
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable and convenient synthesis of highly dispersed Fe–Nx catalysts from graphene-supported Zn–Fe-ZIF for efficient oxygen reduction in acidic media†
Limeng Yang *a and
Zhigang Shaob
*a and
Zhigang Shaob
aSchool of Textile Science and Engineering, Xi'an Polytechnic University, 19 Jinhua Road, Xi'an 710048, China. E-mail: yanglmxpu@163.com
bDalian National Laboratories for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China
First published on 19th December 2019
Abstract
The development of low-cost, efficient and stable electrocatalysts for the oxygen reduction reaction (ORR) is desirable but remains a great challenge. We report a convenient and efficient synthesis approach of highly dispersed Fe–Nx catalysts for ORR. Typically, Fe–Zn-ZIF (zeolitic imidazolate frameworks) nanocrystals cast as precursor and graphene as supports, highly dispersed Fe–Nx species were fabricated with PVP (polyvinyl pyrrolidone) as surfactant via pyrolysis. With the help of graphene and surfactant, the agglomeration of iron particles has been avoided during pyrolysis, and the size and morphology of ZIF particles intercalating into the graphene layers can be regulated precisely as well. The amount of Fe–Nx active sites in C-rGO-ZIF catalyst arrived 4.29%, which is obviously higher than most monodispersed non-precious metal catalysts reported. The obtained C-rGO-ZIF catalyst exhibits a high onset potential of 0.89 V and a half-wave potential of 0.77 V, which is only 30 mV away from Pt/C in acidic media. The active sites of the catalyst was characterized and found to be the highly dispersed Fe–Nx species, large and accessible specific surface area of graphene and abundant active nitrogen atoms. When the C-rGO-ZIF catalyst was applied in the cathode of fuel cell, the power density can reach up to 301 mW cm−2, which highlights a practical application potential on small power supplies.
Introduction
Fuel cells (FCs) are considered as one of the most promising power source candidates. Nevertheless, catalysts at both the anode and cathode of fuel cells are generally based on platinum (Pt) or platinum alloys. The oxygen reduction reaction (ORR) which requires much more Pt than the faster anode hydrogen oxidation is a sluggish and vital reaction happening at the cathode. By virtue of the price of Pt, it's pivotal to design and develop novel superior non-precious metal catalysts (NPMCs) with outstanding activity and durability to replace platinum at fuel cell cathodes.1In the midst of the NPMCs, transition metal doped carbon-supported nitrogen materials (M–Nx/C) are the most promising,2–4 especially Fe–Nx/C catalysts.5–7 There is a point that catalyst's electrocatalytic activity results from the entrapment of metal sites in carbon matrix.8,9 And metal–organic frameworks (MOFs), thanks to their well-defined porous structures and regular arrangement of metal nodes and heteroatoms, are viewed as a brilliant precursor to synthesize new forms of M–Nx/C materials.10–12 While there are still defects obstructing the performance of MOF derived cathode electrocatalysts. One of the defects is the devastation of morphology during carbonization.13 Specifically, the collapse of MOF and the agglomeration of conductive carbon caused a huge surface area decline of MOF precursor.14 In addition, the metal species inevitably agglomerated during calcination which resulted in the lessening of active sites density.
To solve the problem, MOF derived NPMCs are widely prepared into single-atoms or monodispersed recently. In the past three years, MOF derived Co single-atom catalyst, Fe single-atom and Co–Fe double sites catalysts, etc, are developing rapidly.15–17 Combining double solvents methods,16 gas-migration strategy18 or space isolation agent together with high temperature pyrolysis, the collapse of MOF and the agglomeration of the metal species during carbonization can be perfectly prohibited. Li's group applied pyrrole and thiophene copolymer as support via impregnation and thermal reduction method obtaining Fe single atoms in S/N co-doped carbon catalyst in alkaline medium.19 However, the synthesis routes of S/N co-doped support is complex and its conductivity of S/N co-doped carbon could be improved. Moreover, the increase in amount of iron-containing active sites is still a vital challenge.
Herein, we designed a highly dispersed Fe–Nx catalyst derived from electrical conductive graphene supported Fe–Zn-ZIF. With the supports of high conductive graphene, the oxygen reduction electrocatalytic activity of as prepared C-rGO-ZIF catalyst raised a lot. Along with the surfactant polyvinyl pyrrolidone (PVP), it can not only improve the dispersion of ZIFs in graphene oxides solution, but also can achieve the highly dispersion of iron-containing active sites through regulating the particle size of ZIFs. This work innovatively constructed a simple and convenient method to tunably prepare highly dispersed Fe–Nx catalyst with Fe–Zn-ZIF, graphene supports and PVP surfactant. The obtained NPMC shows a half-wave potential of 0.77 V, which is lower than Pt/C by only 30 mV in the acidic medium. The C-rGO-ZIF catalyst further demonstrated optimistic electrocatalytic in a proton exchange membrane fuel cell with a peak power density of 301 mW cm−2. The ORR active sites were characterized and found to be the large and accessible specific surface area of graphene, abundant pyridine-/graphite-N content and highly dispersed Fe–Nx species. Thus, the Fe–Nx catalyst with PVP surfactant assisted and graphene supported would open a new avenue to design high performance NPMC for vital applications in proton exchange membrane fuel cells.
Experimental
Material synthesis
Transmission electron microscopy (TEM) was carried out on JEM-2100 at 120 kV.
Powder X-ray diffraction (XRD) were conducted on a D/MAX2500VB2/PC with a 2θ range from 5° to 90° at a scan rate of 5 °min−1.
The electronic structure and surface species were identified by X-ray photoelectron spectroscopy (XPS) (ESCA Lab250 Xi, Thermo Scientific).
Through inductively coupled plasma atomic emission spectrometry (ICP-AES, PerkinElmer 7300DV), the metal content in the electrocatalysts was determined. Thermogravimetric analysis (TGA) were conducted on a Netzsch-STA 449 F3 system at a heating speed of 10 °C min−1 at a range of 40–1000 °C in N2.
Aiming at exploring the porous features of the catalysts, nitrogen sorption measurements were prceeded on Quantachrome Autosorb-IQ gas adsorption analyzer at 77 K. The catalysts were degassed before tests at 150 °C for 5 h under dynamic vacuum. The pore size distributions of samples were fitted from Quenched Solid Density Functional Theory (QSDFT) method and the BET surface areas were calculated from the adsorption branches.
57Fe Mössbauer spectra were measured at room temperature (295 K) using a 57Co source encapsulated in a rhodium matrix and a constant accelerator spectrometer. The isomer shifts were referenced at test temperature relative to that of α-Fe. At least 100 mg catalyst powder was pressed into pill-shape of ca. 15 mm-diameter and 2 mm-thick.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:9 (v/v/v) to get 2 mg mL−1 ink under sonication. Then the homogenously dispersed ink was syringed onto the disk of the working electrode (0.19625 cm2), which was then left to dry in air under an infrared heat lamp. The procedure was repeated to obtain a catalyst loading of ca. 400 μg cm−2 finally. In contrast, the commercial Pt/C (20 wt%, Johnson Matthey) loading was set to 20 μgPt cm−2. The electrochemical potential in this work was referenced to reversible hydrogen electrode (RHE). The calibrated ΔE was −0.308 V (vs. SCE) and −0.962 V (vs. Ag/AgCl) in Fig. S1.† The cyclic voltammetry (CV) curves were attained after purging N2 or O2 for at least 30 min. The loaded electrode was subjected to cycling scan between 0.05 and 1.2 V (vs. RHE) at 0.1 V s−1 until steady voltammograms were gained. If there isn't denoted particularly in this paper, all potentials are vs. RHE.
:1:9 (v/v/v) to get 2 mg mL−1 ink under sonication. Then the homogenously dispersed ink was syringed onto the disk of the working electrode (0.19625 cm2), which was then left to dry in air under an infrared heat lamp. The procedure was repeated to obtain a catalyst loading of ca. 400 μg cm−2 finally. In contrast, the commercial Pt/C (20 wt%, Johnson Matthey) loading was set to 20 μgPt cm−2. The electrochemical potential in this work was referenced to reversible hydrogen electrode (RHE). The calibrated ΔE was −0.308 V (vs. SCE) and −0.962 V (vs. Ag/AgCl) in Fig. S1.† The cyclic voltammetry (CV) curves were attained after purging N2 or O2 for at least 30 min. The loaded electrode was subjected to cycling scan between 0.05 and 1.2 V (vs. RHE) at 0.1 V s−1 until steady voltammograms were gained. If there isn't denoted particularly in this paper, all potentials are vs. RHE.The background capacitive currents were test in N2-saturated electrolyte at a scan rate of 10 mV s−1 from 0.2 to 1.2 V in rotating disk electrode (RDE) tests. Then linear sweep voltammograms (LSV) were tested at an electrode rotation rate of 1600 rpm in O2-saturated electrolyte. The final O2 reduction current was calibrated by the background capacitive current.
The ring potential was controlled at 1.150 V in HClO4 to oxidize the H2O2 produced on the disk electrode during ORR in rotating ring-disk electrode (RRDE, PINE AFE7R9GCPT) tests. The hydrogen peroxide yield (% H2O2) and the number of transferred electron (n) were calculated using equations below:
 | (1) |
 | (2) |
During the accelerated durability test (ADT), the electrodes were cycled with a scan speed of 0.1 V s−1 between 0.6 and 1.0 V for 10000 cycles in O2 saturated KOH or 5000 cycles in O2 saturated HClO4.
Results and discussion
Optimization of ZIF particle size and amount of graphene supports
First, we attained different sizes of Zn–Fe-ZIF rhombic dodecahedral nanoparticles by modulating the volume of methanol according to the literature.20 It can be found in Fig. 1 that the particle size (marked as the diagonal length of the hexagonal) of the C–Zn–Fe-ZIF-1 is 640 nm, and the average particle size of the C–Zn–Fe-ZIF-2 is 270 nm. It can be seen that the particle size of C–Zn–Fe-ZIF-3 is obviously lower than C–Zn–Fe-ZIF-1 and C–Zn–Fe-ZIF-2. To clarify the particle size of C–Zn–Fe-ZIF-3, the size distribution of C–Zn–Fe-ZIF-3 particles was measured. In the inset of Fig. 2e, it can be found that the particle size distribution of C–Zn–Fe-ZIF-3 is relatively concentrated. Its average particle size is 43 nm. In Fig. 2b, compared with the other two smaller particle samples, there exists a small amount of ferrous metal particles in the larger C–Zn–Fe-ZIF-1 particles. The amplification of it is performed by TEM in the inset of Fig. 2b. The lattice spacing of the crystal is about 0.203 nm along [110] plane of α-Fe. It can be speculated that agglomerated metallic iron particles are more likely to form in larger particle-size ZIFs due to the size-effect during pyrolysis. Considering that ZIFs will be carried on supports, C–Zn–Fe-ZIF-3 with a relatively tiny particle size (43 nm) is selected in order to avoid the agglomeration of Fe metal particles. | ||
| Fig. 1 Synthesis schematic of Zn–Fe-ZIF derived catalyst. | ||
 | ||
| Fig. 2 SEM and TEM images of different-particle-size C–Zn–Fe-ZIF particles: (a and b) C–Zn–Fe-ZIF-1; (c and d) C–Zn–Fe-ZIF-2; (e and f) C–Zn–Fe-ZIF-3. | ||
Next, on the basis of preferred Zn–Fe-ZIF particle size, we investigated the influence of the amount of graphene supports on the morphology of the catalyst. As shown in Fig. 3b, it was found that the graphene appeared curled when the addition amount of graphene was low.
 | ||
| Fig. 3 SEM and TEM images of different graphene loading of C-rGO-ZIF-1# catalyst: (a and b) C-rGO-ZIF-1#; (c and d) C-rGO-ZIF-2#; (e and f) C-rGO-ZIF-3#. | ||
It has been reported that heavy metal cations such as Zn2+, Cu2+, Ag+, etc., can easily pass through the electric double layer, and combine with the surface of GO, then change the surface potential to generate curling of GO.21 During the process of synthesizing C-rGO-ZIF-3#, 2# and 1#, the total quantity of Zn2+ and Fe3+ cations is gradually increasing relative to GO, which may cause the curling of GO sheets in C-rGO-ZIF-3# and 2#. Additionally, the curled graphene couldn't support ZIFs, resulting in a decrease of supports' utilization. As the additon of graphene increased, the curling phenomenon disappeared and the ZIF distribution became uniform. Therefore, the 0.23 mg mL−1 GO concentration of C-rGO-ZIF-3# was selected to apply for C-rGO-ZIF catalysts with PVP as surfactant.
The influence of PVP on particle size of ZIFs
Since that the reactive carboxyl groups on the surface of GO are few, it is insufficient for ZIFs to nucleate. While the surfactant PVP has a negatively charged amide bond and a pyrrolidone ring which has a strong coordination with Zn2+,22 ZIFs can grow on the surface of GO uniformly. Moreover, that the PVP molecular layer on the surface of ZIF particles is believed to prevent particle agglomeration during pyrolysis. Therefore, different concentration of PVP is nominated to regulate the morphology and growth of ZIF on the surface of GO.23,24The insets in Fig. 4a, c and e are the particle size distributions of C-rGO-ZIF-1*/2*/3* catalysts, which are measured by Nano Measurer via at least 60 particles in each graph. It can be found that the ZIF particle size of the three catalysts ranges are: C-rGO-ZIF-1* 90–120 nm; C-rGO-ZIF-2* 200–220 nm; C-rGO-ZIF-3* 110–150 nm. As the addition of PVP increases, the ZIF particle size shows a tendency of increase first and then decrease. From Fig. 4c and e, it can be seen that the graphene sheet of C-rGO-ZIF-2*is more prominently cleaved because of the higher crystallized ZIF particles than those of C-rGO-ZIF-3* catalyst. It indicates that PVP can regulate the ZIF particle size and morphology by the coordination and electrostatic interaction with Zn2+.23,25 The larger particle size ZIF in the C-rGO-ZIF-2* catalyst can better support the graphene sheets. The inset in Fig. 4d is a partially enlarged high-resolution TEM image of the graphene layer in Fig. 4c. It rarely can be seen the aggregated metal particles implanted in the folded graphene layers. Consequently, it can be concluded that metal species of C-rGO-ZIF-2* catalyst is mainly dispersed in atoms or clusters.
 | ||
| Fig. 4 SEM and TEM images of C-rGO-ZIF catalysts with different PVP additions: (a and b) C-rGO-ZIF-1*; (c and d) C-rGO-ZIF-2*; (e and f) C-rGO-ZIF-3*. | ||
Structure characterization
In order to investigate the phase species and porous features of C-rGO-ZIF catalysts with different PVP addition, XRD and nitrogen isothermal adsorption–desorption measurements were executed. First, the phase features of different Zn–Fe-ZIF precursors and C-rGO-ZIF catalysts were investigated. Fig. 5a shows the X-ray patterns of Zn–Fe-ZIF-1, 2 and 3 precursors. It can be seen that the diffraction peaks of them are basically unchanged, indicating the size of the ZIF crystals has no effect on the position and intensity of the diffraction peak. Moreover, the XRD peaks of Zn–Fe-ZIF synthesized here are consistent with those in the literature,20 demonstrating that Zn–Fe-ZIF crystals have been successfully prepared. Fig. 5b displays the XRD patterns of C-rGO-ZIF with different PVP addition amounts and C–Zn–Fe-ZIF-3 catalyst without the addition of GO. The diffraction peaks at 25 and 44° in Fig. 5b can be attributed to the (002) and (101) facet of disordered carbon according to JCPDS card no. 00-034-0567. From the diffraction peaks of C–Zn–Fe-ZIF-3 and three C-rGO-ZIF catalysts in Fig. 5b, it can be found that the peak density of disordered carbon in C–Zn–Fe-ZIF-3 is stronger due to the absence of graphene. Thus it can be concluded that the addition of graphene can increase the order degree of carbon in the catalyst. The peak standing for (002) facet of disordered carbon in C–Zn–Fe-ZIF-3 has a negative shift with respect to 25°. It might be ascribed to the lattice expansion of carbon thanks to the doping of nitrogen into the carbon matrix. Although the addition amount of PVP in synthesizing the C-rGO-ZIF-1*, 2* and 3* catalysts is gradually increasing, the diffraction peaks of them are basically the same in Fig. 5b. It's probably because of the ultralow Fe content in the catalysts. The diffraction peak at 44.6° in Fig. 5b can be assigned to α-Fe according to JCPDS card no. 03-056-4899.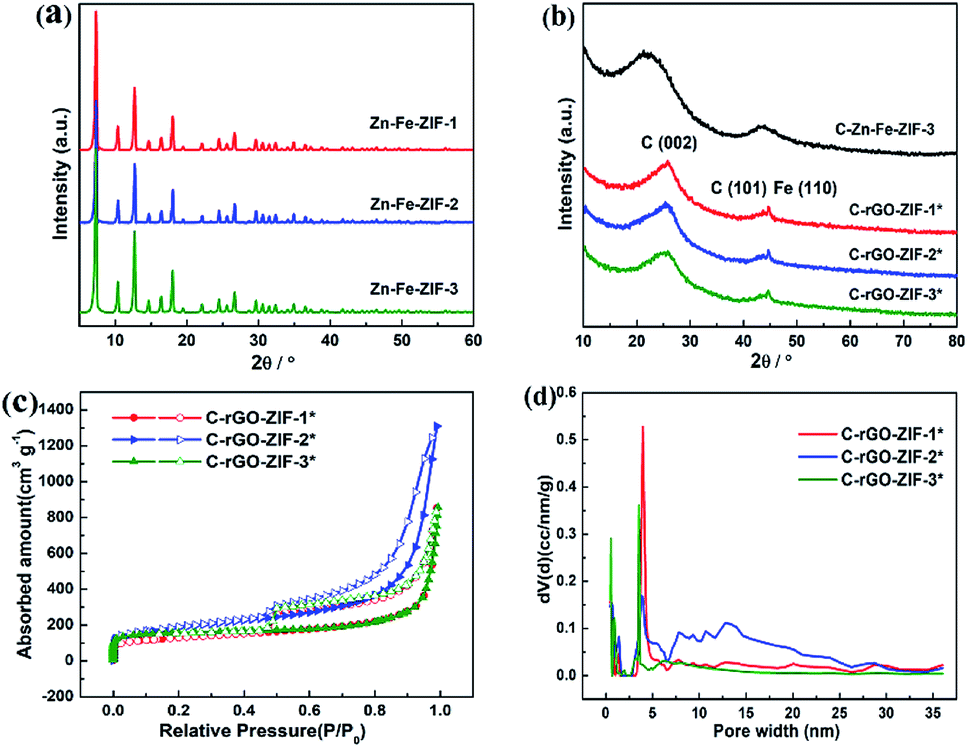 | ||
| Fig. 5 The XRD pattern of the (a) Zn–Fe-ZIF precursor with different sizes and (b) different C-rGO-ZIF catalysts. Porous feature of different C-rGO-ZIF catalysts; (c) N2 sorption isotherms (closed, adsorption; open, desorption); (d) NLDFT pore size distribution. | ||
Next, nitrogen isothermal adsorption–desorption measurements were applied to examine the porous features of the catalysts. It can be seen from Table S1† that the C-rGO-ZIF-2* catalyst has a large BET specific surface area of 650 m2 g−1, while the specific surface area of C-rGO-ZIF-1* and C-rGO-ZIF-3* catalysts is similar. Furthermore, the total pore volume 2.03 cm3 g−1 of the C-rGO-ZIF-2* catalyst is also the highest. As the pore size distribution of the three C-rGO-ZIF catalysts in Fig. 5d shown, there are more pores distributed above 5 nm in C-rGO-ZIF-2* than the other two catalysts which leads to the largest pore volume of C-rGO-ZIF-2*. The better porous features of C-rGO-ZIF-2* may stem from the more prominently cleaved graphene sheet as shown in Fig. 4c. As we all known, ORR occurs on the surface and inside the pores of catalyst particles, so the higher pore volume and BET specific surface area of C-rGO-ZIF-2* indicate a higher catalytic activity.
To investigate the element contents of C-rGO-ZIF catalysts, XPS and ICP-OES were conducted. The relative mass contents of iron in C-rGO-ZIF-1*, 2*, 3* and C–Zn–Fe-ZIF-3 samples were 2.72%, 4.29%, 2.67% and 13.31% characterized by ICP-OES. Because of the adding of graphene supports, the Fe content of all C-rGO-ZIF catalysts is much lower than that of C–Zn–Fe-ZIF-3. Besides, as the relative contents of different elements in each catalyst shown in Table S2,† the quantity of nitrogen in the C-rGO-ZIF-2* is the highest among all catalysts via XPS. Perhaps that the higher crystallized ZIF particles on graphene sheets of C-rGO-ZIF-2* with a larger ZIF particle size lead to a higher iron content compared with other C-rGO-ZIF catalysts. In Fe–N/C catalysts, the nitrogen atom is n-type doped in carbon, thus resulting in the formation of a disordered carbon phase (Fig. 5b).26 Furthermore, the nitrogen atom can give electrons to the bonding orbital in carbon matrix, which can in turn increase its feedback electrons to the π* orbital of the oxygen molecule, causing the splitting of O–O bond.27 Hence, a relatively high doping amount of nitrogen can increase the catalytic activity of ORR to some extent. According to the results in Table S2,† the C-rGO-ZIF-2* catalyst has a higher nitrogen content than other C-rGO-ZIF catalysts, consequently it may deserve a better catalytic performance. The result of C-rGO-ZIF-2*'s highest nitrogen content is also consistent with that of ICP. As well, comparing the contents of Zn in the C-rGO-ZIF catalysts, Zn–Fe-ZIF-3 and the carbonized C–Zn–Fe-ZIF-3, it can be found that the Zn in three C-rGO-ZIF catalysts couldn't be detected. It indicates that the Zn in Zn–Fe-ZIF-3 precursor is completely volatilized during pyrolysis. The volatilized Zn could increase the specific surface area and act as pore forming agent moderately.28
Through XPS characterization, the chemical states of nitrogen in the catalysts were analyzed. Four states of N deconvoluted from N 1s XPS spectra are shown in Fig. 6.29 The peaks at 398.6, 399.0, 400.4, 401.2 and 403.2 eV, which are assigned to the presence of pyridinic-N, Fe–N, pyrrolic-N, graphitic-N and oxidized-N, respectively. Pyridinic and graphitic nitrogen are considered to behave the largest positive effect to ORR catalytic sites.30–32 The relative contents of N elements of different valence types in the C-rGO-ZIF catalysts with three different PVP additions are shown in Table S3.† It can be seen that the N types content of different C-rGO-ZIF catalysts varies. The C-rGO-ZIF-2* obtains the highest total content of pyridinic and graphitic nitrogen. The C-rGO-ZIF-1* catalyst gains a slightly lower total content, and the C-rGO-ZIF-3* deserves the lowest. At the same time, according to Table S3,† it can be concluded that C-rGO-ZIF-2* also obtains the highest Fe–N species content and the Fe–N species content of C-rGO-ZIF-1*and 3* is faintly lower than that of C-rGO-ZIF-2* catalyst. Hence among the three C-rGO-ZIF catalysts, the C-rGO-ZIF-2* catalyst predicts the highest ORR activity. Combined with the electron microscopy and XPS results, it can be demonstrated that the C-rGO-ZIF-2* catalyst may have highly dispersed Fe–Nx active sites. The binding energies between 705 and 735 eV were assigned to Fe 2p peak deconvolution. It is shown in the XPS Fe 2p spectrum of MIL-5 DCD-800 that the peaks centered at 711.2 and 723.8 eV represent Fe(II) 2p3/2 and 2p1/2, respectively, and no Fe(II) peak is observed.33 As for the signal at around 711 eV, it can be assigned to Fe in Fe–Nx configuration.5,34
 | ||
| Fig. 6 High-resolution XPS spectra of N 1s of (a) C-rGO-ZIF-1*, (c) C-rGO-ZIF-2*, (e) C-rGO-ZIF-3* and Fe 2p of (b) C-rGO-ZIF-1*, (d) C-rGO-ZIF-2*, (f) C-rGO-ZIF-3* catalysts. | ||
So as to study the valence and chemical environment of iron in detail, the 57Fe-Mossbauer spectroscopy characterization of C-rGO-ZIF-2* catalyst was carried out. The obtained results are shown in Fig. 7 and Table S4.† The spectrum is deconvoluted to one singlet, two doublets, and one sextet. The sextet with a magnetic field of 33.1 T in Fig. 7 represents α-Fe, which is consistent with the XRD result in Fig. 5b and the singlet with a isomer shift (IS) of −0.10 mm s−1 symbolizes γ-Fe. The isomer shift of doublet-1 is 0.20 mm s−1, and the quadrupole splitting (QS) value is 3.64 mm s−1, which correspond to the typical signatures of a low-spin state FeII–N4. The isomer shift of doublet-2 is 0.42 mm s−1, and the QS value is 1.39 mm s−1, which are the typical signatures of FeII–N4/FeII–N2+2 with a medium-spin state. According to reports,35,36 the doublets of 57Fe in the Mössbauer spectrum mainly comprise three types: doublet-1 represents the non-planar low-spin state FeII–N4 site; doublet-2 represents the moderate spin-state FeII–N4/FeII–N2+2 site with a plane structure; doublet-3 represents the N (axial)-FeIIIN4–O2 site. It is generally believed that the moderate spin-state FeII–N4/FeII–N2+2 sites symbolized by doublet-2 display the weakest ORR activity, especially in acidic media. According to Fig. 7, the Fe of FeII–Nx sites represented by the doublets in the C-rGO-ZIF-2* catalyst are both divalent, which is consistent with the XPS results in Fig. 6d. And the total relative area of the two doublets is 65.9%, indicating that the main form of Fe in C-rGO-ZIF-2* catalyst is FeII–Nx, which means that C-rGO-ZIF-2* contains highly dispersed FeII–Nx sites. The amount of Fe–Nx active sites in C-rGO-ZIF-2* arrived 4.29%, which is obviously higher than most single-atoms or monodispersed NPMCs reported.2,15,16 It demonstrated this work may provide a new viewpoint for the design and preparation of single-atoms or monodispersed NPMCs.
 | ||
| Fig. 7 Experimental 57Fe Mössbauer transmission spectra measured for C-rGO-ZIF-2* and their fittings. | ||
Performance characterization of ORR
To evaluate the effect of PVP addition on the oxygen reduction performance of the catalyst, we investigated the activity of C-rGO-ZIF catalysts with different PVP additions in acidic medium, as shown in Fig. 8a. It can be seen that as the addition amount of PVP increases, the performance of C-rGO-ZIF-1*, 2* and 3* catalysts exists a greatest one. The catalytic activity of C-rGO-ZIF-2* was the highest with a PVP concentration of 1.157 mg mL−1. The onset and half-wave potential was 0.86 V and 0.77 V, and the half-wave potential was only 30 mV lower than the commercial Pt/C catalyst. The superior activity in an acidic medium may be thanks to the non-planar low-spin FeII–N4 site and the possible better mass transfer derived from the more prominently cleaved graphene sheet. It demonstrates the importance of increasing the active site density when designing non-precious metal catalysts. Then, the activity of the C–Zn–Fe-ZIF-3 contrast with same Fe loading was also examined, as shown in Fig. 8b. The half-wave potential of C–Zn–Fe-ZIF-3 is only 0.70 V, i.e. the ORR activity of the C-rGO-ZIF-2* catalyst is much higher than that of the same C–Zn–Fe-ZIF-3 catalyst only without graphene. It indicates that the construction of supports such as the addition of conductive and dispersed graphene, play an important role in the improvement of the catalytic activity. To benchmark our work against state of the art catalysts, ORR performance of NPMCs tested in acid has been summarized in Table S5.† It can be concluded that the onset and half-wave potential of most reported catalysts can't rival with those of C-rGO-ZIF-2* in acidic media. It illustrates that the dispersed Fe–Nx, large, high specific surface area of graphene and active nitrogen atoms actually play an important role in enhancing the ORR property of NPMCs in half-cell tests. | ||
| Fig. 8 RDE polarization curves in O2 saturated 0.1 M HClO4 of (a) different C-rGO-ZIF electrocatalysts and (b) C-rGO-ZIF-2* and C–Zn–Fe-ZIF-3 catalysts sample set. (c) RRDE polarization curves of C-rGO-ZIF-2* and 20% Pt/C catalyst in 0.1 M HClO4. (d) % H2O2 and transferred electron number (n) during ORR process in O2 saturated 0.1 M HClO4. (e) Chronoamperometric response of C-rGO-ZIF-2* and 20% Pt/C at 1600 rpm. (f) The arrow indicates the introduction of 5 vol% methanol. | ||
With the aim of recognizing the transferred electron number (n), an RRDE test was performed on the catalyst. The upper part of Fig. 8c plots is the ring current (Ir) over applied potential when the lower part plots displays the disk current density (jd). The platinum ring can collect H2O2 produced during ORR, thus Ir is closely related with the hydrogen peroxide yield (% H2O2) which can be calculated from eqn (1). In Fig. 8c, both the jd curve and the Ir curve of C-rGO-ZIF-2* are comparable to the curves of Pt/C. It predicts a similar low H2O2 yield below 5% and the calculated number of electron transfer reaches up to 3.8, which is close to 4. Therefore, the ORR on C-rGO-ZIF-2* is governed by a primarily direct 4 e− process same with commercial Pt/C.
The electroactivity degradation was evaluated by an accelerated decay test (ADT) of a potentiostatic sweep as shown in Fig. 8e. The catalytic stability of C-rGO-ZIF-2* and 20% Pt/C was tested in an oxygen saturated HClO4 solution at a given 0.8 V constant potential for 20 h. As Fig. 8e shows, the current density of C-rGO-ZIF-2* is only reduced by 39% after 20 h of constant potential scanning, while the Pt/C catalyst is decreased by 61%. It can be confirmed that the stability of C-rGO-ZIF-2* is higher than Pt/C catalyst.37 The reason of the higher electrochemical stability than 20% Pt/C may be owing to the likely protective effect of the supporting graphene layers on the active sites. The anti-methanol test was carried out by injecting 5 vol% methanol into electrolyte under chronoamperometric technique. Fig. 8f shows that the C-rGO-ZIF-2* catalyst exhibits excellent resistance to methanol in acids.
The C-rGO-ZIF-2* catalyst synthesized in this work was applied on cathode of MEA in fuel cell. The mass ratio of resin to cathode catalyst was 0.5, and the I–V polarization curve and power density curve with different catalyst loading was investigated. As shown in Fig. 9a, as the catalyst loading increases, the open circuit potential (OCP) and the highest power density of the fuel cell also increases gradually. When the cathode catalyst loading is 4 mg cm−2, the fuel cell power density can reach up to 301 mW cm−2. While the peak power density of MEA-2 and MEA-1 is only 162 mW cm−2 and 77 mW cm−2, respectively. The open circuit voltages of MEA-3, MEA-2 and MEA-1 are 0.75 V, 0.67 V and 0.53 V, separately. The OCP increases with the cathode catalyst loading probably because an increase of the number of effective active sites. Besides, when the catalyst loading increased to 6 mg cm−2, the fuel cell performance of MEA-4 such as the open circuit voltage and peak power density began to decline. The relatively inferior OPC of MEA-4 which is below 0.45 V may result from a high internal resistance of the cathodic catalyst layer. Through the optimization of the catalyst loading on fuel cell, it can be verified that the full cell performance of the non-precious metal catalyst is greatly affected by the catalyst loading. Fig. 9b shows the I–V and power density curves for C-rGO-ZIF-2* and Pt/C as the cathodic catalyst. The fuel cell power density of Pt/C exhibits a peak of 1.137 W cm−2. Accordingly, there is still a gap of the fuel cell performance between the commercial Pt/C catalyst and the home-made C-rGO-ZIF-2* catalyst. To benchmark the PEMFC performance of our NPMC against state of the art catalysts, the PEMFCs performance of different reported NPMCs has been listed in Table S6.† It can be found that the maximum power density of home-made C-rGO-ZIF-2* is apparently lower than those of other NPMCs under the same operating conditions, such as catalyst loading, gas back pressure and flow rates. This indicates that the full cell performance of the C-rGO-ZIF-2* catalyst is moderate among state of the arts. The unsatisfactory PEMFC performance may derived from the low quantity of active Fe–Nx species. The optimistic fuel cell performance of the C-rGO-ZIF-2* catalyst on small power density may highlight a practical application potential on small power supplies, such as uninterruptible power systems (UPS).
 | ||
| Fig. 9 The I–V and power density curves for (a) C-rGO-ZIF-2* with different catalyst loading and (b) Pt/C and C-rGO-ZIF-2* with a 4 mg cm−2 loading. (Nafion 211, H2/O2, 1.8 bar, 80 °C.) | ||
Conclusions
In summary, a tunable and convenient synthesis approach of highly dispersed Fe–Nx catalysts for ORR was reported. With the help of graphene and surfactant PVP, the agglomeration of iron particles has been avoided during pyrolysis, and the size and morphology of ZIF particles intercalating into the graphene layers can been regulated precisely as well. The obtained C-rGO-ZIF catalyst showed a respectable ORR performance in acid media, offering comparable onset and half-wave potentials. The ORR active site of the catalyst was characterized and the C-rGO-ZIF catalyst further demonstrated positive electrocatalytic in a proton exchange membrane fuel cell. This general synthetic strategy creates avenues for the rational design of high performance NPMCs for efficient energy conversion and applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Key Research and Development Program of China (No. 2016YFB0101208), the Key Program of National Natural Science Foundations of China (No. 21436003, No. U1508202).Notes and references
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS.
- Z. Yang, B. Chen, W. Chen, Y. Qu, F. Zhou, C. Zhao, Q. Xu, Q. Zhang, X. Duan and Y. Wu, Nat. Commun., 2019, 10, 3734 CrossRef.
- E. Luo, H. Zhang, X. Wang, L. Gao, L. Gong, T. Zhao, Z. Jin, J. Ge, Z. Jiang, C. Liu and W. Xing, Angew. Chem., Int. Ed. Engl., 2019, 58, 12469–12475 CrossRef CAS.
- X. Wan, X. F. Liu, Y. C. Li, R. H. Yu, L. R. Zheng, W. S. Yan, H. Wang, M. Xu and J. L. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- W.-J. Jiang, L. Gu, L. Li, Y. Zhang, X. Zhang, L.-J. Zhang, J.-Q. Wang, J.-S. Hu, Z. Wei and L.-J. Wan, J. Am. Chem. Soc., 2016, 138, 3570–3578 CrossRef CAS PubMed.
- H. Wang, F. X. Yin, N. Liu, R. H. Kou, X. B. He, C. J. Sun, B. H. Chen, D. J. Liu and H. Q. Yin, Adv. Funct. Mater., 2019, 29, 1901531 CrossRef.
- X. Fu, N. Li, B. Ren, G. Jiang, Y. Liu, F. M. Hassan, D. Su, J. Zhu, L. Yang, Z. Bai, Z. P. Cano, A. Yu and Z. Chen, Adv. Energy Mater., 2019, 9, 1803737 CrossRef.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS.
- M. Lefevre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS.
- L. Yang, Y. Zeng, X. Tang, D. Xu, D. Fang, H. Huang, Z. Shao and B. Yi, ChemElectroChem, 2018, 5, 3731–3740 CrossRef CAS.
- Q. Wu, J. Liang, J. D. Yi, D. L. Meng, P. C. Shi, Y. B. Huang and R. Cao, Dalton Trans., 2019, 48, 7211–7217 RSC.
- H. Yang, X. Chen, W. T. Chen, Q. Wang, N. C. Cuello, A. Nafady, A. M. Al-Enizi, G. I. N. Waterhouse, G. A. Goenaga, T. A. Zawodzinski, P. E. Kruger, J. E. Clements, J. Zhang, H. Tian, S. G. Telfer and S. Ma, ACS Nano, 2019, 13, 8087–8098 CrossRef CAS.
- W. Xia, R. Q. Zou, L. An, D. G. Xia and S. J. Guo, Energy Environ. Sci., 2015, 8, 568–576 RSC.
- L. Yang, Y. Bai, H. Zhang, J. Geng, Z. Shao and B. Yi, RSC Adv., 2017, 7, 22610–22618 RSC.
- Z. Yang, Y. Wang, M. Zhu, Z. Li, W. Chen, W. Wei, T. Yuan, Y. Qu, Q. Xu, C. Zhao, X. Wang, P. Li, Y. Li, Y. Wu and Y. Li, ACS Catal., 2019, 9, 2158–2163 CrossRef CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- S. Y. Guo, P. F. Yuan, J. A. Zhang, P. B. Jin, H. M. Sun, K. X. Lei, X. C. Pang, Q. Xu and F. Y. Cheng, Chem. Commun., 2017, 53, 9862–9865 RSC.
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang, C. Zhao, J. Wang, C. Zhao, X. Wang, F. Zhou, Z. Zhuang, Y. Wu and Y. Li, Nat. Catal., 2018, 1, 781–786 CrossRef CAS.
- Q. Li, W. Chen, H. Xiao, Y. Gong, Z. Li, L. Zheng, X. Zheng, W. Yan, W. C. Cheong, R. Shen, N. Fu, L. Gu, Z. Zhuang, C. Chen, D. Wang, Q. Peng, J. Li and Y. Li, Adv. Mater., 2018, 30, 1800588 CrossRef PubMed.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- K. Yang, B. Chen, X. Zhu and B. Xing, Environ. Sci. Technol., 2016, 50, 11066–11075 CrossRef CAS PubMed.
- H.-x. Zhong, J. Wang, Y.-w. Zhang, W.-l. Xu, W. Xing, D. Xu, Y.-f. Zhang and X.-b. Zhang, Angew. Chem., Int. Ed., 2014, 53, 14235–14239 CrossRef CAS PubMed.
- A. Liu, C. Yu, J. Lin, G. Sun, G. Xu, Y. Huang, Z. Liu and C. Tang, Mater. Res. Bull., 2019, 112, 147–153 CrossRef CAS.
- Q. T. Liu, X. F. Liu, L. R. Zheng and J. L. Shui, Angew. Chem., Int. Ed., 2018, 57, 1204–1208 CrossRef CAS PubMed.
- H.-x. Zhong, J. Wang, Y.-w. Zhang, W.-l. Xu, W. Xing, D. Xu, Y.-f. Zhang and X.-b. Zhang, Angew. Chem., Int. Ed., 2014, 53, 14235–14239 CrossRef CAS.
- C. W. B. Bezerra, L. Zhang, H. Liu, K. Lee, A. L. B. Marques, E. P. Marques, H. Wang and J. Zhang, J. Power Sources, 2007, 173, 891–908 CrossRef CAS.
- Q. Liu, H. Zhang, H. Zhong, S. Zhang and S. Chen, Electrochim. Acta, 2012, 81, 313–320 CrossRef CAS.
- B. You, N. Jiang, M. Sheng, W. S. Drisdell, J. Yano and Y. Sun, ACS Catal., 2015, 5, 7068–7076 CrossRef CAS.
- L. Zhang, Z. Su, F. Jiang, L. Yang, J. Qian, Y. Zhou, W. Li and M. Hong, Nanoscale, 2014, 6, 6590–6602 RSC.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS.
- Y. Zhao, L. Yang, S. Chen, X. Wang, Y. Ma, Q. Wu, Y. Jiang, W. Qian and Z. Hu, J. Am. Chem. Soc., 2013, 135, 1201–1204 CrossRef CAS PubMed.
- K.-H. Wu, D.-W. Wang, D.-S. Su and I. R. Gentle, Chemsuschem, 2015, 8, 2772–2788 CrossRef CAS PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, PerkinElmer Corp., 2nd edn, 1992 Search PubMed.
- R. Cao, R. Thapa, H. Kim, X. Xu, M. Gyu Kim, Q. Li, N. Park, M. Liu and J. Cho, Nat. Commun., 2013, 4, 2076 CrossRef.
- J. K. Li, S. Ghoshal, W. T. Liang, M. T. Sougrati, F. Jaouen, B. Halevi, S. McKinney, G. McCool, C. R. Ma, X. X. Yuan, Z. F. Ma, S. Mukerjee and Q. Y. Jia, Energy Environ. Sci., 2016, 9, 2418–2432 RSC.
- K. Singh, F. Razmjooei and J.-S. Yu, J. Mater. Chem. A, 2017, 5, 20095–20119 RSC.
- X. Yuan, X. Zeng, H.-J. Zhang, Z.-F. Ma and C.-Y. Wang, J. Am. Chem. Soc., 2010, 132, 1754–1755 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08867a |
| This journal is © The Royal Society of Chemistry 2019 |