
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron trifluoride etherate promoted microwave-assisted synthesis of antimalarial acridones†
Papireddy Kancharla *a,
Rozalia A. Dodeanab,
Yuexin Liab and
Jane X. Kelly*ab
*a,
Rozalia A. Dodeanab,
Yuexin Liab and
Jane X. Kelly*ab
aDepartment of Chemistry, Portland State University, Portland, Oregon 97201, USA. E-mail: papiredd@pdx.edu; kellyja@ohsu.edu; Tel: +1-503-725-2566
bDepartment of Veterans Affairs Medical Center, Portland, Oregon 97239, USA. Tel: +1-503-220-8262 ext. 54356
First published on 20th December 2019
Abstract
A microwave-assisted, rapid and efficient method using boron trifluoride etherate (BF3.Et2O) for the synthesis of acridones, via an intramolecular acylation of N-phenylanthranilic acid derivatives, has been developed. The reaction proceeds under solvent-free conditions, tolerates a wide range of functional groups, and provides rapid access to a range of acridones in good to excellent yields. Several of the synthesized acridones exhibited potent antimalarial activities against CQ sensitive and multi-drug resistant (MDR) parasites.
Introduction
Acridones are one of the important classes of heteroaromatic compounds and they have ubiquitous structural motifs in biologically active molecules. The natural and synthetic acridone products (Fig. 1) are of worldwide interest because of their wide range of biological properties, including antitumor, anticancer, antimalarial, anti-HIV, antiviral, and antifungal activities.1–13 The intriguing structural features and therapeutic importance of acridone scaffold has attracted the great attention of organic and medicinal chemists to discover new synthetic routes14–18 for the generation of novel acridones to be evaluated for biological activities and structure–activity relationships (SAR).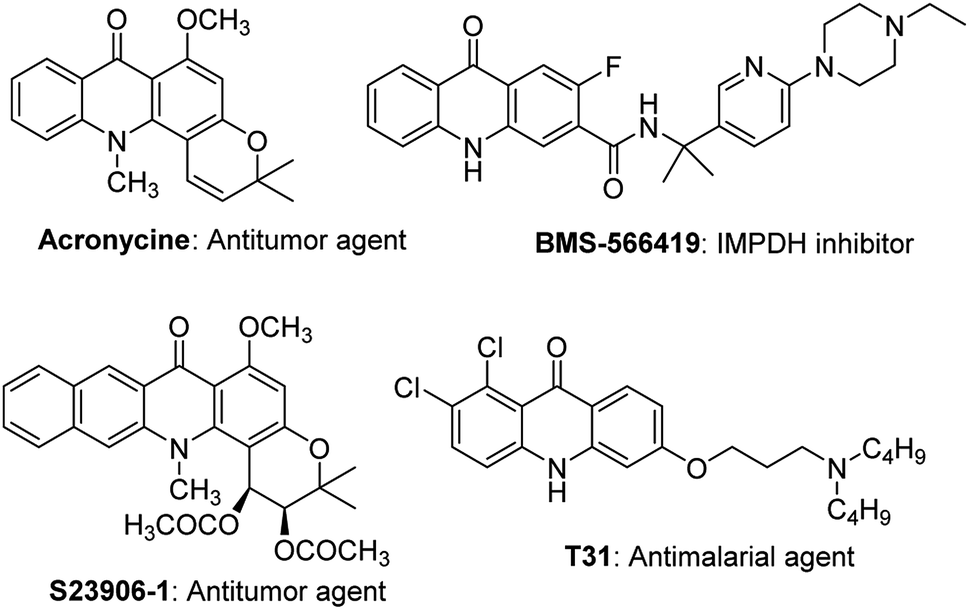 | ||
| Fig. 1 Biologically active natural and synthetic acridones. | ||
As part of an ongoing interest in developing new antiparasitic agents, we recently discovered a novel acridone chemotype that demonstrates efficacy against both liver and blood stage malaria.11,12 Of the many conventional methods available for the synthesis of acridones from N-phenylanthranilic acid and ester derivatives, the most widely used is the strong acid catalyzed intramolecular Friedel–Crafts cyclization in the presence of either sulfuric acid (H2SO4), polyphosphoric acid (PPA), or Eaton's acid (Scheme 1).12,19,20 In 2006, Nadaraj et al. demonstrated the use of several other catalysts (e.g. PTSA, ZnCl2, AlCl3 and PPA) in the synthesis of acridones under microwave irradiation.21 Recently, Zhang et al. reported the intramolecular acylation of N,N-diphenylanthranilic acids in the presence of BF3.Et2O, under conventional conditions (Scheme 1, top panel),22 however, they did not explore the substrate scope of the reaction. To date, there has not been any report of synthetic method for acridones under a non-conventional pathway using microwave irradiation in the presence of BF3.Et2O.
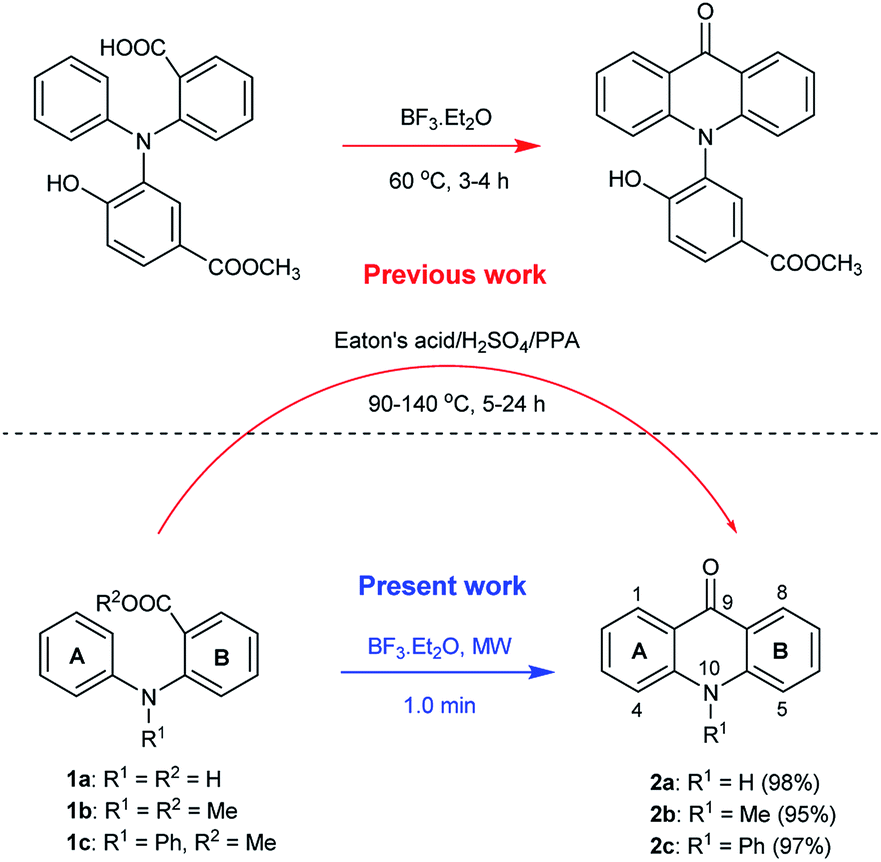 | ||
| Scheme 1 Present and previous synthetic methods for the synthesis of acridones. | ||
The currently available methods for the synthesis of acridones, which lack either the aryl or alkyl moiety at N-10 position of the middle ring, have some limitations, such as low yields, long reaction times, functional group intolerance, harsh reaction conditions, usage of larger amount of catalysts and, solvents and tedious workup procedures. An efficient and improved synthetic method for the synthesis of acridones to overcome the aforementioned disadvantages is highly desirable but remains a challenge for organic chemists. In our ongoing drug discovery program to produce new antimalarial chemotypes, our synthetic efforts focused on the development of new and efficient synthetic methods towards the generation of a large library of antimalarial acridones. In this work we report a facile, versatile, eco-friendly, and cost-effective synthetic method that is microwave-assisted for the synthesis of various acridones. In addition, we report the potent antimalarial activities of the newly generated acridones.
Results and discussion
To evaluate the broad scope of boron trifluoride etherate (BF3.Et2O) as a catalyst/reagent for the synthesis of various acridones, including (10H)-9-acridones, N-alkyl-9-acridones and N-aryl-9-acridones, we started the initial investigation with N-phenylanthranilic acid (1a) as the pilot substrate. First a careful investigation of the reaction conditions, including reaction temperature and solvent selection was carried out without the use of microwave irradiation. No desired acridone 2a formation was observed at room temperature under solvent free conditions, and in 1,4-dioxane, DMF and acetonitrile (Table 1, entries 1–4). However, the acridone 2a was formed with low yield (10%) when the reaction was performed in 1,4-dioxane under reflux conditions for 6 h (Table 1, entry 5). Acridone 2a was produced with 30% yield when the reaction was conducted under solvent free conditions at 80 °C for 24 h (Table 1, entry 6). The yield of 2a was enhanced to 65% when the reaction was performed at 130 °C for 5 h (Table 1, entry 7).| Entry | Reaction conditions | Reagent (no. eq.) | Solvent | Reaction time | Yieldb (%) 2a |
|---|---|---|---|---|---|
| a RT; room temperature.b Isolated yield.c NR; no reaction.d MW; microwave.e Formed black tar. | |||||
| 1 | RTa | BF3.Et2O (2.0) | — | 24 h | NRc |
| 2 | RT | BF3.Et2O (1.0) | 1,4-Dioxane | 24 h | NR |
| 3 | RT | BF3.Et2O (1.0) | DMF | 24 h | NR |
| 4 | RT | BF3.Et2O (1.0) | Acetonitrile | 24 h | NR |
| 5 | 100 °C | BF3.Et2O (1.0) | 1,4-Dioxane | 6 h | 10 |
| 6 | 80 °C | BF3.Et2O (2.0) | — | 24 h | 30 |
| 7 | 130 °C | BF3.Et2O (2.0) | — | 5 h | 65 |
| 8 | MWd, 100 °C | BF3.Et2O (1.0) | 1,4-Dioxane | 1.0 min | 10 |
| 9 | MW, 130 °C | BF3.Et2O (1.0) | 1,4-Dioxane | 1.0 min | 20 |
| 10 | MW, 150 °C | BF3.Et2O (1.0) | 1,4-Dioxane | 1.0 min | 45 |
| 11 | MW, 150 °C | BF3.Et2O (1.0) | DMF | 1.0 min | NR |
| 12 | MW, 150 °C | BF3.Et2O (1.0) | Acetonitrile | 1.0 min | 33 |
| 13 | MW, 100 °C | BF3.Et2O (2.0) | — | 1.0 min | 25 |
| 14 | MW, 130 °C | BF3.Et2O (2.0) | — | 1.0 min | 60 |
| 15 | MW, 150 °C | BF3.Et2O (2.0) | — | 1.0 min | 98 |
| 16 | MW, 150 °C | SnCl4 (2.0) | — | 3.0 min | NR |
| 17 | MW, 150 °C | SnCl4 (2.0) | 1,4-Dioxane | 3.0 min | Tracee |
| 18 | MW, 150 °C | TiCl4 (2.0) | — | 3.0 min | NR |
| 19 | MW, 150 °C | TiCl4 (2.0) | 1,4-Dioxane | 3.0 min | Tracee |
| 20 | MW, 150 °C | FeCl3 (2.0) | 1,4-Dioxane | 3.0 min | NR |
| 21 | MW, 150 °C | Pd(OAc)2 (2.0) | 1,4-Dioxane | 3.0 min | NR |
| 22 | MW, 150 °C | PdCl2 (2.0) | 1,4-Dioxane | 3.0 min | NR |
| 23 | MW, 150 °C | I2 (2.0) | 1,4-Dioxane | 3.0 min | NR |
Encouraged by these results, we next evaluated this reaction under microwave irradiation using 1,4-dioxane, DMF and acetonitrile as solvent and solvent free conditions at various temperatures (Table 1, entries 8–15). Great improvement was observed as acridone 2a was obtained with 98% yield in the presence of BF3.Et2O (2.0 eq.), with a short reaction time (1.0 min) (Scheme 1 and Table 1, entry 15). Remarkably, this reaction was very efficient under solvent free conditions and generated no side products. Having now changed several reaction parameters, we wished to determine whether BF3.Et2O was still the best reagent for the model reaction, so we then screened the reaction with other Lewis acids SnCl4, TiCl4, FeCl3, Pd(OAc)2, PdCl2 and I2 under microwave irradiation at 150 °C (Table 1, entries 16–23). All these reactions failed to provide the desired product 2a even at longer reaction time (3.0 min). A trace amount of the acridone 2a was observed with SnCl4 and TiCl4 in 1,4-dioxane (Table 1, entries 17 and 19), however, in both cases, the reaction mixture turned into a black tar. On the basis of all of these experiments, the optimal reaction conditions were identified as no added solvent at 150 °C for 1.0 min under microwave irradiation mediated by BF3.Et2O (2.0 eq.).
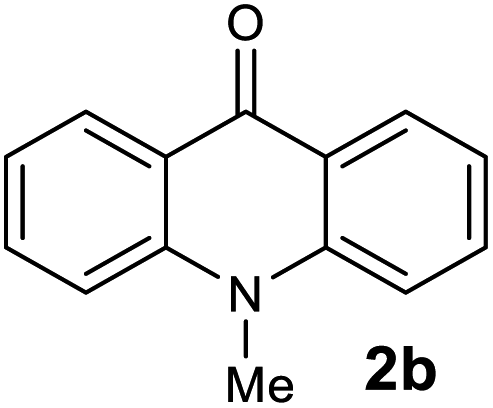
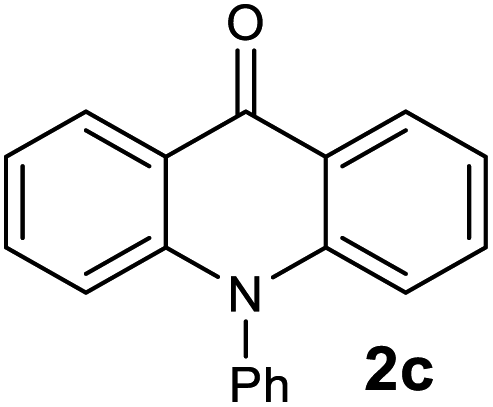
The scope of the reaction was further explored with the newly optimized conditions. Under the same reaction conditions (as in Table 1, entry 15), N-methyl-N-phenylanthranilic acid methyl ester (1b) and N,N-diphenylanthranilic acid methyl ester (1c) also provided the N-methyl-9-acridone (2b) and N-phenyl-9-acridone (2c), respectively, in high yields (Scheme 1 and Table 2, entries 2 and 3). It is noteworthy that this is a unique synthetic strategy to generate various acridones, such as (10H)-9-acridones, N-alkyl-9-acridones and N-aryl-9-acridones.
| Entry | Substrate | Product | Yielda (%) |
|---|---|---|---|
| a Isolated yield.b NR; no reaction, recovered the substrate at 150 °C, but decomposed at 200 °C.c Trace amount of 2y was observed along with other side products at 170 °C, and decomposed at 200 °C. | |||
| 1 | 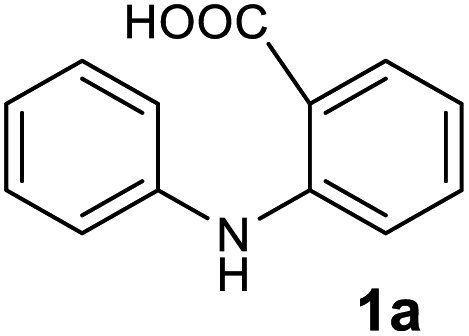 |
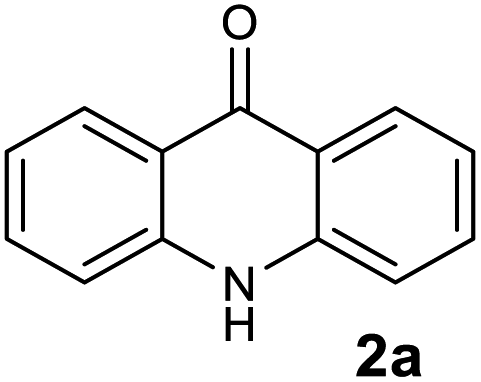 |
98 |
| 2 | 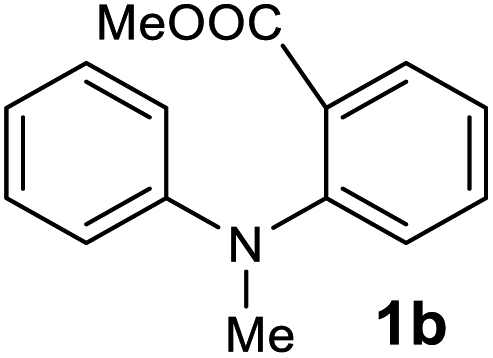 |
 |
95 |
| 3 | 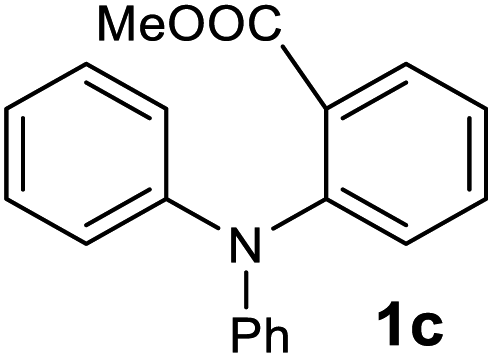 |
 |
97 |
| 4 | 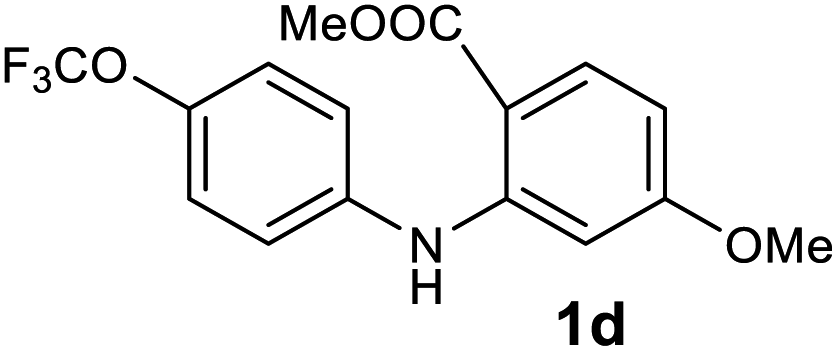 |
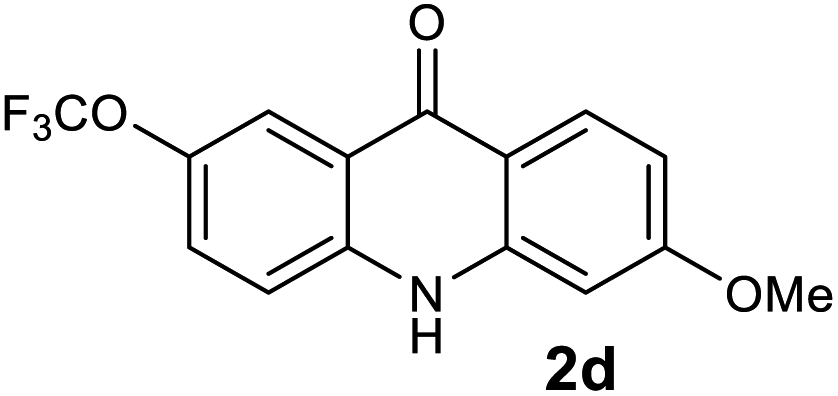 |
97 |
| 5 | 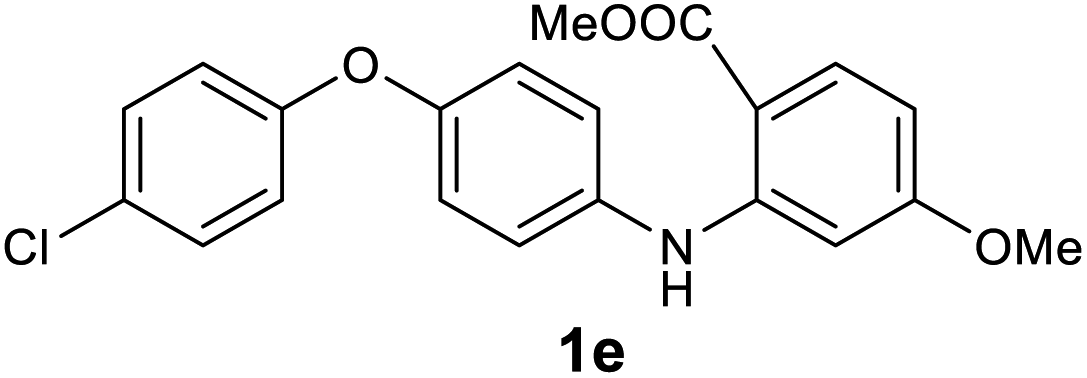 |
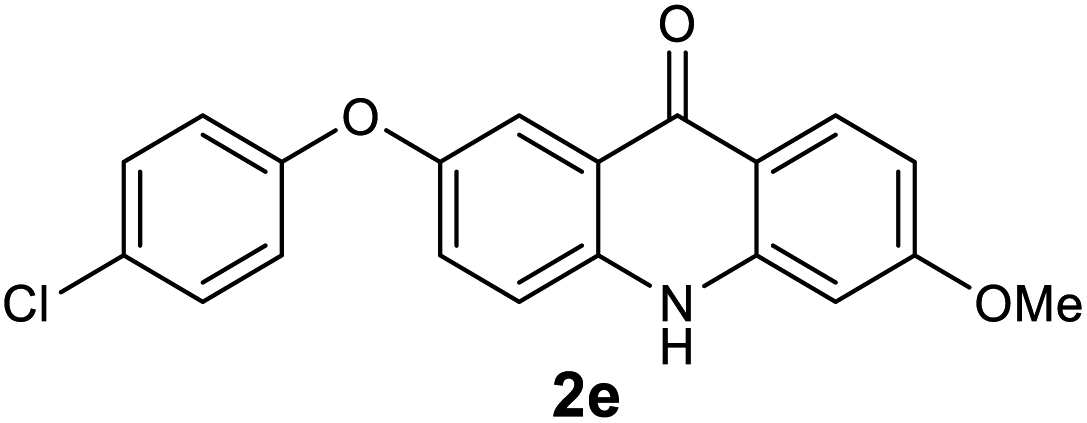 |
96 |
| 6 | 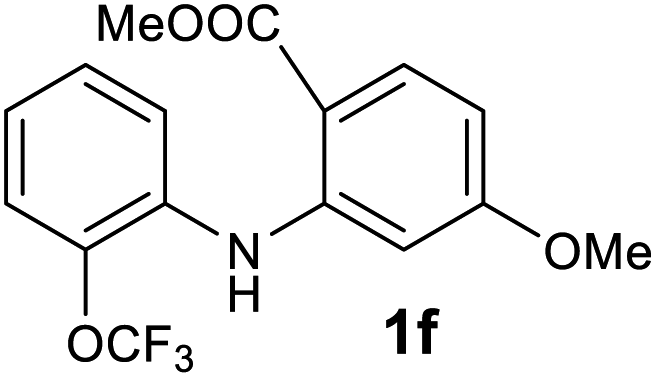 |
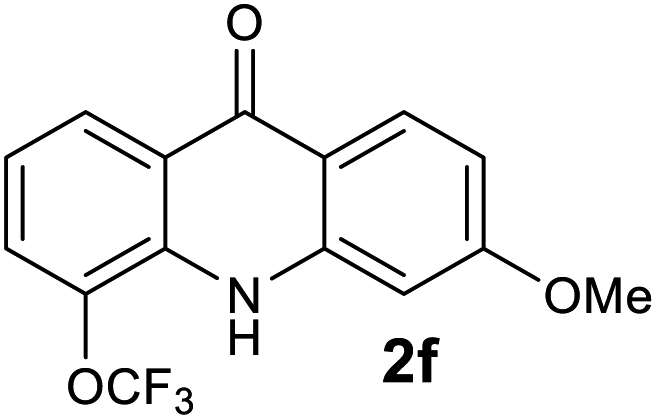 |
95 |
| 7 | 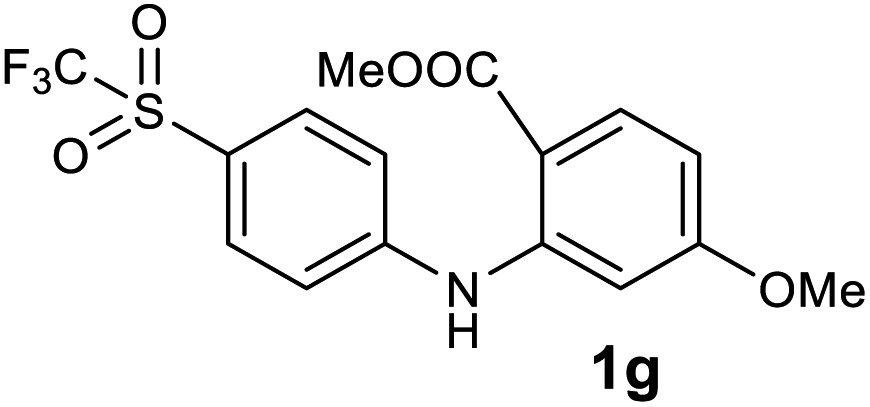 |
 |
97 |
| 8 | 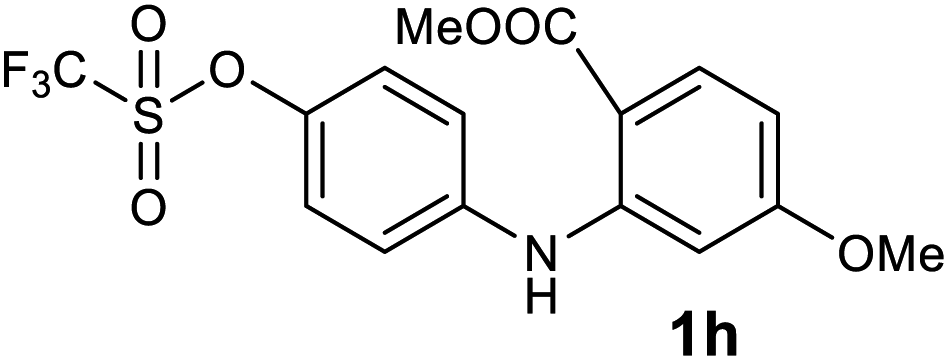 |
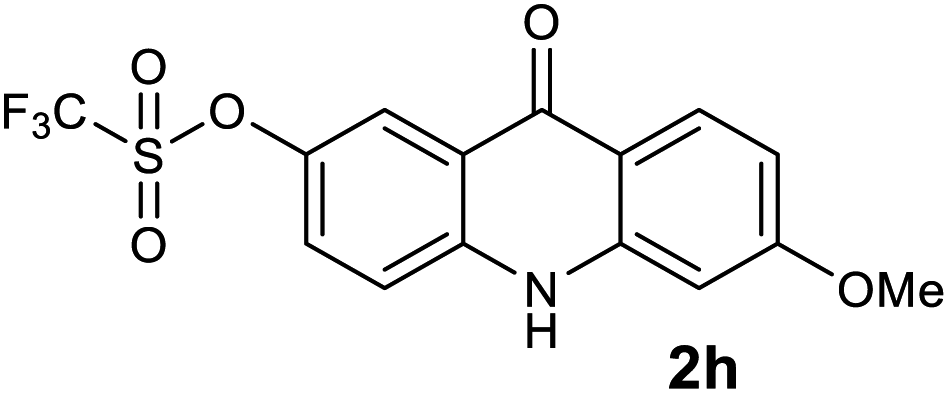 |
98 |
| 9 | 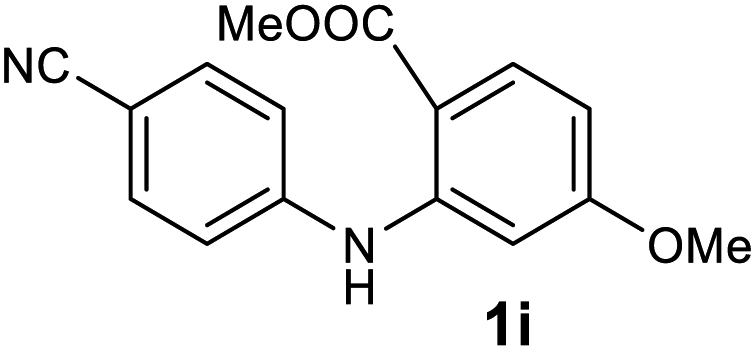 |
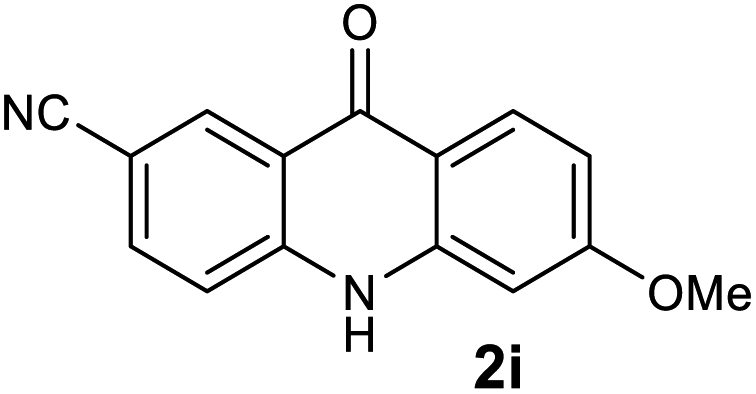 |
97 |
| 10 | 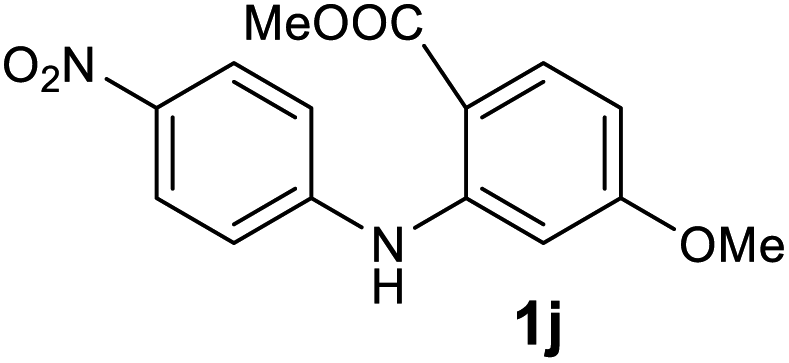 |
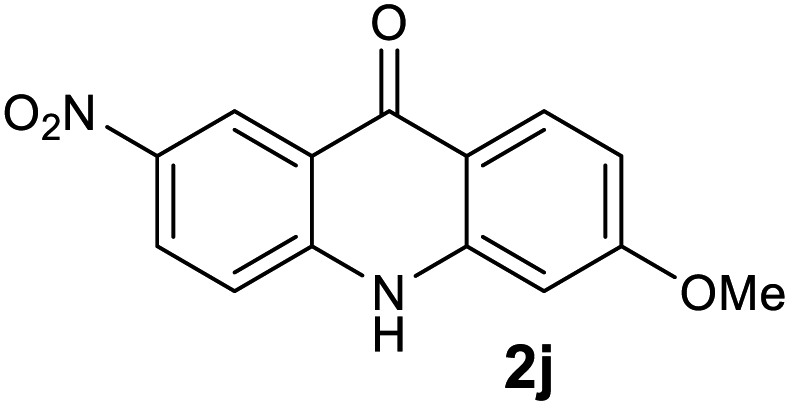 |
96 |
| 11 | 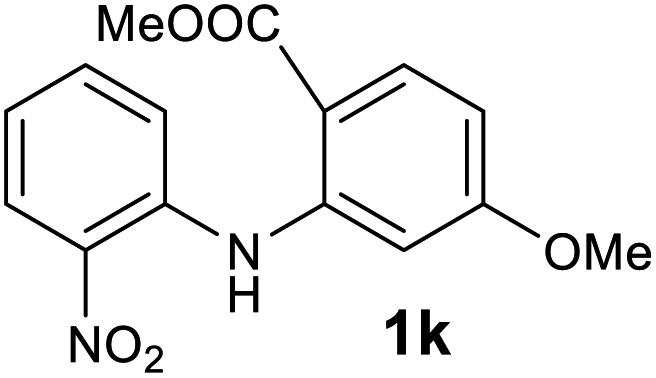 |
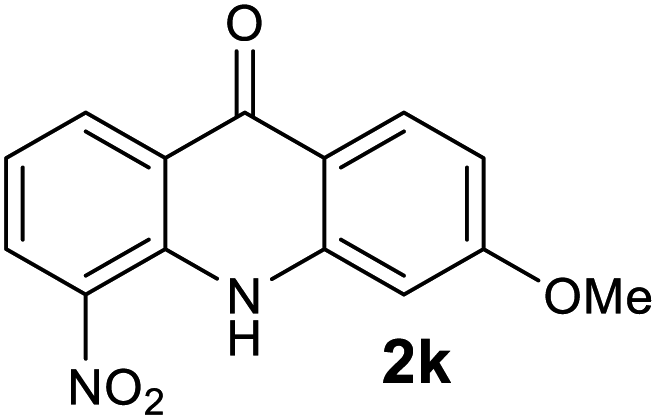 |
95 |
| 12 | 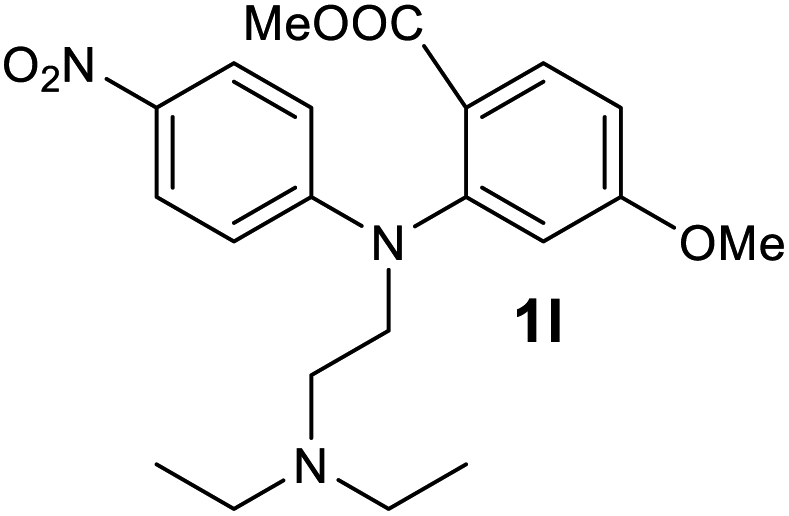 |
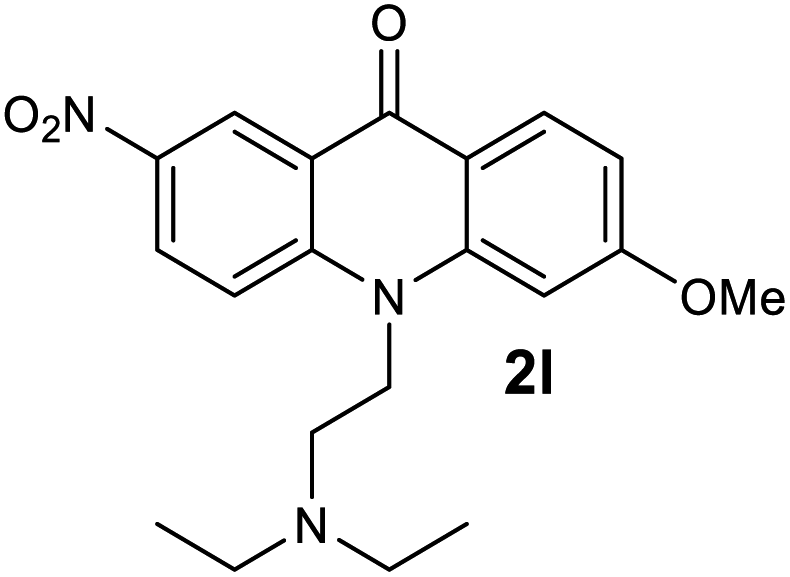 |
94 |
| 13 | 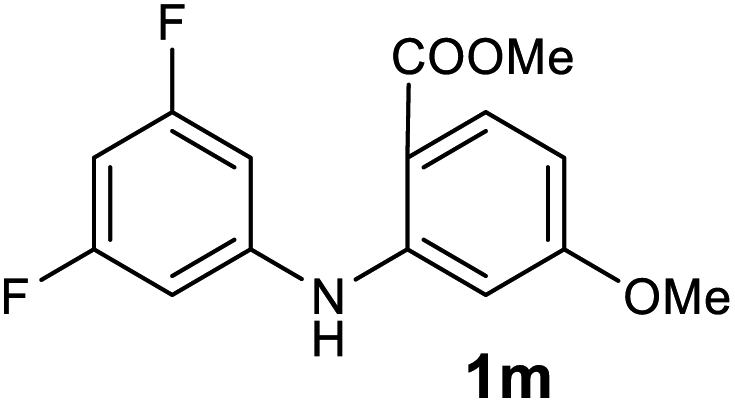 |
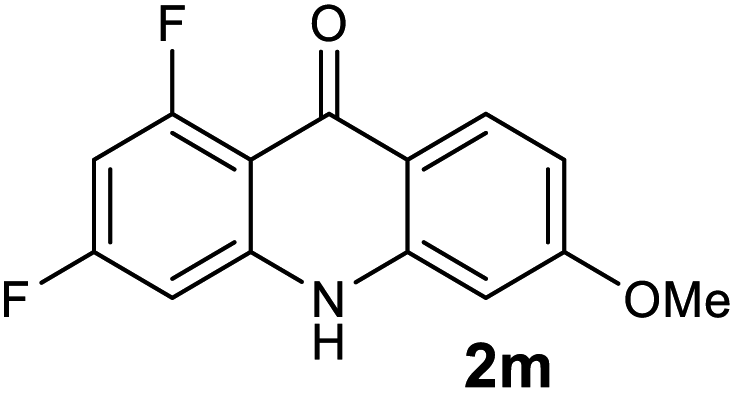 |
85 |
| 14 | 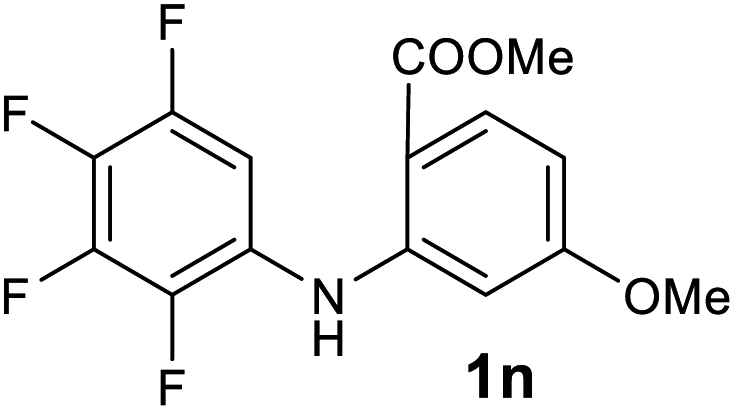 |
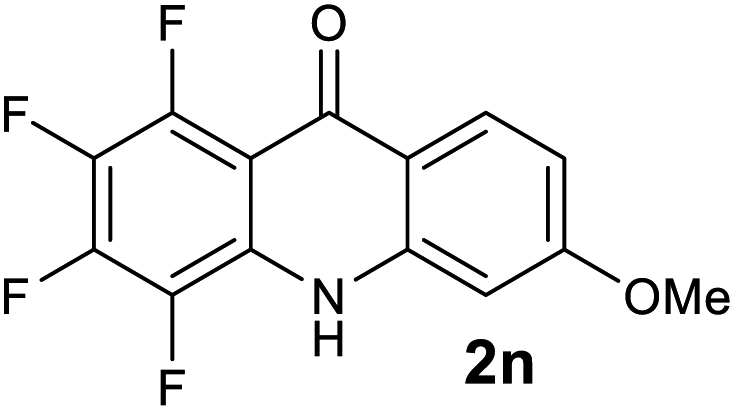 |
67 |
| 15 | 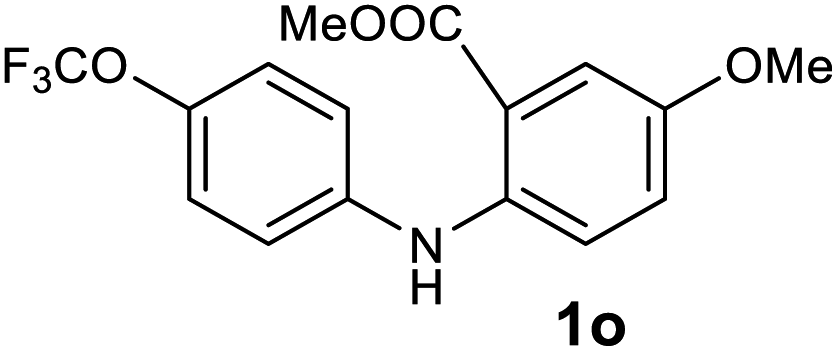 |
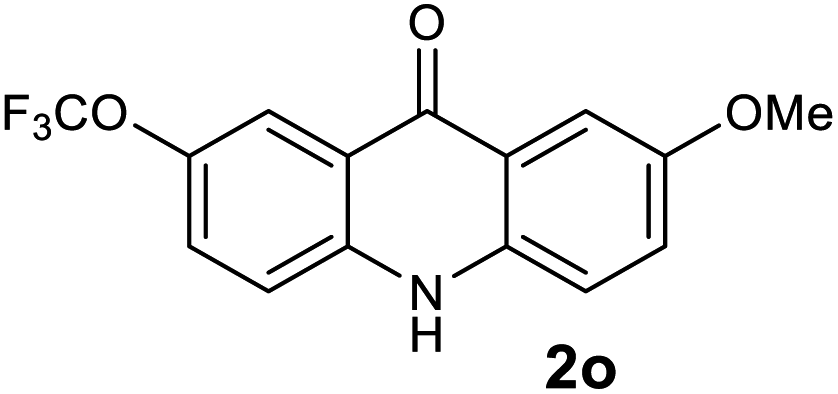 |
69 |
| 16 |  |
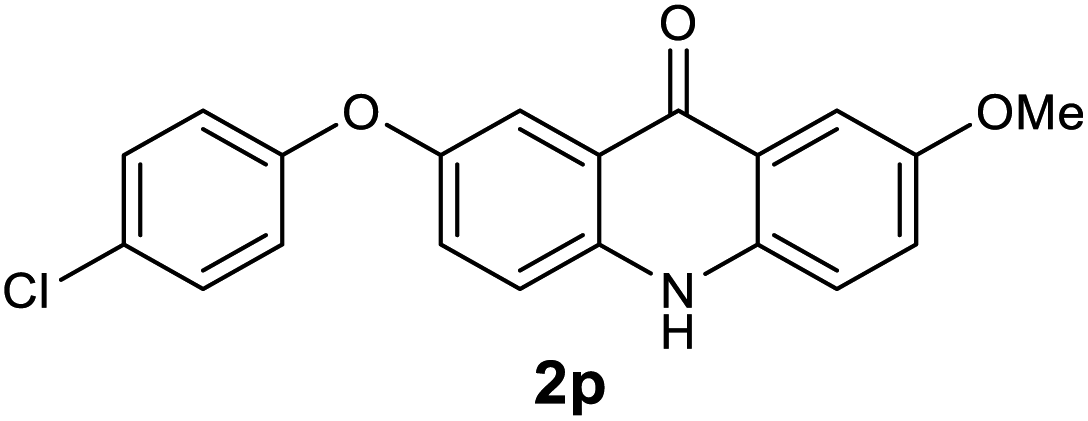 |
56 |
| 17 | 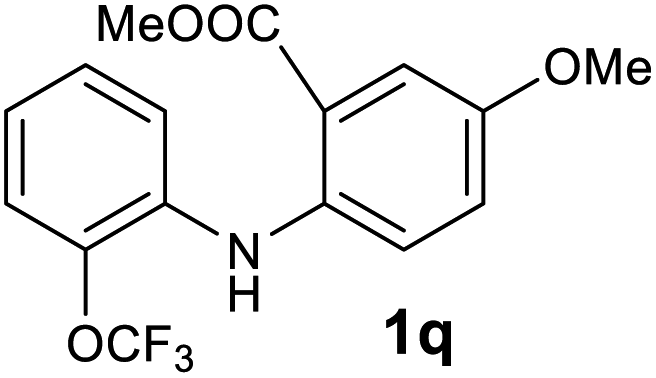 |
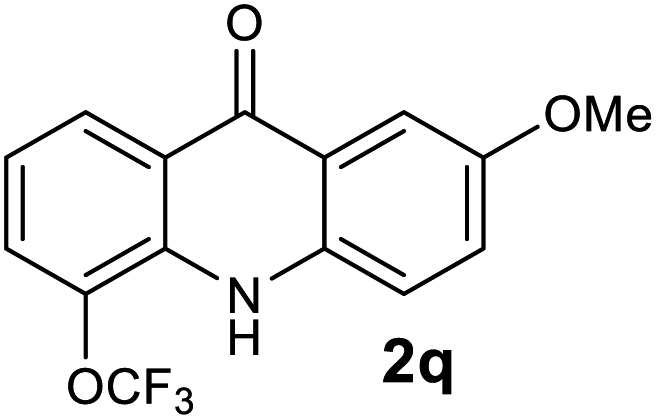 |
65 |
| 18 | 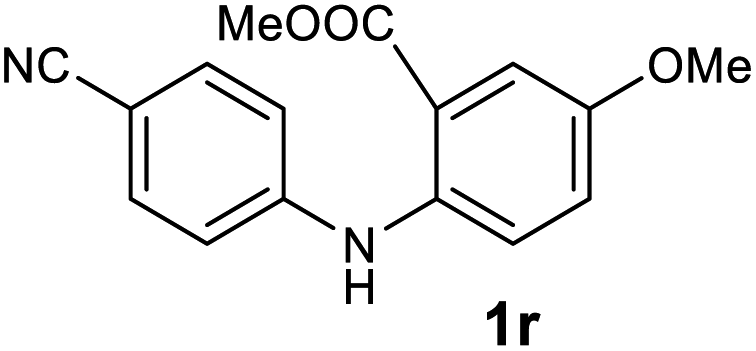 |
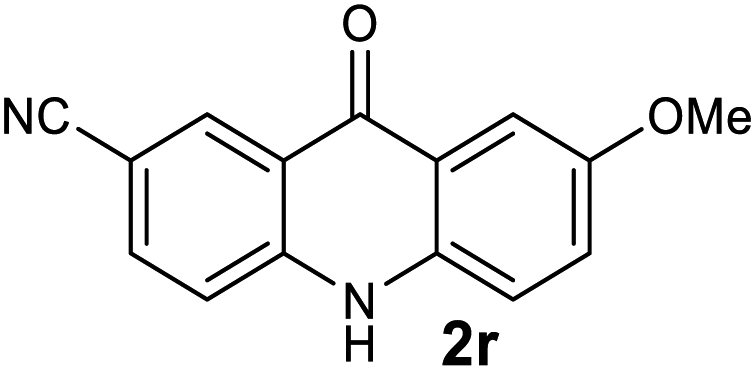 |
60 |
| 19 | 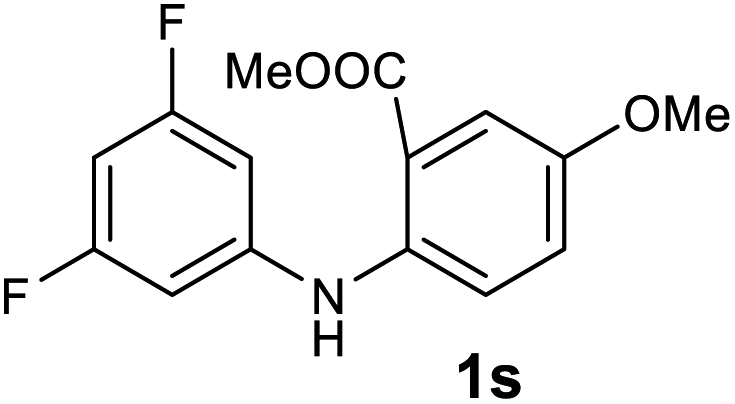 |
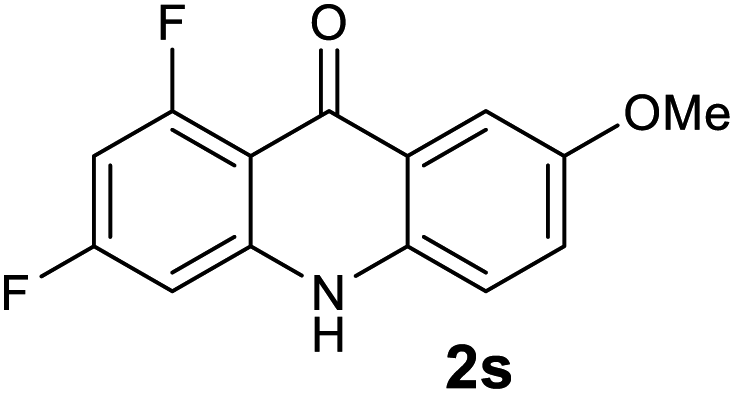 |
64 |
| 20 | 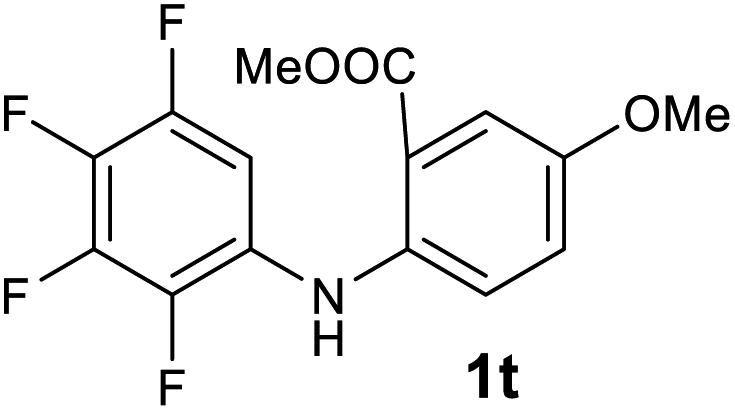 |
 |
32 |
| 21 | 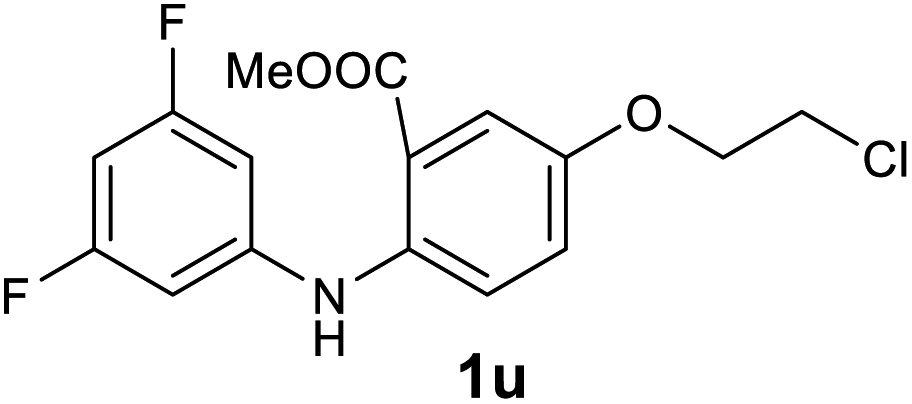 |
 |
72 |
| 22 | 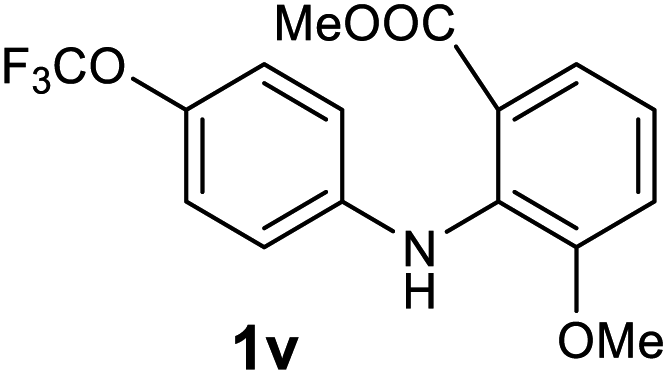 |
 |
71 |
| 23 | 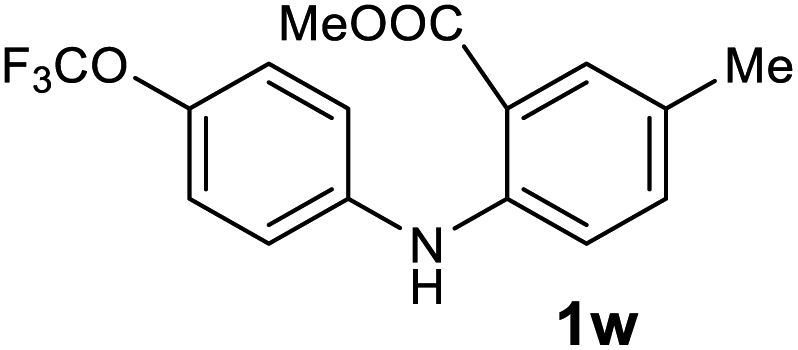 |
 |
86 |
| 24 | 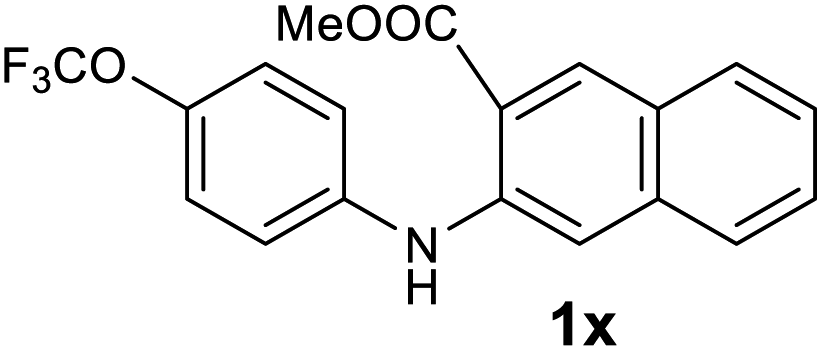 |
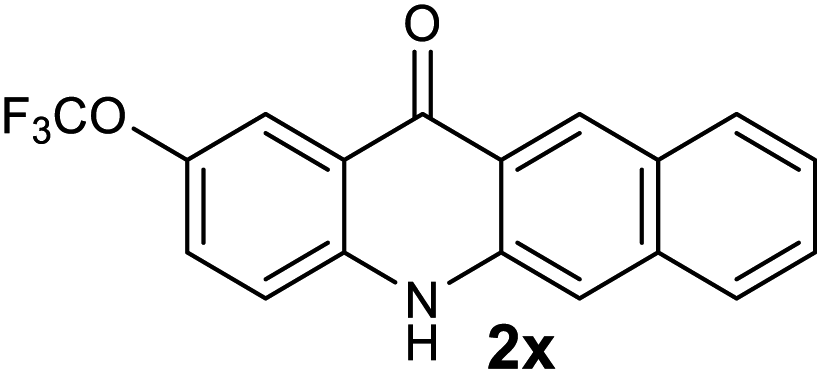 |
45 |
| 25 | 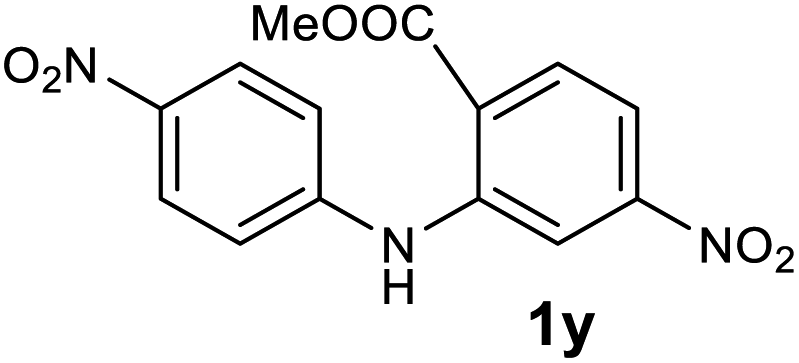 |
— | NRb |
| 26 | 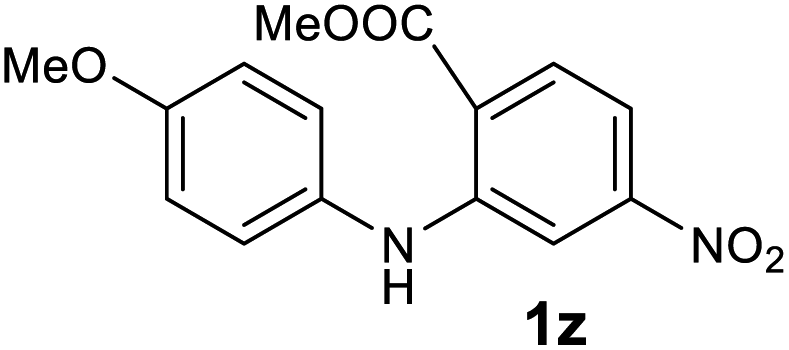 |
— | NRb |
| 27 | 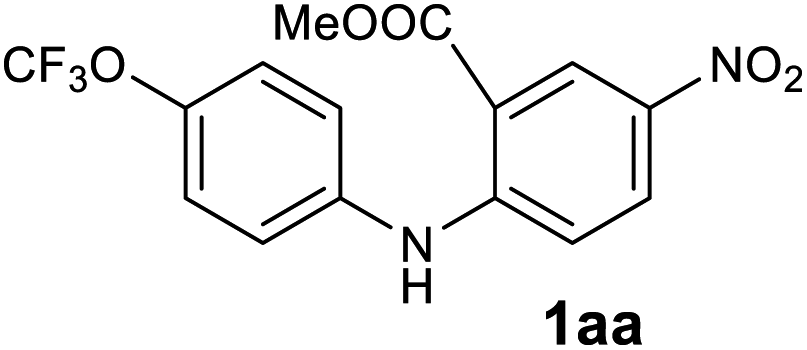 |
 |
Tracec |
We next examined the scope of the reaction by verifying the functional groups positioned on both aryl rings (A and B) (Table 2). N-phenylanthranilic acid analogues 1d–1k (Table 2, entries 4–11) bearing an electron-donating methoxy group at the para-position to the methyl ester on ring-B, and various electron-donating and electron-withdrawing groups on ring-A produced the corresponding acridones 2d–2k in excellent yields (95–98%). Notably, N-phenylanthranilic acid methyl ester 1l (Table 2, entry 12) bearing (diethylamino)ethyl moiety at N-10 position of the middle ring also responded well under the optimized conditions to provide the desired acridone 2l in excellent yield (94%). Together, it was demonstrated that the N-phenylanthranilic acid analogues bearing an alkyl, aryl and alkyl-amine moieties at N-10 position of the middle ring, and electron-donating and electron-withdrawing groups on ring-A are well tolerated producing desired products with great efficiency. A decrease in yield was observed in the case of acridone 2m and 2n (Table 2, entries 13 and 14) with N-phenylanthranilic acid methyl ester bearing multiple fluoro substitutions on ring-A as a substrate.
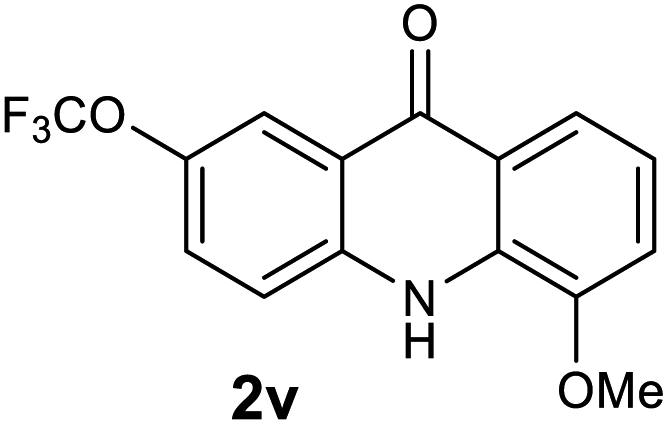
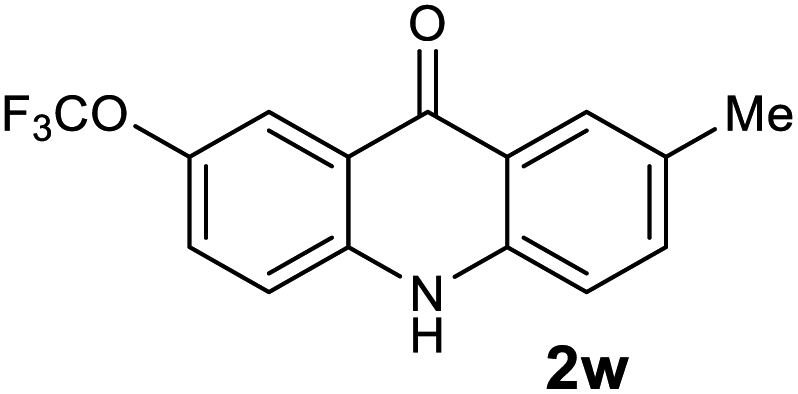
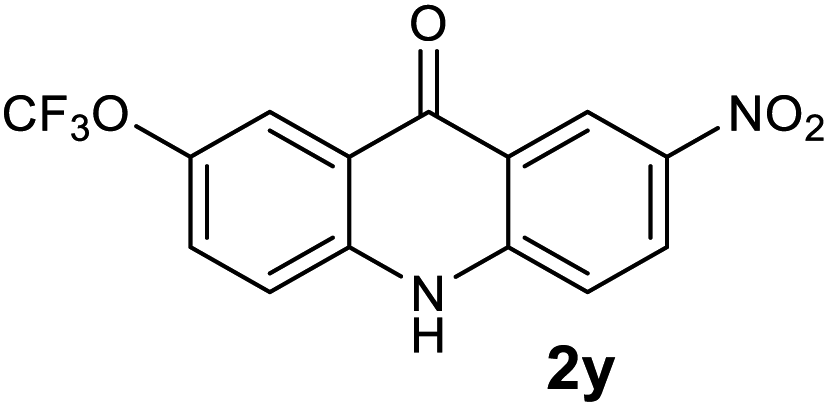
Next, the effect of substituents at meta-position to the methyl ester on ring-B was studied. N-phenylanthranilic acid analogues 1o–1v (Table 2, entries 15–22) bearing an electron-donating groups at meta-position to the methyl ester on ring-B and various electron-donating and electron-withdrawing groups on ring-A were converted to their corresponding acridones with good yields. However, the yields of acridones 2o–2v were roughly 25% lesser than the corresponding positional acridones 2d–2n. In contrast, the N-phenylanthranilic acid analogue (1w) with the methyl instead of methoxy group provided the corresponding acridone 2w in higher yield (86%). Of note, during the reaction with substrates 1o–1w small amount of side products were observed (not isolated). The extended conjugation on ring-B as with substrate 1x significantly reduced the yield of acridone 2x (45%). Disappointingly, when substrates 1y and 1z bearing a strong electron-withdrawing nitro group at 6 positon of the ring-B were used, no desired acridones were obtained even at elevated temperatures (170 °C and 200 °C) and longer reaction time (3.0 min) (Table 2, entries 25 and 26). However, a trace amount of the acridone 2y was observed (not isolated) along with other side products at 170 °C, when substrate 1aa containing a nitro group at 7 positon of the ring-B, was used. All these results demonstrated that the electron-withdrawing groups (EWGs) on the ring-B, are not tolerated in this methodology.
The possible mechanistic pathway involves an intramolecular Friedel–Crafts cyclization, and we propose that initially the BF3.OEt2 coordinates with the oxygen of a carbonyl of the substrate 3 to form the key intermediate 4. The following intramolecular Friedel–Crafts cyclization proceeds via the intermediates 5 and 6, and the subsequent hydrolysis provides the desired acridone 2 (Scheme 2). In fact, it appears that the electron-donating group (EDG) on the ring-B of 3 significantly enhances the nucleophilic nature at the oxygen of the carbonyl group, which facilitates toward the formation of the BF3 complex 4. Conversely, electron-withdrawing group (EWG) on the ring-B of 7 diminishes the nucleophilic nature at the oxygen of the carbonyl group, which prevents the formation of BF3 complex (Scheme 2). It is noteworthy that the formation of key intermediate 4 is vital in this transformation.
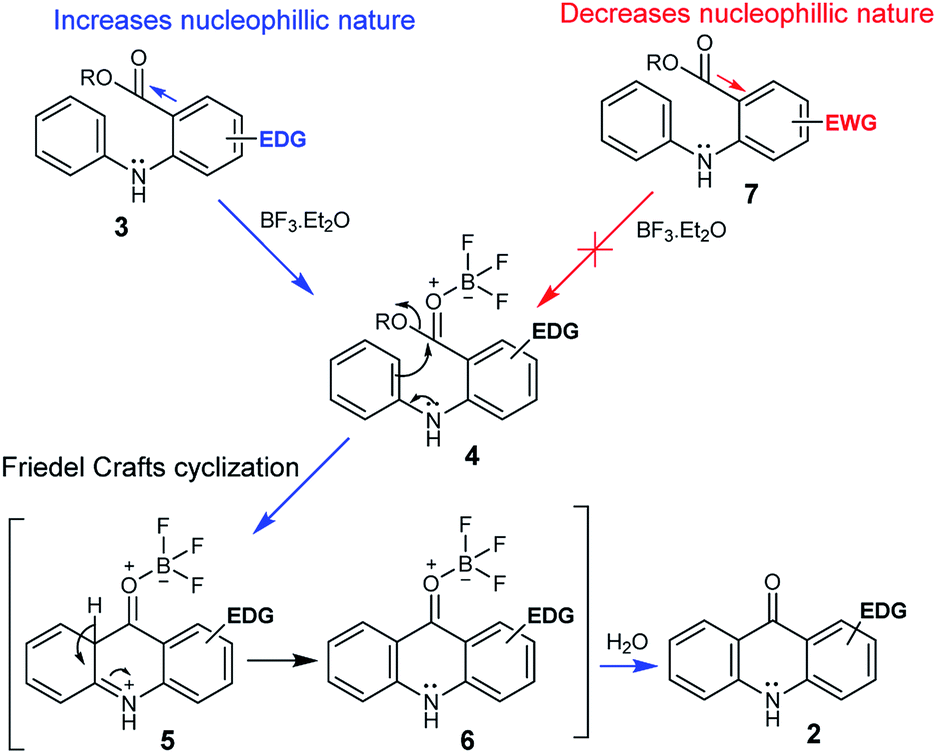 | ||
| Scheme 2 Possible reaction mechanism for BF3.Et2O mediated Friedel–Crafts cyclization. | ||
To demonstrate the feasibility of the present protocol, a larger-scale reaction (5 g × 5) was performed with 1a. Significantly, our reaction has shown excellent efficiency even at a larger scale reaction with >95% yield. This reaction was also carried out under domestic microwave conditions obtaining the corresponding acridones with excellent yields.
All newly synthesized acridones were then evaluated for their in vitro antimalarial blood stage activity against a panel of Plasmodium falciparum malaria parasites with different geographic and genetic backgrounds using a SYBR Green based assay.23 Standard antimalarials chloroquine (CQ) and atovaquone (ATV) were used as reference drugs. In parallel, the cytotoxicity of acridones was tested against human hepatic HepG2 cancer cell line.24 The results are summarized in Table 3. Majority of the acridones have shown great antimalarial activity (IC50 < 100 nM). Significantly, acridones 2d, 2e, 2g, 2s, 2u, 2v and 2x exhibited excellent activity against all tested strains with a high therapeutic index (the ratio of cytotoxicity to antimalarial therapeutic efficacy), as demonstrated in Table 3. In general, the activity and structure–activity relationship (SAR) analyses demonstrate that the electron donating groups at 2 position of ring-A, and 6 position of ring-B are well tolerated. Conversely, the electron withdrawing groups on ring-A have an adverse effect on antimalarial potency.
| Compd | In vitro activitya (IC50 nM) | Cytotoxicitya (nM) vs. HepG2 | ||
|---|---|---|---|---|
| D6 | Dd2 | 7G8 | ||
| a IC50 values are the average of at least three determinations, each carried out in triplicate (±10%). D6: CQ-sensitive; Dd2: MDR strain with Old World genetic background; 7G8: MDR strain with New World genetic background; ATV: atovaquone; CQ: chloroquine. | ||||
| 2d | 0.103 | 1.10 | 1.51 | >200000 |
| 2e | 8.84 | 11.7 | 13.5 | >200000 |
| 2f | 782 | 753 | 1138 | >200000 |
| 2g | 11.6 | 11.6 | 22.2 | >200000 |
| 2h | 57.7 | 60.7 | 56.6 | >200000 |
| 2i | 100 | 177 | 228 | >200000 |
| 2j | 84.1 | 98.1 | 35.0 | >200000 |
| 2l | 405 | 684 | 462 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 296 296 |
| 2n | 257 | 325 | 41.8 | >200000 |
| 2o | 632 | 425 | 437 | >200000 |
| 2p | 573 | 513 | 911 | >200000 |
| 2q | 1493 | 1678 | >2500 | >200000 |
| 2r | 983 | 932 | >2500 | >200000 |
| 2s | 0.05 | 0.29 | 0.20 | >200000 |
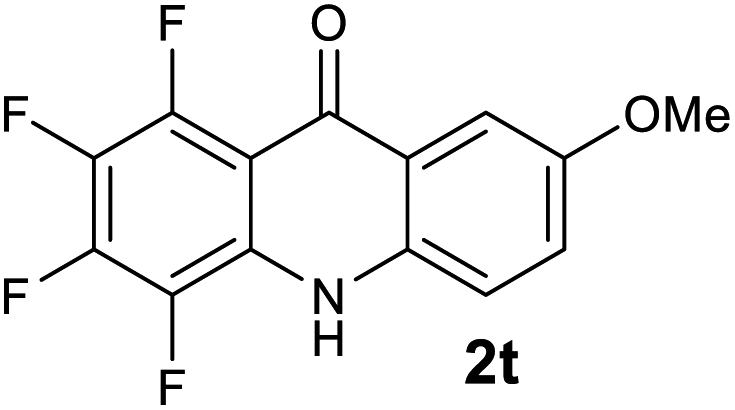
| 2t | >2500 | 1954 | 393 | >200000 |
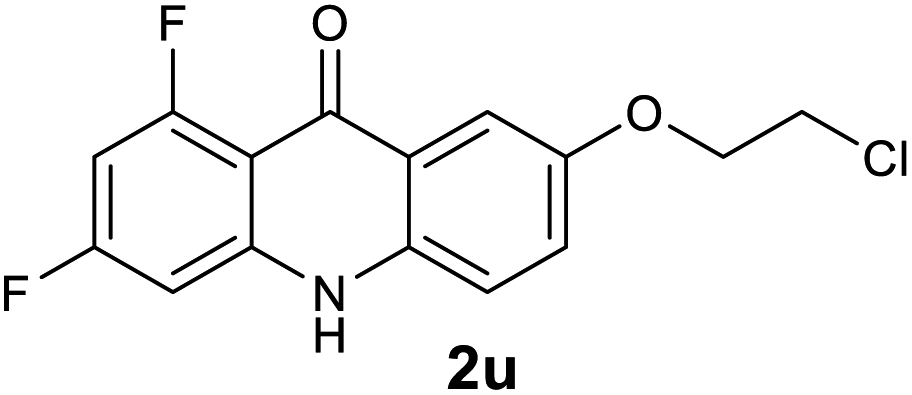
| 2u | 11.6 | 0.91 | 1.22 | >200000 |
| 2v | 27.7 | 15.5 | 46.3 | 139435 |
| 2w | 63.1 | 49.2 | 109 | >200000 |
| 2x | 7.03 | 0.563 | 16.5 | >200000 |
| ATV | 0.10 | 0.10 | 0.20 | 23160 |
| CQ | 15.0 | 163 | 172 | 37577 |
Conclusions
In conclusion, we developed an efficient, simple and convenient synthetic method for the synthesis of acridones in good to excellent yields for the first time in the presence BF3.Et2O under microwave irradiation conditions. The advantages of this method are: simple experimental and workup procedures, short duration of reaction (1.0 min), proceeds under solvent free conditions, the use of mild and low cost catalyst/reagent, replacement of the strong acid catalysts, and tolerance of large variety functional groups. This synthetic method has the potential to be carried out on a large scale and is suited for the generation of an extensive library of acridones and their precursors. Significantly, a number of newly generated acridones exhibited potent antimalarial activity with great selectivity. The preparation of a large library containing structurally diversified second generation acridones is currently underway in our laboratory.Experimental section
General
NMR spectra were recorded on Bruker AMX-400 spectrometer at 400 MHz (1H) and 100 MHz (13C). Experiments were recorded in CDCl3 and DMSO-d6 at 25 °C. Chemical shifts are given in parts per million (ppm) downfield from internal standard Me4Si. High-resolution mass spectrometry (HRMS) (electrospray ionization (ESI)) were recorded on a high-resolution (140000) Q Exactive Orbitrap mass spectrometer. The microwave reactions were conducted using Biotage® Initiator+ microwave synthesizer and domestic microwave. Unless otherwise stated, all reagents and solvents were purchased from commercial suppliers and used without further purification. Reactions that required the use of anhydrous, inert atmosphere techniques were carried out under an atmosphere of argon/nitrogen. Chromatography was executed on CombiFlash instrument, using silica gel (230–400 mesh) as the stationary phase and mixtures of ethyl acetate (EtOAc) and hexane or dichloromethane (DCM) and methanol as eluents. Substrate 1a was obtained from commercial sources and 1b was synthesized from 1a via standard methylation reaction. Substrate 1c was synthesized from methyl 2-bromobenzoate via a copper-mediated substitution reaction with diphenylamine.25 Substrates 1d–1z, and 1aa were synthesized using Buchwald–Hartwig cross-coupling methods.12,26
Representative procedure for the synthesis of acridin-9(10H)-one (2a)21
BF3.Et2O (580 μL, 4.69 mmol) was added to a microwave reaction vial containing N-phenylanthranilic acid (1a) (500 mg, 2.34 mmol). Then the reaction mixture was exposed to microwave irradiation for 1.0 min at 150 °C. After cooling to room temperature, the reaction mixture was poured into water (25 mL) and allowed to stir for 5 min. The solid material was filtered by a sintered funnel and washed with water (50 mL) and methanol (5 mL) to afford the desired acridone 2a (448 mg. 98%). 1H NMR (DMSO-d6, 400 MHz) δ 11.74 (s, 1H), 8.23 (dd, J = 8.2, 1.4 Hz, 2H), 7.73 (m, 2H), 7.54 (d, J = 8.2 Hz, 2H), 7.26 (m, 2H); 13C NMR (DMSO-d6, 100 MHz) δ 177.2, 141.4 (2C), 133.9 (2C), 126.5 (2C), 121.5 (2C), 120.9 (2C), 117.8 (2C); HRMS (ESI) calcd for C13H10N1O1 (M + H)+ 196.0757, found 196.0754.In vitro blood stage antimalarial activity assay
In vitro antimalarial activity was determined by the Malaria SYBR Green I-based Fluorescence (MSF) assay described previously with minor modifications, and expressed as the compound concentration inhibiting growth by 50% (IC50).HepG2 cytotoxicity assay
The general cytotoxic effect of acridones on host mammalian cells was assessed by functional assay as described previously, using hepatic HepG2 cells. Drugs were dissolved in DMSO to make 10 mM stock solutions. Human hepatocarcinoma cells (HepG2) were maintained on RPMI-1640 medium supplemented with 10% fetal bovine serum at 37 °C in a humidified 5% CO2 atmosphere. Cells were seeded at a density of 2 × 104 per well in 96-well flat-bottom tissue culture plates containing complete medium in a total volume of 160 μL per well. The cells were allowed to attach at 37 °C overnight. On the following day, drug solutions (40 μL per well) were serially diluted with complete culture medium across the plate. The plates were then incubated at 37 °C and 5% CO2 for another 24–36 h. Afterward, the medium was aspirated and replaced with complete RPMI medium (200 μL per well), and the plates were incubated for an additional 24 h at 37 °C and 5% CO2. An aliquot of a stock solution of resazurin (Alamar Blue, prepared in 1 × PBS) was then added at 20 μL per well (final concentration 10 μM), and the plates were returned to the incubator for 3 h. After this period, fluorescence in each well, indicative of cellular redox activity was measured in a Gemini EM plate reader with excitation wavelength at 560 nm and emission wavelength at 590 nm. IC50 values were determined by nonlinear regression analysis of logistic concentration–fluorescence intensity curves (GraphPad Prism software).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Oregon Health & Science University Medicinal Chemistry Core Department (Portland, OR) for the use the Biotage® Initiator+ microwave synthesizer. This project was supported by NIH/NIAID (award 1R01AI093784) and DOD/PRMRP (award PR160693/W81XWH-17-2-0041).References
- G. H. Svoboda, G. A. Poore, P. J. Simpson and G. B. Boder, J. Pharm. Sci., 1966, 55, 758 CrossRef CAS PubMed
.
- J. H. Scarffe, A. R. Beaumont and D. Crowther, Cancer Treat. Rep., 1983, 67, 93 CAS
- R. T. Dorr, J. D. Liddil, D. D. Von Hoff, M. Soble and C. K. Osborne, Cancer Res., 1989, 49, 340 CAS
- N. Costes, H. LeDeit, S. Michel, F. Tillequin, M. Koch, B. Pfeiffer, P. Renard, S. Léonce, N. Guilbaud, L. KrausBerthier, A. Pierré and G. Atassi, J. Med. Chem., 2000, 43, 2395 CrossRef CAS PubMed
- N. Guilbaud, L. Kraus-Berthier, F. Meyer-Losic, V. Malivet, C. Chacun, M. Jan, F. Tillequin, S. Michel, M. Koch, B. Pfeiffer, G. Atassi, J. Hickman and A. Pierré, Clin. Cancer Res., 2001, 7, 2573 CAS
- I. Antonini, P. Polucci, A. Magnano, B. Gatto, M. Palumbo, E. Menta, N. Pescalli and S. Martelli, J. Med. Chem., 2003, 46, 3109 CrossRef CAS PubMed
- S. H. Watterson, P. Chen, Y. Zhao, H. H. Gu, T. G. Dhar, Z. Xiao, S. K. Ballentine, Z. Shen, C. A. Fleener, K. A. Rouleau, M. Obermeier, Z. Yang, K. W. McIntyre, D. J. Shuster, M. Witmer, D. Dambach, S. Chao, A. Mathur, B. C. Chen, J. C. Barrish, J. A. Robl, R. Townsend and E. J. Iwanowicz, J. Med. Chem., 2007, 50, 3730 CrossRef CAS PubMed
- O. Tabarrini, G. Manfroni, A. Fravolini, V. Cecchetti, S. Sabatini, E. De Clercq, J. Rozenski, B. Canard, H. Dutartre, J. Paeshuyse and J. Neyts, J. Med. Chem., 2006, 49, 2621 CrossRef CAS PubMed
- A. Stankiewicz-Drogon, B. Dörner, T. Erker and A. M. Boguszewska-Chachulska, J. Med. Chem., 2010, 53, 3117 CrossRef CAS PubMed
- I. B. Taraporewala, J. W. Cessac, T. C. Chanh, A. V. Delgado and R. F. Schinazi, J. Med. Chem., 1992, 35, 2744 CrossRef CAS PubMed
- J. X. Kelly, M. J. Smilkstein, R. Brun, S. Wittlin, R. A. Cooper, K. D. Lane, A. Janowsky, R. A. Johnson, R. A. Dodean, R. Winter, D. J. Hinrichs and M. K. Riscoe, Nature, 2009, 459, 270 CrossRef CAS PubMed
- R. A. Dodean, P. Kancharla, Y. Li, V. Melendez, L. Read, C. E. Bane, B. Vesely, M. Kreishman-Deitrick, C. Black, Q. Li, R. J. Sciotti, R. Olmeda, T. L. Luong, H. Gaona, B. Potter, J. Sousa, S. Marcsisin, D. Caridha, L. Xie, C. Vuong, Q. Zeng, J. Zhang, P. Zhang, H. Lin, K. Butler, N. Roncal, L. Gaynor-Ohnstad, S. E. Leed, C. Nolan, S. J. Huezo, S.
A. Rasmussen, M. T. Stephens, J. C. Tan, R. A. Cooper, M. J. Smilkstein, S. Pou, R. W. Winter, M. K. Riscoe and J. X. Kelly, J. Med. Chem., 2019, 62, 4375 CrossRef PubMed
- D. B. C. de Oliveira, L. B. Silva, B. V. da Silva, T. C. Borges, B. C. Marques, M. B. Dos Santos, L. F. de Oliveira, V. S. Bolzani, A. R. A. Rodrigues, L. O. Regasini and A. A. Andrade, J. Appl. Microbiol., 2019, 127, 1362 CrossRef CAS PubMed
- X. A. Li, H. L. Wang and S. D. Yang, Org. Lett., 2013, 15, 1794 CrossRef CAS PubMed
- Z. Zheng, L. Dian, Y. Yuan, D. Zhang-Negrerie, Y. Du and K. Zhao, J. Org. Chem., 2014, 79, 7451 CrossRef CAS PubMed
- J. Wen, S. Tang, F. Zhang, R. Shi and A. Lei, Org. Lett., 2017, 19(1), 94 CrossRef CAS PubMed
- H. Wu, Z. Zhang, Q. Liu, T. Liu, N. Ma and G. Zhang, Org. Lett., 2018, 20, 2897 CrossRef CAS PubMed
- J. Song, K. Ding, W. Sun, S. Wang, H. Sun, K. Xiao, Y. Qian and C. Liu, Tetrahedron Lett., 2018, 59, 2889 CrossRef CAS
- C. F. H. Allen and G. H. W. McKee, Org. Synth., 1939, 19, 6 CrossRef CAS
- L. C. Speight, A. K. Muthusamy, J. M. Goldberg, J. B. Warner, R. F. Wissner, T. S. Willi, B. F. Woodman, R. A. Mehl and E. J. Petersson, J. Am. Chem. Soc., 2013, 135, 18806 CrossRef CAS PubMed
- V. Nadaraj, S. Kalaivani and S. T. Selvi, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2006, 45, 1958 Search PubMed
- E. Zhang, X. Zhang, W. Wei, D. Wang, Y. Cai, T. Xu, M. Yan and Y. Zou, RSC Adv., 2015, 5, 5288 RSC
- M. Smilkstein, N. Sriwilaijaroen, J. X. Kelly, P. Wilairat and M. Riscoe, Antimicrob. Agents Chemother., 2004, 48, 1803 CrossRef CAS PubMed
- M. Ferrari, M. C. Fornasiero and A. M. Isetta, J. Immunol. Methods, 1990, 131, 165 CrossRef PubMed
- Y. K. Kim, S. J. Lee and K. H. Ahn, J. Org. Chem., 2000, 65, 7807 CrossRef CAS PubMed
- J. Louie, M. Driver, B. Hamann and J. Hartwig, J. Org. Chem., 1997, 62, 1268 CrossRef CAS
- W. Zhou, Y. Yang, Y. Liu and G.-J. Deng, Green Chem., 2013, 15, 76 RSC
- A. K. Sukhomlinov and V. P. Maksimets, Chem. Heterocycl. Compd., 1965, 1, 66 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C-NMR spectra of the final products. See DOI: 10.1039/c9ra09478d |
| This journal is © The Royal Society of Chemistry 2019 |