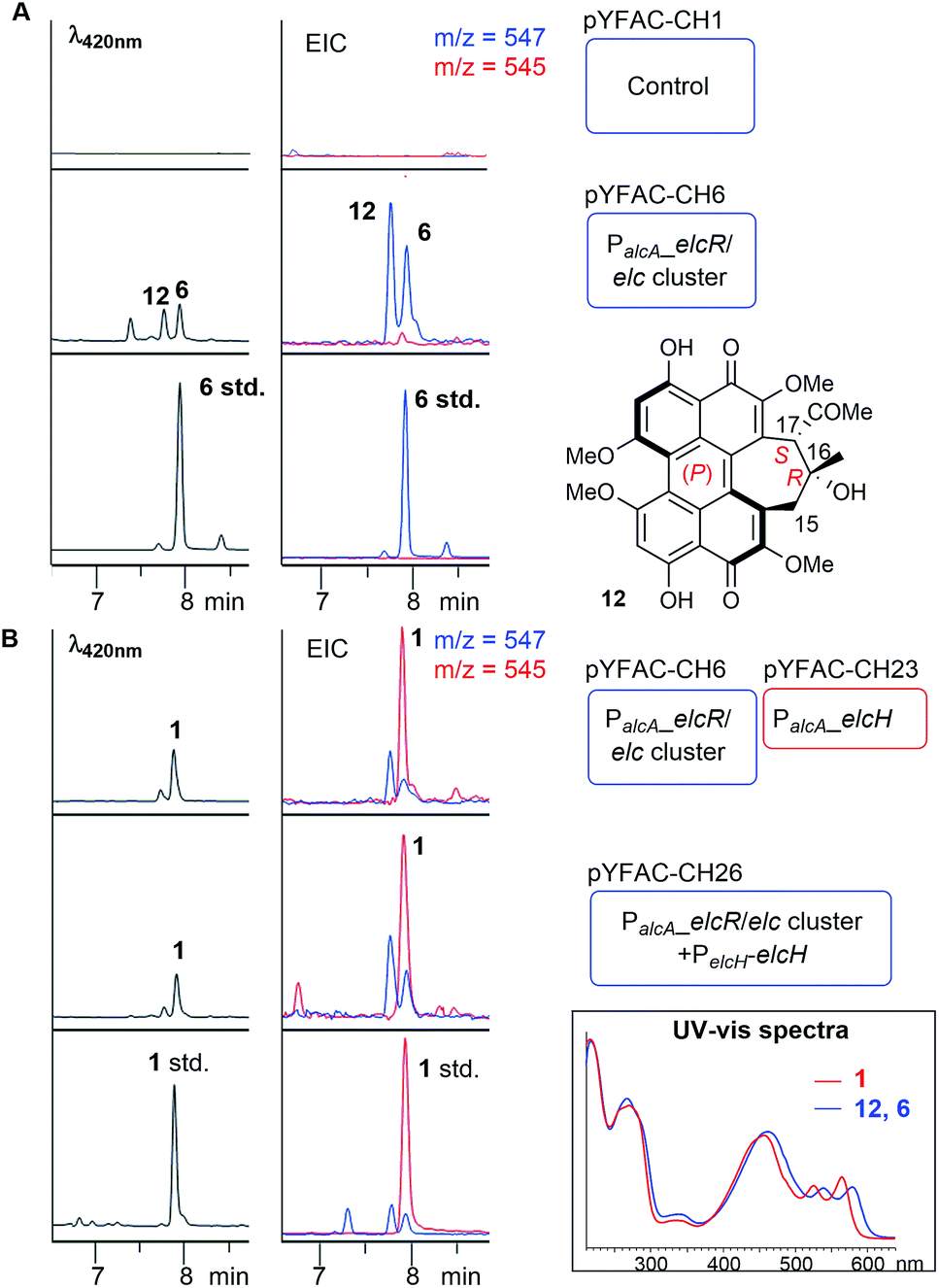 | ||
| Fig. 3 Heterologous biosynthesis of 12, 6 (A) and 1 (B). LC-DAD chromatograms and extracted ion chromatograms (EIC) of A. nidulans cultures harbouring the different YFAC constructs. Blue and red rounded rectangles represent YFAC vector backbones pYFAC-CH1 and -CH2, respectively. (Box) UV-vis spectra comparison of 12, 6 and 1. | ||
Compound 12 eluted slightly earlier than 6 on LC-DAD-MS (Fig. 3A). The minor 1H NMR chemical shift differences for the signals around the C16–C17 stereogenic centres suggested that 12 is a diastereomer of 6. We performed further 1D NOESY experiments on 12 to deduce its relative configuration (Table S6, Fig. S4 and S19†). Key NOESY correlations narrowed down the structure of 12 to a pair of enantiomers – either 16R and 17S for the (P) helicity or 16S and 17R for the (M) helicity (Fig. S4†). ECD spectroscopic analysis showed that 12 shared almost identical Cotton effects with 6 between 240 nm and 600 nm, except at around 295 nm, which had an opposite Cotton effect to that of 6 (Fig. S3†). Cotton effects at the 250–600 nm region have been associated with the helicity of perylenequinones.16 This suggested that 12, like 6, adopts the (P) helicity, but with the opposite configuration (16R and 17S) on the 7-membered ring compared to 6, and is thus an atropisomer of 5 (Fig. 3A). The elucidation of 12 and 6 as members of hypocrellins verified that the putative elc cluster contains all the genes required for the construction of the perylenequinone core but not 1–3, suggesting that there may be still genes missing in the elc cluster on the pYFAC-CH6.
A previously unidentified flavin-dependent monooxygenase gene elcH encodes elsinochrome synthase
Reviewing previous in planta transcriptomic data of P. nodorum17,18 revealed that another gene, SNOG_08601 encoding a putative flavin-dependent monooxygenase (FMO), located approximately 8 kbp upstream of the elc cluster was co-upregulated with other genes in the cluster (Fig. S5†). Interestingly, a homolog of SNOG_08601, HYP7, can be found in the HYP cluster (Fig. 2A and Table S4†). To determine the role of SNOG_08601 (herein named elcH) in the biosynthesis of 1–3, pYFAC-CH23 carrying only elcH under PalcA regulation was constructed and co-expressed with pYFAC-CH6 (containing P. nodorum elc cluster with PalcA:elcR). LC-DAD-MS analysis of the four-day culture extract detected a new peak with m/z 545 [M + H]+ and a small amount of 12 and 6 (Fig. 3). The new compound matched the tR and ion mass of the standard for 1. 1H, 13C, and 2D NMR analysis of the new compound confirmed its structure to be 1 (Table S8, Fig. S22–S25†). As expected, the ECD spectrum of 1 was identical to that of the 1 standard and nearly identical to that of 3 isolated from P. nodorum (Fig. S3†), thus confirming its (M) helicity. In a parallel experiment, the elcH gene with its native P. nodorum promoter region was inserted into pYFAC-CH6, resulting in pYFAC-CH26. Compound 1 could be detected as the major product in the post-induction culture of A. nidulans pYFAC-CH27, along with trace amounts of 12 and 6. This result indicates that elcH is indeed co-regulated with other elc genes by the pathway-specific transcriptional regulator ElcR.Heterologous expression of elcA/B/D/E/F/G reconstituted the biosynthesis of 12 and 6 and yielded novel naphthol derivatives
To elucidate the biosynthesis, we initially developed an in vivo YFAC editing technique in yeast based on the available yeast CRISPR-Cas9 system (Fig. S6†).19 Homology modelling by Phyre2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 showed that ElcE is a flavo-oxidase belonging to the berberine bridge enzyme-like oxidase (BBEO) family (Fig. S7†), which are known to catalyse C–C bond formation.21 On the other hand, elcG encodes a LMCO known to catalyse radical coupling reactions.21 Thus, we proposed elcE and elcG to be involved in the dimerisation of the naphthol 10 and targeted the two genes. We successfully disrupted elcE by CRISPR-Cas9 editing on the pYFAC-CH6 (pYFAC-CH9) and constructed a truncated version of pYFAC-CH6 without elcG (pYFAC-CH7) (Table S3†). LC-DAD-MS analysis of A. nidulans culture extracts for both mutant constructs showed that the production of 12 and 6 was completely abolished (Fig. S8†), suggesting that elcE and elcG are important for the biosynthesis of perylenequinones. However, there was no pathway intermediate accumulated. Similar results were observed when elcG was deleted in the P. nodorum elcR-OE strain producing 3 in the previous work (data not shown)11 and in the most recent study where the elcG homolog in the 4 pathway, CTB12, was deleted in C. beticola.13
20 showed that ElcE is a flavo-oxidase belonging to the berberine bridge enzyme-like oxidase (BBEO) family (Fig. S7†), which are known to catalyse C–C bond formation.21 On the other hand, elcG encodes a LMCO known to catalyse radical coupling reactions.21 Thus, we proposed elcE and elcG to be involved in the dimerisation of the naphthol 10 and targeted the two genes. We successfully disrupted elcE by CRISPR-Cas9 editing on the pYFAC-CH6 (pYFAC-CH9) and constructed a truncated version of pYFAC-CH6 without elcG (pYFAC-CH7) (Table S3†). LC-DAD-MS analysis of A. nidulans culture extracts for both mutant constructs showed that the production of 12 and 6 was completely abolished (Fig. S8†), suggesting that elcE and elcG are important for the biosynthesis of perylenequinones. However, there was no pathway intermediate accumulated. Similar results were observed when elcG was deleted in the P. nodorum elcR-OE strain producing 3 in the previous work (data not shown)11 and in the most recent study where the elcG homolog in the 4 pathway, CTB12, was deleted in C. beticola.13
Since no further biosynthetic insight could be gained from the gene disruption approach, we took an alternative bottom-up approach reconstructing the elc cluster by cloning each gene in the elc cluster under the regulation of A. nidulans alcohol-inducible promoters22 on a tripartite YFAC system. pYFAC-CH2 with a pyrG marker was constructed to contain a PalcA promoter, a bidirectional PalcS/M promoter and a PaldA along with terminators in between (Fig. S1†). The system was expanded to two other auxotrophic markers, riboB (pYFAC-CH3) and pyroA (pYFAC-CH4), respectively. Together, the tripartite YFAC system is capable of inducible expression of up to 12 genes and has been used recently for genome mining of novel phytotoxic α-pyrones from P. nodorum.23
An elcA expression plasmid pYFAC-CH10, which produced 7 in A. nidulans, was already constructed in previous work.11 Here, a new peak with m/z 273 [M + H]+ was detected in the A. nidulans pYFAC-CH10/14 culture co-expressing elcA and elcB-MT (truncated elcB encoding only the O-methyltransferase domain), which was isolated and confirmed by NMR to be toralactone 8 (Fig. 4A-i and S26†). This is in agreement with the previous in vitro work on CTB3-MT (ElcB homolog).7 Co-expression of intact elcB with elcA resulted in complete conversion of 8 to three new compounds, 13–15, in the culture (Fig. 4A-ii). Compound 15 was found to have an m/z of 277 [M + H]+, which has and an identical mass to cercoquinone A and D observed previously.7 However, due to poor stability and yield, we were unable to obtain sufficient quantities of 15 for NMR analysis.
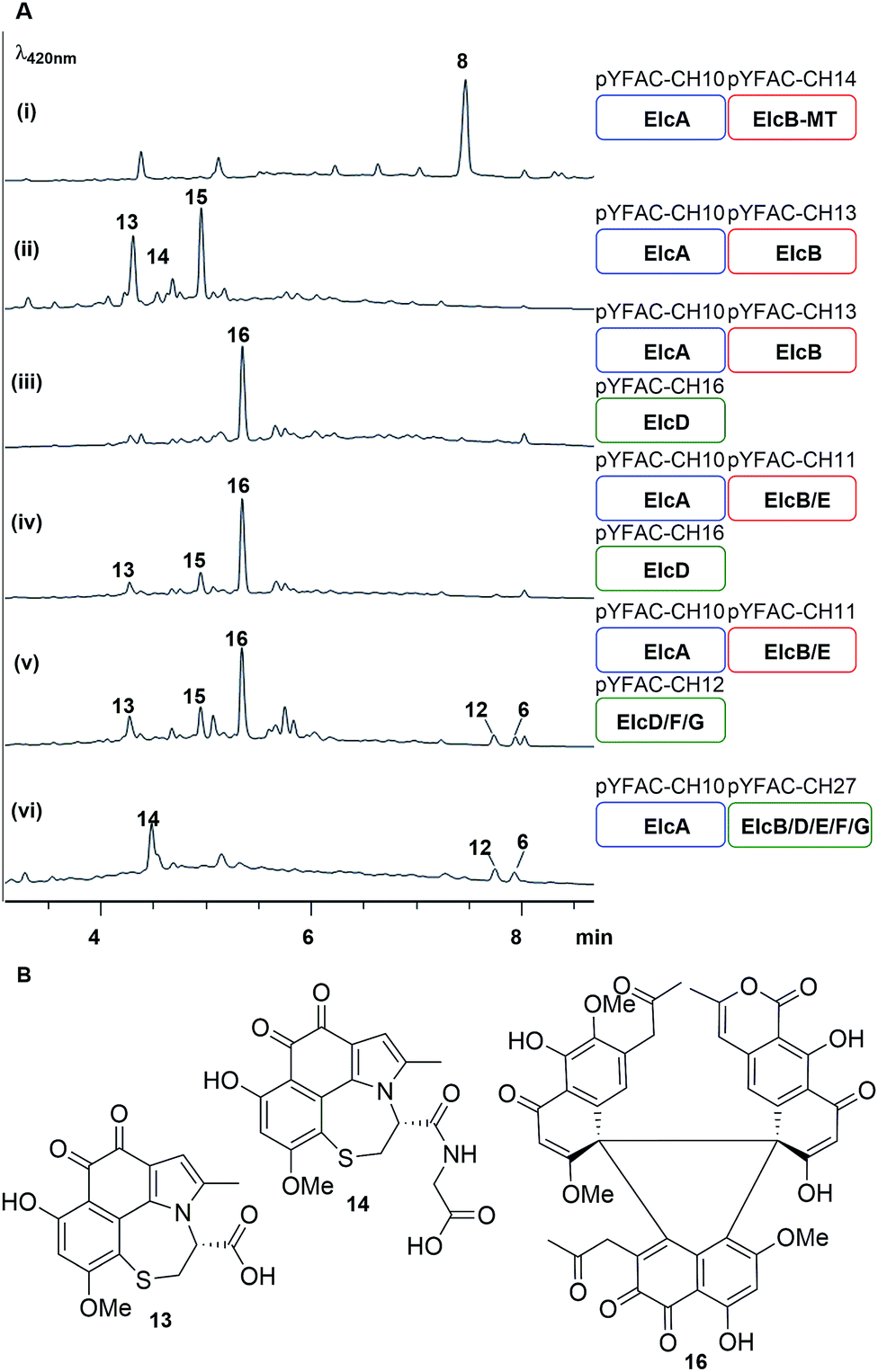 | ||
| Fig. 4 Bottom-up heterologous reconstruction of pathway to the hypocrellins 12 and 6. (A) LC-DAD chromatograms of A. nidulans strain harbouring the various constructs (i–vi) (also see Fig. S10†). Blue, red and green rounded rectangles represent the different YFAC vector backbone pYFAC-CH1-3, respectively. (B) Compounds isolated from these A. nidulans cultures. | ||
Compounds 13 and 14 possessed identical UV-vis spectra and their mass ions corresponded to m/z of 360 [M + H]+ and 417 [M + H]+, respectively. The two compounds were isolated as water-soluble purple pigments. The observed high-resolution mass ions for 13 were 360.0525 [M + H]+/314.0501 [M − H − CO2]−, which corresponds to the molecular formula C17H13O6NS (calculated m/z 360.0536 [M + H]+/314.0501 [M − H − CO2]−) (Fig. S9†). The high resolution mass for 14 was measured as m/z 417.0742 [M + H]+/415.0623 [M − H]−, which is consistent with C19H16O7N2S (calculated m/z 417.0751 [M + H]+/415.0605 [M − H]−) (Fig. S9†). Based on extensive analysis of 1D and 2D NMR data for 13 in D2O (Table S9, Fig. S27–S31†) and 14 (Table S11, Fig. S32–S37†) in DMSO-d6, we identified the presence of a cysteine moiety bridging C1 and C8 of the naphthol unit in both 13 and 14, with 14 containing an additional glycine residue connected to the cysteine via an amide bond (Fig. 4B). The proposed structures of 13 and 14 were supported by density functional theory (DFT) calculations (B3LYP/def-TZVP) of the 13C NMR shielding tensors for 13, which showed an excellent correlation with all experimental 13C chemical shifts (Table S10†). We reasoned that 13 and 14 were derived from the proposed naphthol intermediate 9 as a result of the glutathione S-transferase (GST) detoxification pathway endogenous to the A. nidulans host.
Next, pYFAC-CH16 carrying elcD was co-expressed with pYFAC-CH10 and pYFAC-CH13. Interestingly, this combination of genes (elcA/B/D) resulted in the accumulation of one major new compound 16, which has an m/z of 789 [M + H]+, almost three times the mass of the expected dimethoxy-naphthol 10 (Fig. 4a-iii). The high-resolution mass ions of 16 were consistent with the molecular formula C43H32O15 (observed m/z 789.1788 [M + H]+/787.1685 [M − H]−, calculated 789.1814 [M + H]+/787.1668 [M − H]−) (Fig. S9†). The NMR data for 16 in DMSO-d6 and D2O revealed the presence of three distinct naphthol units (Table S12, Fig. S38–S50†), which were likely derived from 7, 9 and 10 (Fig. 4B). The NMR spectra also clearly showed that one of the three naphthol units was methylated at 6-OH (corresponding to 10), supporting the function of ElcD as an O-methyltransferase. Although the structure of 16 could not be unequivocally confirmed due to the presence of multiple contiguous non-protonated carbons, detailed analysis of the 1D and 2D NMR data in both D2O and DMSO-d6 allowed us to propose a tentative structure in which the three naphthol units are linked via a central 5-membered ring (Fig. 4B). A similar cyclic trimeric construct was reported for the fungal antibiotic PF1158A, which is comprised of three semivioxanthin monomers.24 The experimental 13C NMR shifts for 16 were also in close agreement with DFT-calculated shielding tensors for the proposed structure (Table S13†).
We next co-expressed elcE with elcA/B/D (pYFAC-CH10/11/16) to test its potential role in oxidative coupling as previously proposed. However, this resulted in no major change in metabolic profile compared to A. nidulans expressing elcA/B/D only (Fig. 4A-iv). The production of the perylenequinone-like 12 and 6 could only be reconstituted when later elcF and elcG were introduced together with elcA/B/D/E in A. nidulans (Fig. 4-v). In the absence of either elcE or elcG, there was no production of 12 and 6 (Fig. S10A†). On the other hand, the results from omitting elcF only varied between transformants and batches (Fig. S10B†). In some cases, we observed reduced production of 12 and 6, while in others 12 and 6 production was completely abolished. Addition of elcC encoding major facilitator superfamily transporter also had no major effects on the yield of 12 and 6 (Fig. S10A†).
Finally, we included all the genes elcA/B/D/E/F/G in two instead of three YFACs and this resulted in increased yield of 12 and 6 and reduced production of shunt intermediates (Fig. 4-vii). This suggested that the accumulation of more shunt intermediates when the genes are split into three YFACs could be due to the poor stability of the intermediates and the episomal nature of the YFACs. As was observed for the episomal expression system in yeast,25 some fungal cells may not contain all three YFACs.
Cognate pairing of ElcE/CTB5 and ElcG/Cz_CTB12 is required for perylenequinone formation
The accumulation of shunt products and the lack of an oxidative coupling intermediate with a single C–C bridge between two naphthol monomers observed in A. nidulans expression strains without either elcE or elcG suggested that ElcE and ElcG could work concurrently, perhaps in the form of a protein complex, to catalyse the two putative oxidative coupling steps. To investigate this possibility, we turned to the homologs of elcE and elcG from the biosynthetic pathway of 4, CTB5 and CTB12, respectively. As no genome sequence for C. nicotianae was available when the experiment was carried out, we switched to another plant pathogen of the same genus, Cercospora zeae-maydis, for which the genome sequence is available and has been reported to produce 4.26 A BLAST search using ElcF and ElcG against the C. zeae-maydis genome identified homologs of both proteins (Cerzm1/106448 and Cerzm1/43209, herein referred to as Cz_CTB11 and Cz_CTB12), which are located ∼15 kbp downstream of the corresponding CTB cluster (Fig. 2A). This is consistent with the most recent report of CTB11 and CTB12.13 We synthesised the two genes based on the C. nicotianae CTB5 and C. zeae-maydis Cz_CTB12 sequences and introduced them into the YFAC expression system. pYFAC-CH25 was constructed by replacing elcE in pYFAC-CH11 with CTB5, and pYFAC-CH26 was constructed by replacing elcG in pYFAC-CH12 with Cz_CTB12. The constructs above were used in different combinations to test if the elc homologs could be complemented by the CTB counterpart. The results showed that when either Cz_CTB12 or CTB5 was used to replace their elc counterpart individually, production of 12 and 6 was abolished. However, when they were both introduced together replacing elcE and elcG, two new peaks 19 and 20, both with a perylenequinone-like UV-vis spectrum and m/z of 533 [M + H]+, were detected, along with trace amount of 12 and 6 (Fig. 5A). The 19 and 20 pair is reminiscent of the 12 and 6 stereoisomers. Due to low yield, the 19 and 20 structures were not fully characterised. However, 1H-NMR spectra of both 19 and 20 and the HMBC correlations suggested that they have the same cyclohepta(ghi)perylenequinone framework as 12 and 6, but lack the methyl group on 6-OH (Table S14, Fig. S51–S58†). HR-MS of 19 and 20 revealed ions with m/z 533.1410 [M + H]+/531.1292 [M − H]− and 533.1414 [M + H]+/531.1290 [M − H]−, respectively. The predicted formula C29H24O10 (calculated m/z 533.1414 [M + H]+/531.1290 [M − H]−) (Fig. S9†) also corresponds to the loss of one methyl group. Collectively, the results showed that cognate pairing of ElcE/CTB5 and ElcG/Cz_CTB12 is required for the double coupling, supporting the hypothesis for protein–protein interaction between the two different classes of oxidases.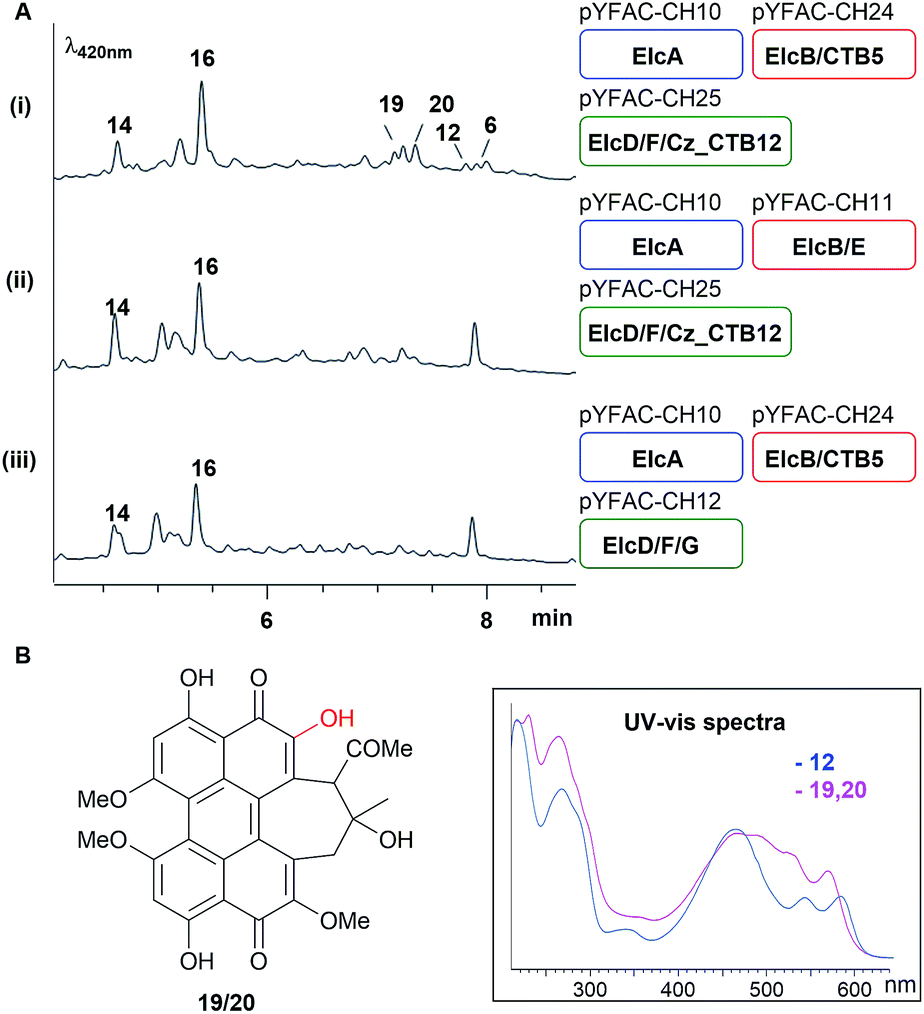 | ||
| Fig. 5 Homologous gene swapping (elcE with CTB5, elcG with Cz_CTB12 or both together) revealed the requirement for cognate pairing in perylenequinone production. (A) LC-DAD chromatograms of A. nidulans culture with YFAC constructs with different gene combination and (B) the structure and UV-vis spectra of 19/20. | ||
Discussion
The oxidative double coupling step, which forges the two C–C bridges between two highly functionalised naphthol units to complete the perylenequinone core that confers photochemical properties, has proven to be challenging to untangle. As in previous studies,7,13 this current work involving gene deletion of candidate oxidase genes resulted in no tractable intermediate. Thus, we adopted both top-down (intact whole gene cluster) and bottom-up (gene-by-gene pathway reconstruction) heterologous reconstitution approaches to shed light onto perylenequinone biosynthesis. The modular nature of the tripartite YFAC Aspergillus expression system allowed the efficient construction of A. nidulans strains carrying YFACs with different gene combinations. In total, we constructed 22 A. nidulans strains, analysed the metabolite profiles and isolated three novel shunt intermediates plus two side hypocrellin products.In light of all the results, we propose a revised biosynthetic pathway (Fig. 6). As in previous studies,11,12 the first isolatable intermediate is the product of the PKS, nor-toralactone 7, common to all Class B perylenequinone pathways. The subsequent steps, methylation of 7 followed by an oxidative ring cleavage and decarboxylation by ElcB (CTB3 homolog) to form 9, are in agreement with the literature.7 Previous in vitro enzymatic experiments suggested that 9 is highly reactive and spontaneously oxidised to naphthoquinones.7 Likewise, we were unable to detect 9 when co-expressing elcA and intact elcB, however, the A. nidulans strain produced 13–15. Compound 15 was hypothesised to be one of the cercoquinones, while 13 and 14 are both novel compounds containing an unusual fusion between a 1,2-naphthoquinone, a 2-methylpyrrole and thiazepane. The presence of an additional glycyl moiety in 14 suggests that 13 and 14 are likely derived from a glutathione adduct of 9 formed via the common GST detoxification pathway in eukaryotes. An example of secondary metabolite thiolation mediated by GST is the gliotoxin pathway.27 We propose that O-methylation of 9 to 10 by ElcD is the next step in perylenequinone biosynthesis after ElcB. Even though we did not directly detect 10 in the A. nidulans expressing elcA/B/D, the strain produced a new heterotrimeric compound 16 as an unusual adduct of 7, 9, and 10 (Fig. 6), likely catalysed by endogenous oxidases in the host. The presence of a 6-OMe group on the naphthol unit corresponding to 10 on 16 demonstrates ElcD catalyse the 6-O-methylation as expected.
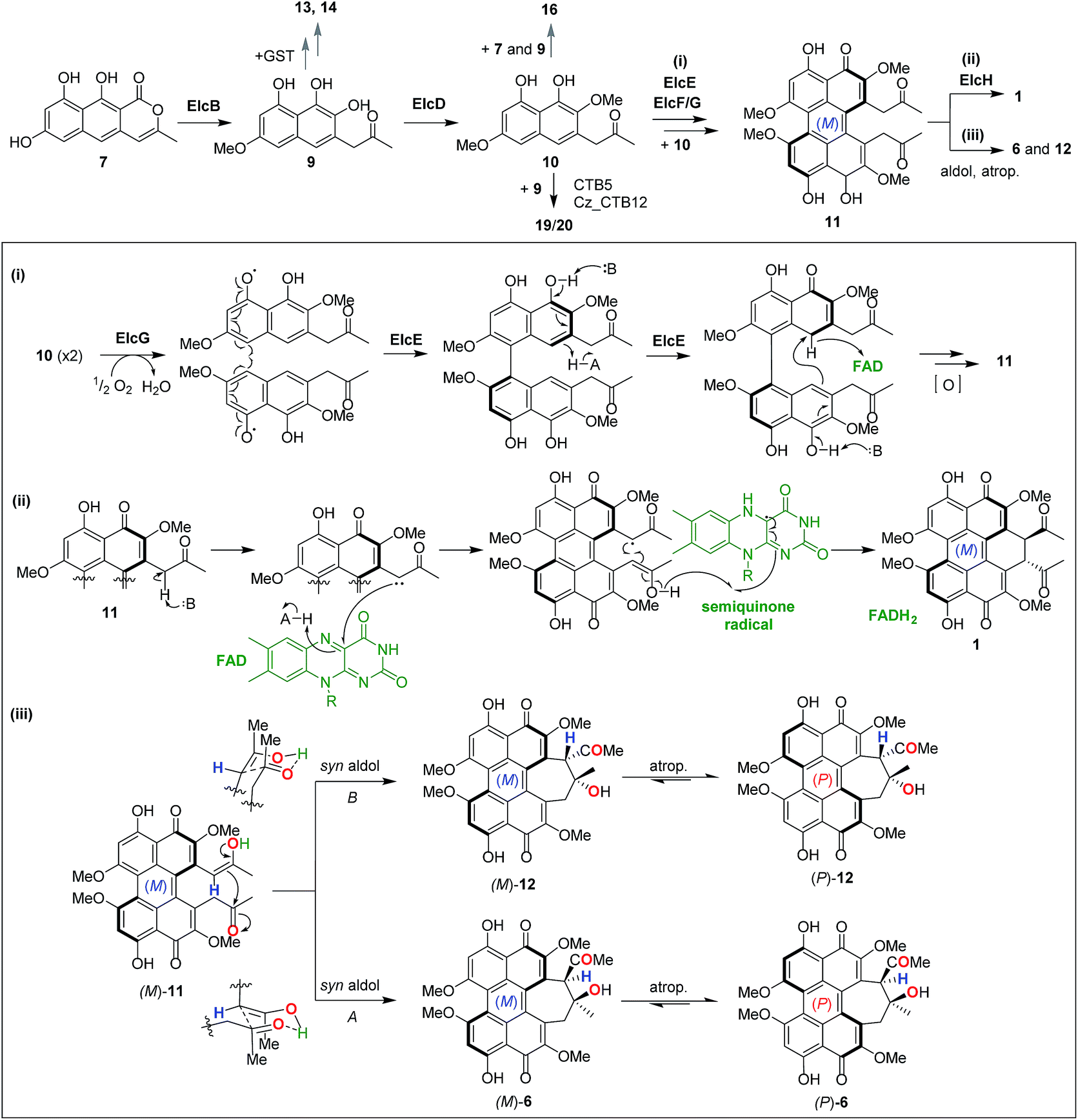 | ||
| Fig. 6 Revised biosynthetic pathway for dihydrobenzo(ghi)perylenequinone 1 and cyclohepta(ghi)perylenequinones 6 and 12. (Box) Proposed mechanisms for the naphthol double coupling to forge the pentacyclic perylenequinone core (i), the enolate coupling to form the dihydrobenzo(ghi)perylenequinone in 1 (ii), and the two syn aldol reactions followed by atropisomerisation that afford 6 and 12. | ||
Stereo- and regio-selective oxidative coupling of polycyclic aromatic compounds have been shown to be catalysed by P450s in fungi.28–30 More recently, a LMCO from the fungus Talaromyces pinophilus that could catalyse the regioselective coupling of naphthopyranone was discovered.31 The involvement of LMCOs in regioselective oxidative coupling of naphthopyrones have also been reported.32,33 The oxidative coupling of the two monomeric naphthol units in perylenequinone biosynthesis was originally proposed to be catalysed by the ElcE homolog, C. nicotianae CTB5, which encodes a BBEO.7 In the most recent study based on gene deletion results,13 it was proposed that the LMCO CTB12 (ElcG homolog) catalyses the oxidative dimerisation instead. Here, we demonstrate conclusively by heterologous biosynthesis that both BBEO and LMCO are required for the double coupling step of the putative intermediate 10 to afford the pentacyclic perylenequinone system in 11, which was converted to 12 and 6 in A. nidulans. By gene swapping with CTB homologs, we further demonstrated that cognate pairing of the BBEO and the LMCO is required for their catalytic function and that it could involve the formation of a protein complex. Interestingly, a new pair of hypocrellins 19 and 20 were observed in A. nidulans expressing CTB5/Cz_CTB12 in place of ElcE/ElcG. From limited 1H and 2D NMR data (due to poor yield), we deduced that 19 and 20 originated from coupling between 9 and 10, while 12 and 6 are dimers of 10. As there was no prior report of the equivalent of 19 and 20, it is possible that this was an artefact of CTB5 and Cz_CTB12 operating in ElcA/B/D/F background and suggests there may be other protein–protein interactions involved (e.g. ElcF), which requires further investigation (Fig. S11†).
Given that there are several precedents for LMCOs involved in intermolecular phenolic coupling,31–33 we propose that the ElcG LMCO catalyses the first intermolecular coupling of 10 in a regio- and stereo-selective manner via a phenol radical coupling mechanism (Fig. 6-i). On the other hand, the ElcE BBEO could forge the second C–C bond intramolecularly via a hydride transfer mechanism similar to other BBEOs then further oxidation to 11 (Fig. 6-i). However, there are no reports of LMCOs that can catalyse stereo-selective oxidate coupling to date. In plants, the regio- and stereo-selective coupling reactions by LMCOs have been demonstrated to be facilitated by a dirigent protein.34,35 It is unknown if protein–protein interactions between the ElcE and ElcG or the fasciclin protein ElcF could play a role in the regio- and stereo-selective coupling of perylenequinones. Although BBEOs are known to catalyse intramolecular C–C bond formation,21,36 their involvement in such intramolecular phenolic coupling is unprecedented. Nonetheless, future in vitro investigation of the enzymes with compatible synthetic substrates is required to resolve these questions.
The last piece of the puzzle in the biosynthesis of 1 is the additional annulation by enolate coupling to afford the dihydrobenzo(ghi)perylenequinone system, which we showed to involve the FMO ElcH. Although enolate coupling is widely exploited in synthesis, there is little if any precedent in biosynthesis so far. Addition of elcH to A. nidulans expressing the elc cluster (elcA/B/C/D/E/F/G) resulted in the production of 1 and loss of production of 12 and 6. We propose that 1 is formed through the putative perylenequinone intermediate 11via a radical mechanism initiated via a single electron transfer from an enolate at the side chain to the FAD in ElcH (Fig. 6-ii). In the absence of ElcH, the hypocrellins 12 and 6 were formed, suggesting that 12 and 6 are most likely derived from the putative intermediate 11via a transannular aldol reaction.
A dilemma emerged when we solved the configuration and helicity of 12 (atrop-5) and 6, which is opposite to 1 and 3 that adopt an (M) conformation. We propose that in the formation of the two (P) hypocrellins 12 and 6, (M)-11 is first converted to (M)-12 and (M)-6via the two possible transannular syn aldol reactions followed by spontaneous atropisomerisation to afford (P)-12 and (P)-6, respectively (Fig. 6-iii). The facile atropisomerisation of 6 has been reported in several studies;1,37,38 which demonstrated that the seven-membered ring in 6 reduced the stability of perylenequinone axial chiralities and allowed atropisomerisation at room temperature. By contrast, higher energy is required for atropisomerisation of pentacyclic perylenequinones, e.g. calphostin.3 Indeed, our DFT calculations supported this (Fig. S12†). We found that the conversion of (M)-11 to (M)-12 and (M)-6 is associated with Gibbs free reaction barrier heights of 11.8 and 74.8 kJ mol−1, respectively (Fig. S12-i†), which indicate that both pathways are kinetically accessible at the temperature the experiments were carried out (30 °C). The (P)-12 and (P)-6 products lie 11.2 and 19.7 kJ mol−1 below the energy of the (M)-11 reactant, respectively, indicating a thermodynamic driving force for the conversion of (M)-12 and (M)-6 to atropisomerism to the (P) form. We have also observed that leaving 12 and 6 in methanol for prolonged periods resulted in partial conversion of the two (P) hypocrellins into what appear to be their corresponding (M) atropisomers (Fig. S13†). Thus, ElcH essentially locks the perylenequinone in the (M) helical conformation by converting 11 to 1.
Nonetheless, we cannot rule out that the aldol reaction could involve endogenous A. nidulans enzymes, especially given that the energy barrier for (M)-11 to (M)-12 is higher than to (M)-6, and that (P)-12 has never been reported previously. The transannular aldol reaction followed by atropisomerisation is reminiscent of the dynamic stereochemical transfer approach employed by Kozlowski et al. in the total synthesis of (P)-6, which is derived from (P)-11 in their synthetic route instead of (M)-11.37 Interestingly, we did not isolate any shiraiachrome A or hypocrellin 5, which adopt the (M) helicity (Fig. 1); it is to be determined if the aldol reaction to these two compounds are enzymatically catalysed. A Shiraia sp. strain was previously reported to produce both 1 and 6, along with 2, 3 and hypocrellin B–C.39 It is possible that the (P)-hypocrellins in this Shiraia strain were derived from an (M) perylenequinone precursor 11 like we have observed here.
In summary, using a heterologous pathway reconstruction approach we demonstrated the involvement of two different classes of oxidases (LMCO and BBEO) in the synthesis of the pentacyclic core common to all perylenequinones. The isolation of different shunt products 13, 14 and 16 that trapped the highly reactive naphthol intermediates provided insights into the biosynthetic step and suggests that A. nidulans is an excellent strain for biotransformation to generate novel naphthol derivatives. Finally, the identification of the elsinochrome synthase ElcH and the isolation of hypocrellin side products 6 and 12 allowed us to establish the biosynthetic link between the three subclasses of perylenequinones (Fig. 1).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by an Australian Research Council (ARC) Discovery Project (DP170100228). Y-HC, AK and AMP are ARC Future Fellows (FT160100233, FT170100373, FT130100142). NMR and HR-MS analyses were performed at the UWA Centre for Microscopy, Characterisation and Analysis (CMCA). JH and HL are both recipients of Australian International Postgraduate Research Scholarship. We thank Prof. Berl Oakley for A. nidulans strain LO7890 and Prof. Kenji Watanabe for the pKW20088 plasmid.References
- C. A. Mulrooney, B. J. Morgan, X. Li and M. C. Kozlowski, J. Org. Chem., 2009, 75, 16–29 CrossRef PubMed.
- M. Olivo and W. W. Chin, J. Environ. Pathol., Toxicol. Oncol., 2006, 25, 223–237 CrossRef CAS.
- B. J. Morgan, S. Dey, S. W. Johnson and M. C. Kozlowski, J. Am. Chem. Soc., 2009, 131, 9413–9425 CrossRef CAS PubMed.
- M. E. Daub, S. Herrero and K.-R. Chung, Antioxid. Redox Signaling, 2013, 19, 970–989 CrossRef CAS PubMed.
- S. Kuyama and T. Tamura, J. Am. Chem. Soc., 1957, 79, 5726–5729 CrossRef CAS.
- H.-Q. Chen, M.-H. Lee and K.-R. Chung, Microbiology, 2007, 153, 2781–2790 CrossRef CAS PubMed.
- A. G. Newman and C. A. Townsend, J. Am. Chem. Soc., 2016, 138, 4219–4228 CrossRef CAS PubMed.
- C. Yu, T. Huang, Z. Ding, X. Gao and Z. Zhang, Encyclopedia of Chinese Medicines, Chinese Medicinal Science and Technology Press, Beijing, People's Republic of China, 1993 Search PubMed.
- H. Yang, Y. Wang, Z. Zhang, R. Yan and D. Zhu, Genome Announc., 2014, 2, e00011–00014 Search PubMed.
- H. Deng, R. Gao, X. Liao and Y. Cai, J. Biotechnol., 2017, 259, 228–234 CrossRef CAS PubMed.
- Y. H. Chooi, G. Zhang, J. Hu, M. J. Muria-Gonzalez, P. N. Tran, A. Pettitt, A. G. Maier, R. A. Barrow and P. S. Solomon, Environ. Microbiol., 2017, 19, 1975–1986 CrossRef CAS PubMed.
- A. G. Newman, A. L. Vagstad, K. Belecki, J. R. Scheerer and C. A. Townsend, Chem. Commun., 2012, 48, 11772–11774 RSC.
- R. de Jonge, M. K. Ebert, C. R. Huitt-Roehl, P. Pal, J. C. Suttle, R. E. Spanner, J. D. Neubauer, W. M. Jurick, K. A. Stott, G. A. Secor, B. P. Thomma, Y. Van de Peer, C. A. Townsend and M. D. Bolton, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5459–E5466 CrossRef PubMed.
- Y. Tsunematsu, N. Ishikawa, D. Wakana, Y. Goda, H. Noguchi, H. Moriya, K. Hotta and K. Watanabe, Nat. Chem. Biol., 2013, 9, 818 CrossRef CAS PubMed.
- Y. M. Chiang, M. Ahuja, C. E. Oakley, R. Entwistle, A. Asokan, C. Zutz, C. C. Wang and B. R. Oakley, Angew. Chem., 2016, 128, 1694–1697 CrossRef.
- H. Wu, X.-F. Lao, Q.-W. Wang, R.-R. Lu, C. Shen, F. Zhang, M. Liu and L. Jia, J. Nat. Prod., 1989, 52, 948–951 CrossRef CAS.
- Y.-H. Chooi and P. S. Solomon, Front. Microbiol., 2014, 5, 640 Search PubMed.
- S. V. Ipcho, J. K. Hane, E. A. Antoni, D. Ahren, B. Henrissat, T. L. Friesen, P. S. Solomon and R. P. Oliver, Mol. Plant Pathol., 2012, 13, 531–545 CrossRef CAS PubMed.
- J. E. DiCarlo, J. E. Norville, P. Mali, X. Rios, J. Aach and G. M. Church, Nucleic Acids Res., 2013, 41, 4336–4343 CrossRef CAS PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. Sternberg, Nat. Protoc., 2015, 10, 845 CrossRef CAS PubMed.
- M.-C. Tang, Y. Zou, K. Watanabe, C. T. Walsh and Y. Tang, Chem. Rev., 2016, 117, 5226–5333 CrossRef PubMed.
- S. Fillinger and B. Felenbok, Mol. Microbiol., 1996, 20, 475–488 CrossRef CAS PubMed.
- H. Li, J. Hu, H. Wei, P. S. Solomon, D. Vuong, E. Lacey, K. A. Stubbs, A. M. Piggott and Y.-H. Chooi, Org. Lett., 2018, 20, 6148–6152 CrossRef CAS PubMed.
- M. Hatsu, N. Takei, F. Someya, T. Yaguchi, M. Nagasawa, S. Ishii and Y. Kurata, Japan Pat., JP 08208677, 1996.
- N. B. Jensen, T. Strucko, K. R. Kildegaard, F. David, J. Maury, U. H. Mortensen, J. Forster, J. Nielsen and I. Borodina, FEMS Yeast Res., 2014, 14, 238–248 CrossRef CAS PubMed.
- W.-B. Shim and L. D. Dunkle, Physiol. Mol. Plant Pathol., 2002, 61, 237–248 CrossRef CAS.
- D. H. Scharf, P. Chankhamjon, K. Scherlach, T. Heinekamp, K. Willing, A. A. Brakhage and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 11092–11095 CrossRef CAS PubMed.
- A. Präg, B. r. A. Grüning, M. Häckh, S. Lüdeke, M. Wilde, A. Luzhetskyy, M. Richter, M. Luzhetska, S. Günther and M. Müller, J. Am. Chem. Soc., 2014, 136, 6195–6198 CrossRef PubMed.
- C. Gil Girol, K. M. Fisch, T. Heinekamp, S. Günther, W. Hüttel, J. Piel, A. A. Brakhage and M. Müller, Angew. Chem., Int. Ed., 2012, 51, 9788–9791 CrossRef CAS PubMed.
- S. Griffiths, C. H. Mesarich, B. Saccomanno, A. Vaisberg, P. J. De Wit, R. Cox and J. Collemare, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6851–6856 CrossRef CAS PubMed.
- M. Kawaguchi, T. Ohshiro, M. Toyoda, S. Ohte, J. Inokoshi, I. Fujii and H. Tomoda, Angew. Chem., 2018, 130, 5209–5213 CrossRef.
- W. Fang, S. Ji, N. Jiang, W. Wang, G. Y. Zhao, S. Zhang, H. M. Ge, Q. Xu, A. H. Zhang, Y. L. Zhang, Y. C. Song, J. Zhang and R. X. Tan, Nat. Commun., 2012, 3, 1039 CrossRef PubMed.
- R. J. Frandsen, C. Schütt, B. W. Lund, D. Staerk, J. Nielsen, S. Olsson and H. Giese, J. Biol. Chem., 2011, 286, 10419–10428 CrossRef CAS PubMed.
- L. B. Davin, H.-B. Wang, A. L. Crowell, D. L. Bedgar, D. M. Martin, S. Sarkanen and N. G. Lewis, Science, 1997, 275, 362–366 CrossRef CAS PubMed.
- T. Wezeman, S. Bräse and K.-S. Masters, Nat. Prod. Rep., 2015, 32, 6–28 RSC.
- J. A. Baccile, J. E. Spraker, H. H. Le, E. Brandenburger, C. Gomez, J. W. Bok, J. Macheleidt, A. A. Brakhage, D. Hoffmeister and N. P. Keller, Nat. Chem. Biol., 2016, 12, 419–424 CrossRef CAS PubMed.
- E. M. O'Brien, B. J. Morgan, C. A. Mulrooney, P. J. Carroll and M. C. Kozlowski, J. Org. Chem., 2009, 75, 57–68 CrossRef PubMed.
- A. Smirnov, D. B. Fulton, A. Andreotti and J. W. Petrich, J. Am. Chem. Soc., 1999, 121, 7979–7988 CrossRef CAS.
- Z. Tong, L. Mao, H. Liang, Z. Zhang, Y. Wang, R. Yan and D. Zhu, J. Liq. Chromatogr. Relat. Technol., 2017, 40, 536–540 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02870b |
| This journal is © The Royal Society of Chemistry 2019 |