
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of pyrrolo[3′,2′:4,5][1,3]diazepino[2,1,7-cd]pyrrolizine derivatives from dicyanovinylene-bis(meso-aryl)dipyrrin†
Ji-Young Shin *
*
Department of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: jyshin@chembio.nagoya-u.ac.jp; Fax: +81-52-747-6771
First published on 4th December 2019
Abstract
Pyrrolo[3′,2′:4,5][1,3]diazepino[2,1,7-cd]pyrrolizine derivatives 2 and 3 were synthesized from dicyanovinylene-bis(meso-aryl)dipyrrin in the presence of either BF3·OEt2 or InBr3, where 2 was readily oxidized in aerobic conditions to be 3. It was understood that the fully elongated π-conjugation of 3 is achieved via the conformation of 2. Crystal structures of 2 and 3 were elucidated by X-ray diffraction analysis. Furthermore, two redox states, 3ox and 3red were observed in the chemical redox processes.
Introduction
Dipyrrins are useful molecular units to produce neutral metal-chelating complexes and also enables pigments to build up enlarged π-electron conjugations.1 Bisdipyrrins, two-dipyrrin-linked molecules, have been employed as appropriate building blocks to form assembling molecules by non-covalent bonding interfaces like metal-chelating and hydrogen bonding interactions, whose subsequent assemblies often stimulated the formation of structures, such as helixes, zigzag ribbons, 2D sheets, and tunnels.2 There is ongoing research about how solvent molecules or gas-phase molecules act as guest molecules within the assembled structures. Furthermore, some of the structures have been promoted to devise materials for ion/proton channels due to their efficient electrochemical behaviors.3 Bisdipyrrins have been also induced in enlarged π-conjugation molecules like expanded porphyrinoids exhibiting useful unique macrocyclic skeletons, whose molecular functions are capable of a myriad of practical applications such as in chemosensing4 and for medicinal sciences.5Unique DDQ-adducts are formed in the oxidation of dipyrromethanes containing electron-withdrawing meso-aryls, with DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone)6 and further treatment of the DDQ adducts with Lewis base results in compound 1 (molecular structure in Fig. 1), which was previously reported.7 It has been reported that the NiII-metallation of dicyanovinylene-bis(meso-aryl)dipyrrin 1 under refluxing toluene conditions results in the formation of a bicyclic pyrrolizine ring which affords two isomeric bisNiII expanded porphyrinoid complexes (reaction shown in Fig. 1).8 It was deliberated that the NiII acetate functions as a Lewis acid and a metalizing source in the reaction process. Furthermore, bisNiII complexes have exhibited routine diatropic ring currents of aromatic expanded porphyrinoids. Since then the creation of the macrocycle was associated with the formation of the pyrrolizine platform followed by metal chelation, the uses of nontransition metallic Lewis acids were considered for forming free-base expanded porphyrinoids. A semi-metallic Lewis acid, BF3·OEt2 that has often been used as a convenient Lewis-acid, was chosen first. The structure of the resulting product was elucidated by X-ray diffraction analysis, where the formation of a sizeable macrocyclic structure was not found. In contrast, a ring-closing to create pyrrolodiazepinopyrrolizine group arose by the nucleophilic displacement of pyrrole-βH by its neighbouring imine.
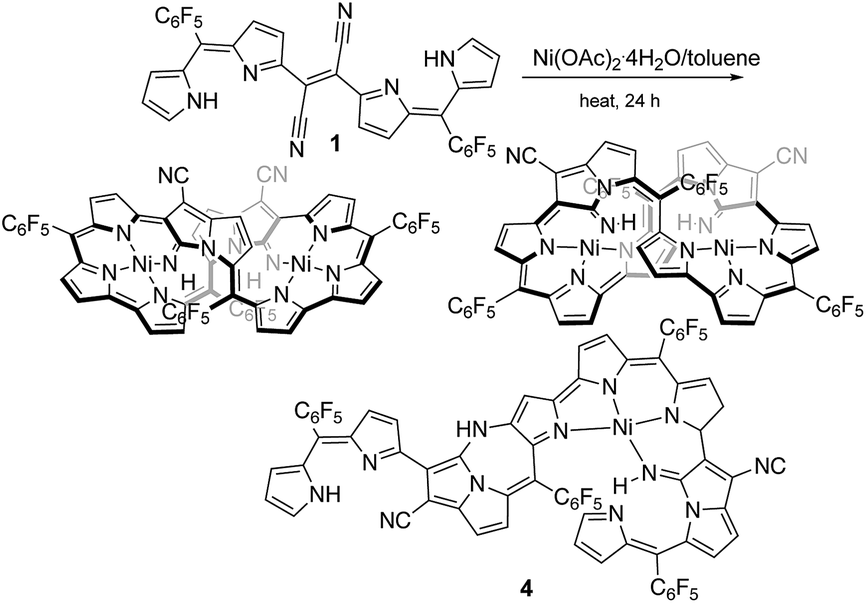 | ||
| Fig. 1 Formation of stereoisomers of expanded porphyrinoid bisNiII complexes from 1 and a minor product 4. | ||
Furthermore, a precursor of the final product compound 2 was obtained when a basic metal Lewis acid, InBr3 replaced the Lewis acid (BF3·OEt2), which contains a dipyrromethane segment instead of dipyrrin. The molecular structure was also elucidated by X-ray diffraction analysis. The details are reported herein.
Results and discussion
The previous reaction rationalized the formation of expanded porphyrinoid bisNiII complexes (Fig. 1), where a by-product was isolated in an ignorable amount.5 Nonetheless, the assignment of the molecular structure was successful by crystallographic analysis, which coincided with the structure of compound 4. From the structure, it was deduced that the generation of pyrrolodiazepinopyrrolizine groups results in incomplete macrocycles. The advanced condensation and metallation were not conducted in the presence of pyrrolo-diazepinopyrrolizine group whose formation drove the two terminal pyrroles apart. Competition between the formations of pyrrolodiazepino-pyrrolizine and the large macrocycles happens simultaneously in the synthesis, which probably was the primary reason for the formation of the products. Alternating metal chelation was the other essential factor, which prevented the formation of the unlikely fused rings. In the reaction of 1 with BF3·OEt2, compound 3 (Scheme 1) was obtained as almost the quantitative product of the reaction. Both compounds 3 and 4 resulted from the formation of the fused rings (Fig. 2 and S12†).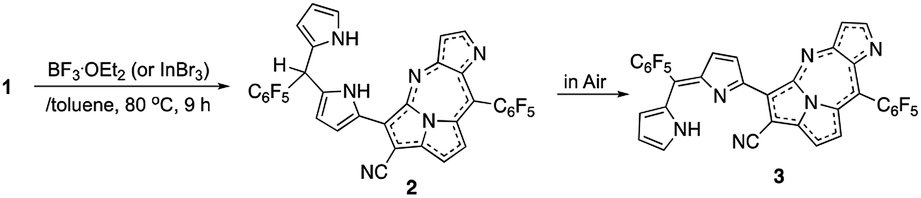 | ||
| Scheme 1 Formation of 3 from 1 in the presence of selected Lewis acids (BF3·OEt2 and InBr3). | ||
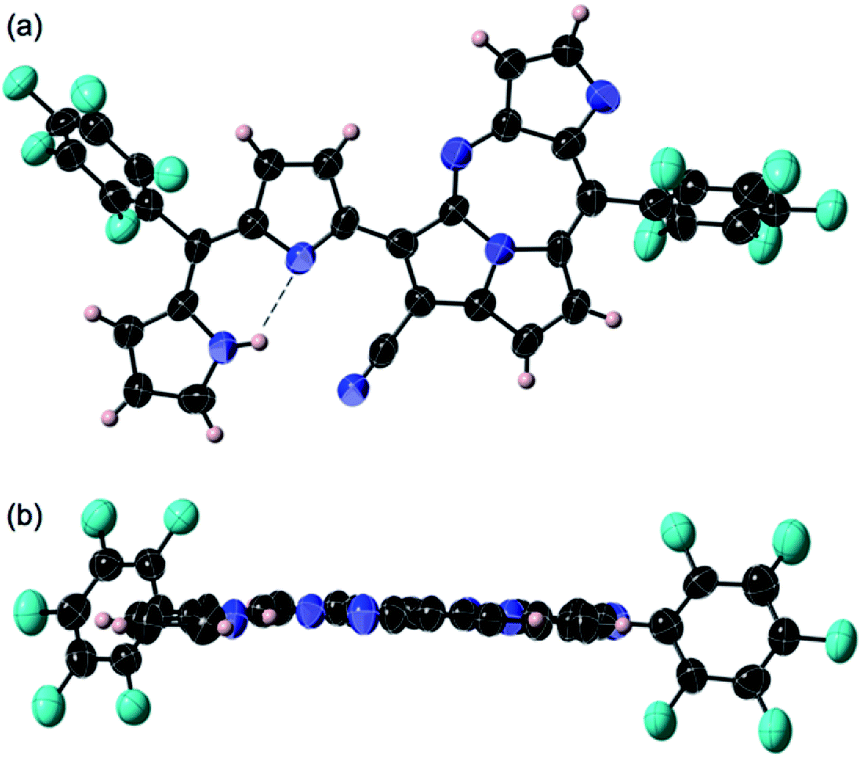 | ||
| Fig. 2 Crystal structure of 3: top view (a) and side views (b). Thermal ellipsoids are scaled at the 50% probability level. Two molecules having slightly different planarity and bond alternations were found in the unit and each pair of the enantiomers was observed in the unit cell. | ||
BF3·OEt2 was added to a solution of 1 in a mixed medium of toluene and THF to manage the solubility of both the Lewis acid and 1. The solution was then refluxed for 1 h under N2 and then cooled down. Blue metallic solids appeared during the refluxing and the formation of solids completed in an hour. The solution colour almost disappeared with the appearance of the precipitates. The liquid phase was removed by decantation to separate the metallic solids. The solids were then dissolved in a co-solvent of MeOH and CH2Cl2. Column chromatography on silica gel gave a polar, blue colour fraction as the major product of the reaction. The growth of the single crystal was successful in a CH2Cl2 solution of 3 with a reversible vapour diffusion of CH2Cl2 and MeOH. The structure of 3 was elucidated by X-ray diffraction analysis, which exhibited one amino-hydrogen providing an intramolecular hydrogen-bond (Fig. 2). Furthermore, compound 2 was isolated in a reasonable amount (ca. 30% yield) succeeded in the reaction proceeding with three molar equivalent InBr3 and the growth of single crystals for X-ray diffraction analysis (Fig. 3).
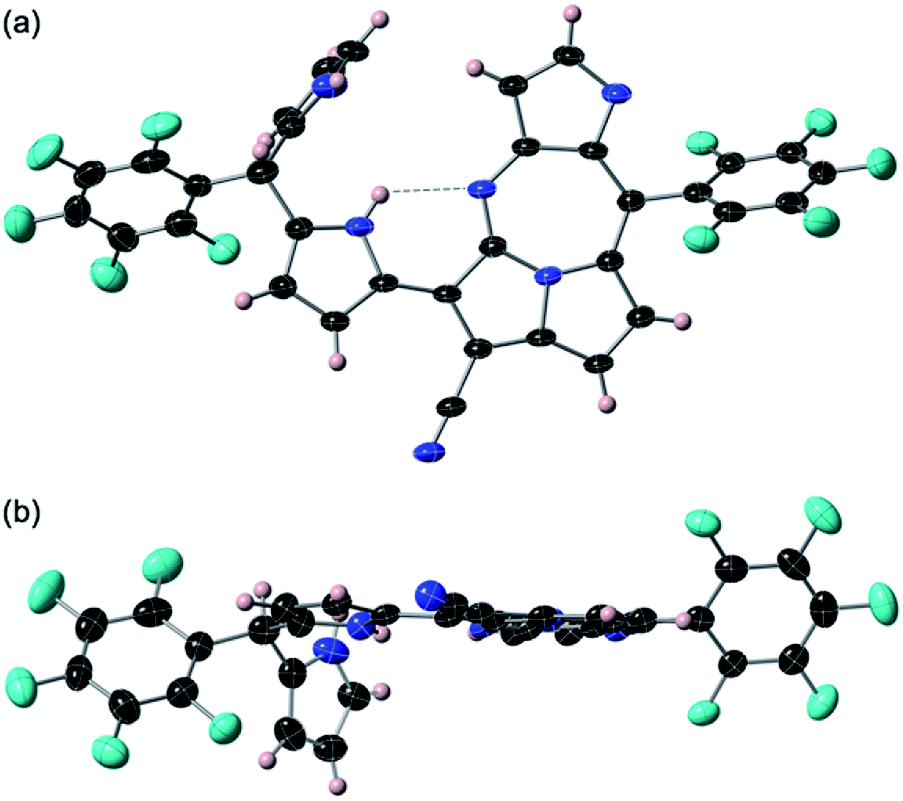 | ||
| Fig. 3 Crystal structure of 2: top view (a) and side views (b). Thermal ellipsoids are scaled at the 50% probability level. A pair of enantiomers was observed in the unit cell. The angle between the mean plane of pyrrolo-diazepinopyrrolizine and second pyrrole ring of the dipyrrin and the plane of terminal pyrrole plane placing perpendicularly were measured in 82.60°. NH–N length of the hydrogen bond shown with dashed line is measured in 2.843 Å. | ||
Both crystal structures of 2 and 3 do not have any amino-H on the tetrafused rings. The crystal structure of 2 showed distortion of planarity due to meso-sp3C bridge while 3 exhibited high planarity. In the molecular structure of 3, an intramolecular hydrogen bond between the amino-group (donor) and the imino-group (acceptor) of the dipyrrin unit was found, which enhanced the planarity of the dipyrrin. Close orientations of the nitrile-N toward the internal hydrogen bond and αC–H of pyrrole toward imino-N of the seven-membered ring were also satisfactory for stabilizing the whole molecular conformation and increasing the planarity. The unit-packing diagram of 3 comprised two types of enantiomeric pairs. The crystallographic analysis of 2 was awarded conclusively in a CH2Cl2 solution by vapour diffusion of hexane, in which thin block crystals of 2 were obtained. Nevertheless, 2 was readily oxidized in aerobic conditions to 3. The terminal pyrrole ring is orthogonal to the mean plane over the pyrrolodiazepino-pyrrolizine and internal pyrrole groups due to the repulsion between two hydrogens of amino-groups at the dipyrrin unit, whose angle between the two planes was measured to be 82.60°. The intramolecular hydrogen bond seen at the dipyrrin unit in the structure of 2, shifted to between an imino-N of the fused linkage created with cyano-origin and the neighbouring pyrrole-NH, which probably is an important factor in the competition for the final molecular stability. The positionally shifted hydrogen bond in the molecular structure of 2 weakened the strength of the hydrogen bonding interaction and the NH–N distance of 2 was measured as 2.843 Å (the distance is relatively longer than that in 3). An enantiomeric pair was found in the unit cell of 2 since the structure is chiral.
Fig. 4 is the 1H NMR spectra of 1, 2, and 3. As the deliberated formation of a strong intramolecular hydrogen bond between imino-N and the amino-NH in the structure of 3, a proton peak is shown at a largely downfield area (13.46 ppm) of the spectrum (Fig. 4a). The corresponding proton peak of 2 appeared at a relatively up-field shifted area (11.74 nm), as reflecting a weakened hydrogen bond. Furthermore, the other two amino-NHs of pyrrolodiazepinopyrrolizine group resonated with the proton peaks at 8.41 and 6.12 ppm for pyrrolo-NH and diazepino-NH, respectively (Fig. 4b). Since 2 was readily converted to 3, a set of the proton peaks of 3 was seen in the 1H NMR spectrum of 2 (the peaks for 3 was distinguished with black dots in the 1H NMR spectrum 4b). Compared to 3, αCH of the external pyrrole of 2 resonated the proton peak at a largely up-field shifted area. The αCH peaks were assigned at 8.12 ppm for 3ox and 6.40 ppm for 2.
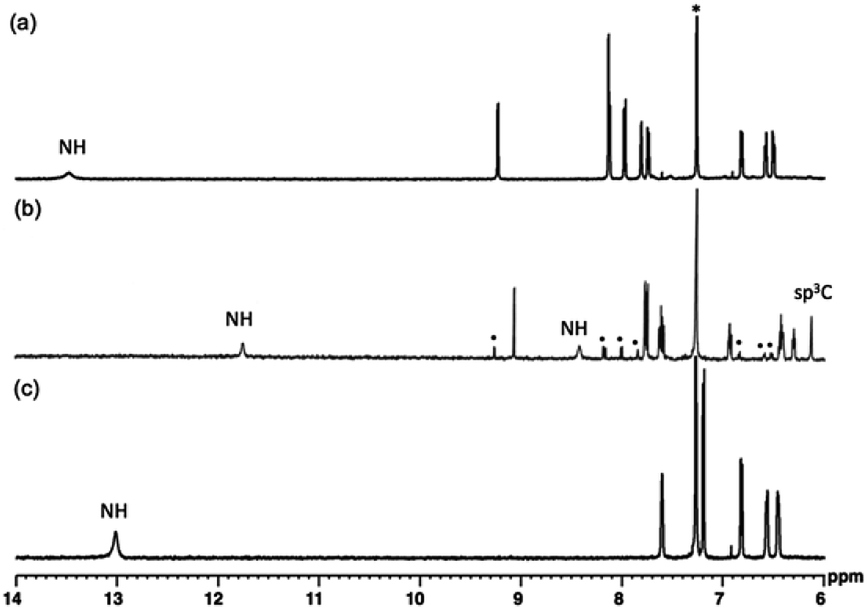 | ||
| Fig. 4 1H NMR spectra of 3 (a), 2 (b) and 1 (c) in CDCl3. * are peaks of solvent molecules. Also, the H-peaks distinguished with black dots are for its oxidation state, 3. | ||
Reversible conversions between the two redox-states were observed with absorption spectroscopy engaged with the redox processes (Fig. 5). An irreversible oxidation potential was found at 0.701 V (vs. Fc/Fc+) and one weak and one strong reduction potentials were found in the cyclic voltammogram of 3ox. Since two redox states of compound 3 were considered, the reduction potential of −1.403 V was assigned as the reduction conversion from 3ox to 3red.
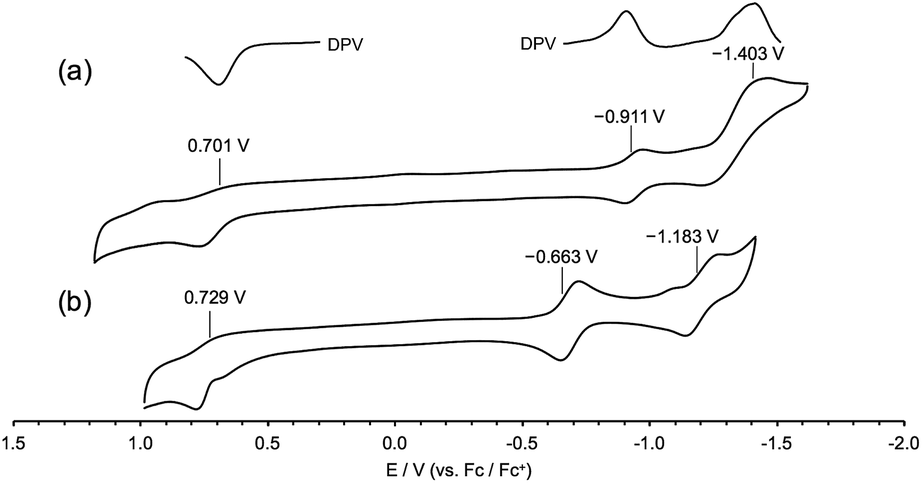 | ||
| Fig. 5 Cyclic voltammograms of 3 (a) and its precursor 1 (b) in CH2Cl2 (0.1 M nBu4NPF6). Working electrode: glassy carbon, counter electrode: Pt, reference cell: Ag/AgCl, and Fc/Fc+: ferrocene/ferrocenium couple. | ||
3 was expected to have two stable redox states, 3red and 3ox, as shown in Fig. 6. Since 3red was readily converted to 3ox in solution as well as solid states by oxygen, complete isolation of 3red failed most of the time. While any significant change of the absorption band of 3ox was not observed in chemical oxidation with DDQ (Fig. S17†), the absorption spectrum of 3ox was drastically changed in a chemical reduction with NaBH4 (Fig. 7). In the absorption spectrum of 3red, the absorption band of 3ox at around 650 nm was slightly shifted to a longer wavelength and broadened as well as weakened, which appears to contain an absorption shoulder that reaches ca. 830 nm. The significant color change of the solution by the reduction was observed from green to purplish-blue, as presented in the corner of Fig. 7.
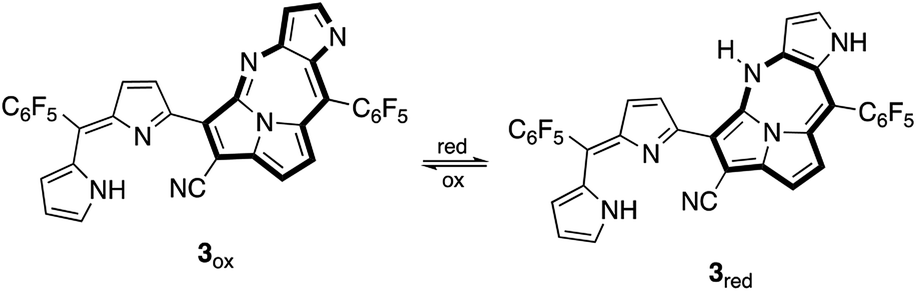 | ||
| Fig. 6 Redox conversions of 3: reduced form (3red) and oxidized form (3ox). | ||
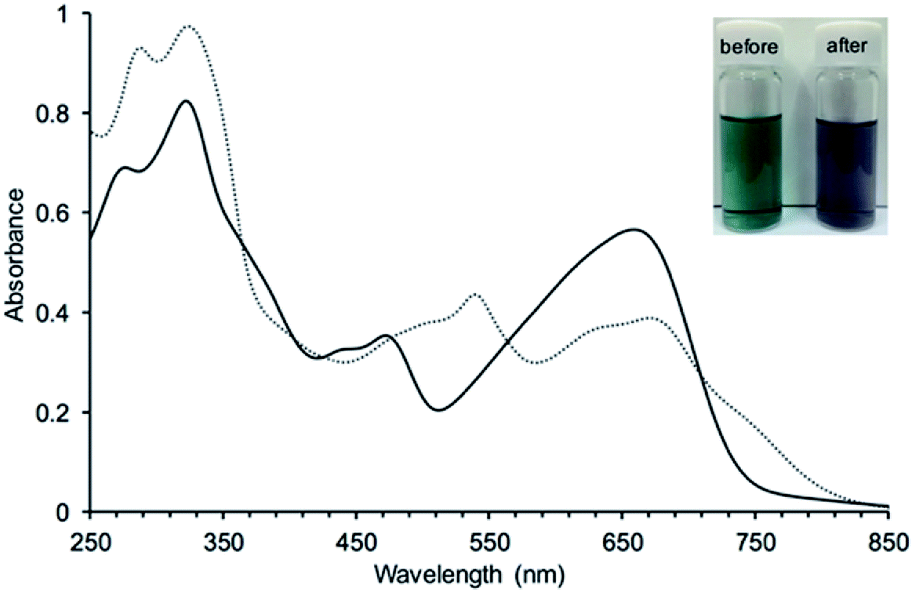 | ||
| Fig. 7 UV absorption spectral change of 3 in CH3OH by a reduction with NaBH4: before (—) and after (⋯) addition of NaBH4. A photograph of the solutions before and after reduction were added in the corner of the spectra as indicating the significant absorption change. | ||
Theoretical calculation results of the density functions for 3ox and 3red were given in Fig. 8. The HOMO energy level of 3ox (−6.00 eV) is lower than that of 3red (−4.94 eV) and a larger HOMO–LUMO gap is found for 3ox (1.78 eV and 2.18 eV are the values of HOMO–LUMO gaps for 3red and 3ox, respectively), thus 3red is converted to 3ox rapidly due to the high stability of 3ox. The small HOMO–LUMO gap of 3red supports why its absorption band appears more bathochromic shifted.
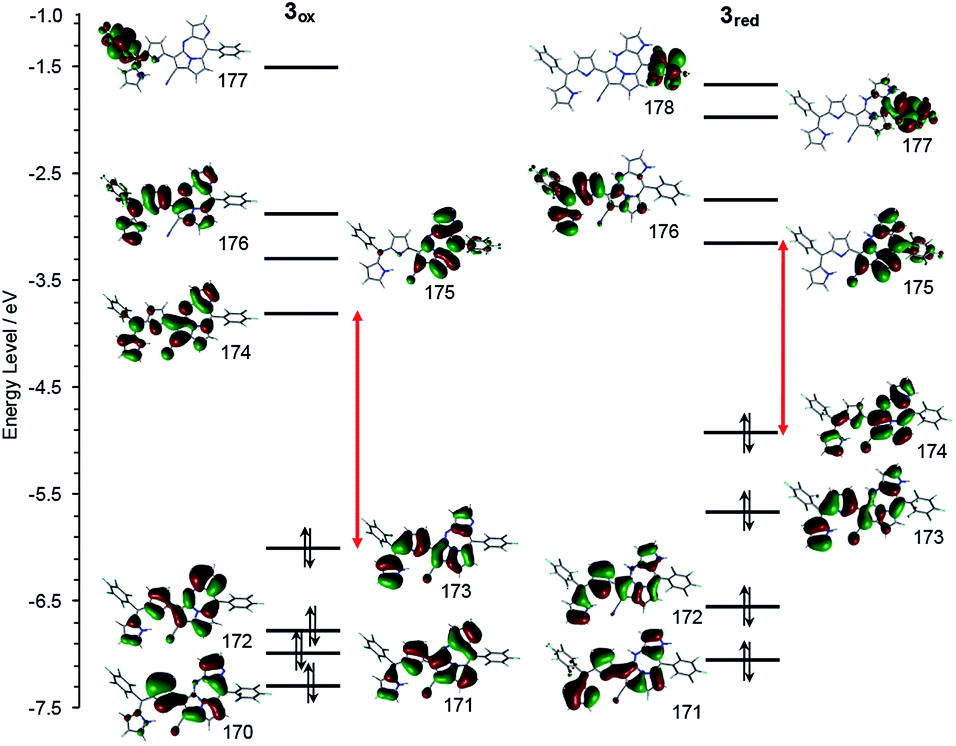 | ||
| Fig. 8 Frontier MO energy levels of 3ox (left) and 3red (right) calculated at B3LYP/6-311+g(d) level.9 | ||
Experimental
Materials and methods
All chemicals were purchased from commercial suppliers and used without further purification. Chromatographic separations were carried out by silica gel column chromatography (Silica gel 300) and gel permeation chromatography. NMR data of the compounds were measured with Bruker Avance 300 and 500 MHz NMR spectrometers in CDCl3 solvent, with the use of an internal standard of TMS. The standard Bruker software was used for homonuclear 1D and homonuclear as well as heteronuclear 2D experiments. Cyclic voltammograms were recorded on an ALS electrochemical analyzer 612C. A 0.1 M solution of tetrabutyl-ammonium hexafluorophosphate in CH2Cl2 was prepared as the CV electrolyte, and a three-electrode system (a glassy carbon working electrode, a Pt counter electrode, and Ag/AgClO4 reference electrode) was used for the CV measurement. All CV potentials were counted with the reference potential of ferrocene/ferrocenium ion couple. Single crystal diffraction data were collected and integrated, using the Rigaku CCD diffractometer (Saturn 724 with MicroMax-007) with Varimax Mo optics using graphite monochromated Mo-Kσ radiation (Nagoya University). The structure was solved by using direct methods with SHELXS97 and refined by using SHELXL97. All hydrogen atoms were placed in the calculated positions, respectively. All theoretical calculations were provided using the Gaussian 09 program.9 Optimization of the structures was performed with Becke's three-parameter hybrid exchange functional and Lee–Yang–Parr correlation functional basis sets (B3LYP/6-311+g(d)).General procedure for the synthesis of compounds 2 and 3
(Method A) BF3·OEt2 (100 μL) was added into a toluene (25 mL) solution containing the precursor 1 (50 mg, 0.07 mmol). The reaction mixture was heating overnight in anaerobic condition at 80 °C. While heating, blue precipitates appeared in the solution and the solution became transparent. The resulting solution was then cooled to room temperature and the solvent was removed by decantation. The precipitates were dissolved in CHCl3 and passed through an alumina gel short column with acetone as additional eluent to dissolve the retained precipitates completely. After the initial separation, the second column chromatography was employed using silica gel with CHCl3. The major fraction, green in color, was determined to be 3 (green, 9.8 mg, 20% yield). (Method B) InBr3 (20 mg) was added into a toluene solution of 1 (50 mg, 0.07 mmol) instead of BF3·OEt2 with the above reaction condition and the resulting solution was heated overnight at 80 °C. The resulting product solution was washed with water, extracted with CH2Cl2, and dried over Na2SO4. After removing the solvent by rotary evaporation, the residue was loaded on silica gel and column chromatographed using neat CH2Cl2. Green (first) and blue (second) fractions were then separated to obtain 3 and 2, respectively (27.5 mg (57% yield) and 14.7 mg (30% yield)). The growth of a crystal of 2 was succeeded conclusively in a completely closed system after many times challenges. The identity of the compounds was determined by a 1H, 19F and 13C-NMR analysis using a CDCl3 solvent and X-ray diffraction analysis of each of crystals. 3 in general organic solvents showed green in colour (dark green powder in the solid-state). 2 in general organic solvents showed blue in colour (dark blue powder in the solid-state).Spectral data of 2‡
λmax (nm (logε), CH2Cl2) 281.5 (4.37), 330.0 (4.44), 475.0 (3.92), 663.5 (4.15); δH (300 MHz, 298 K, CDCl3) 11.74 (bs, 1H, NH), 9.06 (d, 1H, J = 1.8, βCH), 8.41 (bs, 1H, NH), 7.77 (d, 1H, J = 1.8, βCH), 7.75 (d, 1H, J = 4.5, βCH), 7.62 (dd, 1H, dJ = 4.2, ddJ = 1.5, βCH), 7.58 (d, 1H, J = 4.5, βCH), 6.93 (dd, 1H, dJ = 4.5, ddJ = 1.8, βCH), 6.42 (dd, 1H, dJ = 5.4, ddJ = 2.7, βCH), 6.40 (bs, 1H, αCH), 6.29 (t, 1H, J = 3.9, αCH), 6.12 (s, 1H, sp3CH): the 1H NMR of 2 showed a small amount of 3 converted by air oxidation even after freshly purified by column chromatography.; δF (282.38 MHz, 298 K, CDCl3, calibrated with external CF3COOH) 137.59 (t, 2F, tJ = 19.8, o-CF), 141.33 (d, 2F, dJ = 19.8, o-CF),151.21 (t, 1F, J = 19.8, p-CF), 154.31 (t, 1F, J = 19.8, p-CF), 160.33 (td, 2F, tJ = 22.6, tdJ = 8.47, m-CF), 160.55 (td, 2F, tJ = 22.6, tdJ = 8.47, m-CF); m/z MALDI TOF MS, calcd. for C34H12F10N6 ([M]+): 694.096; found: 694.138.Spectral data of 3§
λmax (nm (logε), CH2Cl2) 275.5 (4.39), 324.0 (4.47), 477.5 (4.12), 668.5 (4.33); δH (300 MHz, 298 K, CDCl3) 13.46 (bs, 1H, NH), 9.23 (d, 1H, J = 1.8, βCH), 8.13 (d, 1H, J = 1.7, βCH), 8.12 (d, 1H, J = 4.6, αCH), 7.97 (d, 1H, J = 4.7, βCH), 7.81 (bs, 1H, αCH), 7.74 (d, 1H, J = 4.7, βCH), 6.83 (d, 1H, J = 4.7, βCH), 6.58 (dd, 1H, dJ = 4.3, ddJ = 1.7, βCH); δF (282.38 MHz, 298 K, CDCl3, calibrated with external CF3COOH) 137.27 (dd, 2F, dJ = 19.8, ddJ = 5.6, o-CF), 137.87 (dd, 2F, dJ = 19.8, ddJ = 5.6, o-CF), 150.85 (t, 1F, J = 19.8, p-CF), 151.50 (t, 1F, J = 19.8, p-CF), 160.37 (td, 2F, tJ = 19.8, tdJ = 2.8, m-CF), 160.44 (td, 2F, tJ = 19.8, tdJ = 2.8, m-CF); δC (125.8 MHz, 298 K, CDCl3) 160.56 (sp2CH), 151.63, 145.95, 143.86, 143.48, 137.59, 137.44, 134.05, 132.94, 131.00, 130.49 (sp2CH), 126.40 (sp2CH), 125.01, 124.31 (sp2CH), 123.72 (sp2CH), 121.60, 120.08 (sp2CH), 117.61 (sp2CH), 117.50 (sp2CH), 115.38, 111.38, 99.88 (13C peaks of phenyl groups are broaden due to C–F couplings); m/z MALDI TOF MS, calcd. for C34H10F10N6 ([M + H]+): 693.088, found: 693.127; m/z APCI HRMS, calcd. for C34H10F10N6 ([M + H]+): 693.0972, found: 693.0880.Conclusions
Novel pyrrolodiazepinopyrrolizine derivatives 2 and 3 were prepared from a toluene solution of dicyanovinylenebis(meso-aryl)dipyrrin 1 at 80 °C, in the presence of specific Lewis acids (BF3·OEt2 and InBr3). Spontaneous oxidation in aerobic conditions readily happens, where complete isolation of the reduced form (2) was not successful. However, the reparation of 2 succeeded by the use of InBr3. Elucidation of the structures of both 2 and 3 was accomplished by X-ray diffraction analysis. The structure of 2, shifted to between an imino-N of the fused linkage created with cyano-origin and the neighboring pyrrole-NH, which probably is an important factor in competitions for the stability of 2. The crystal structures of 2 and 3 showed two distinct pairs of enantiomers in each of the crystal units. π-electron delocalization happens over the pyrrolodiazepino-pyrrolizine part of 3, strongly. Two different redox states 3ox and 3red were observed by chemical oxidation with UV/vis absorption spectroscopy. Synthetic investigations towards novel π-conjugation and their characterizations are ongoing in recent research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JSPS KAKENHI Grant Number JP18K04925. J.-Y. Shin thanks Prof. Hiroshi Shinokubo for his support of experimental and analytical instruments and his advice. J. Y. Shin also acknowledges the G30 program foundation of Nagoya University for the support of this work.Notes and references
-
(a) C. Brückner, V. Karunaratne, S. J. Rettig and D. Dolphin, Can. J. Chem., 1996, 74, 2182 CrossRef
; (b) C. Brückner, Y. Zhang, S. J. Rettig and D. Dolphin, Inorganica Chim. Acta, 1997, 263, 279 CrossRef
-
(a) H. Maeda, H. Hasegawa, T. Hashimoto, T. Kakimoto, S. Nishio and T. Nakanishi, J. Am. Chem. Soc., 2006, 128, 10024 CrossRef CAS
-
(a) J. R. Pankhurst, N. L. Bell, M. Zegke, L. N. Platts, C. A. Lamfsus, L. Maron, L. S. Natrajan, S. Sproules, P. L. Arnold and J. B. Love, Chem. Sci., 2017, 8, 108 RSC
-
(a) Y. Ding, W.-H. Zhu and Y. Xie, Chem. Rev., 2017, 117, 2203 CrossRef CAS
-
(a) J. J. Hu, N.-K. Wong, S. Ye, X. Chem, M.-Y. Lu, A. Q. Zha, Y. Guo, A. C.-H. Ma, A. Y.-H. Leung and J. Shen, J. Am. Chem. Soc., 2015, 137, 6837 CrossRef CAS PubMed
-
(a) D. Walker and J. D. Hiebert, Chem. Rev., 1967, 67, 153 CrossRef CAS
- J.-Y. Shin, B. O. Patrick and D. Dolphin, Org. Biomol. Chem., 2009, 7, 2032 RSC
- N. H. Faialaga, S. Ito, H. Shinokubo, Y. Kim, K. Kim and J.-Y. Shin, Dalton Trans., 2017, 46, 10802 RSC
-
(a) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Milliam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Danels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian. Inc., Wallingford CT, 2009
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, mass, and absorption spectra as well as X-ray packing diagrams. CCDC 1936172 and 1952976. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra08850d |
| ‡ Crystallographic data of 2 (CCDC 1936172): C34H12F10N6 Mr = 694.50, T = 93(2) K, crystal size = 0.14 × 0.09 × 0.012 mm3, Mo radiation, monoclinic, space group P21/c (#14), a = 17.8640(10) Å, b = 7.5650(4) Å, c = 20.6477(8) Å, α = 90°, β = 100.169(5)°, γ = 90°, V = 2746.5(2) Å3, Z = 4, Pcalcd. = 1.680 g cm−1, R1(F) = 0.0799 (I > 2(l)), wR2(F2) = 0.2476 (all), GoF = 1.029. |
§ Crystallographic data of 3 (CCDC 1952976): (C34H10F10N6)2 Mr = 1384.96, T = 93 K, crystal size = 0.21 × 0.03 × 0.01 mm3, Mo radiation, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (#2), a = 7.2510(3) Å, b = 18.4847(8) Å, c = 22.9606(10) Å, α = 66.844(4)°, β = 85.123(3)°, γ = 87.644(3)°, V = 2897.0(2) Å3, Z = 2, Pcalcd. = 1.588 g cm−1, R1(F) = 0.0747 (I > 2(l)), wR2(F2) = 0.2040 (all), GoF = 0.949. Squeeze method was used to subtract the electron densities for disordered solvent molecules. The area volume for potential solvent molecules (263.9 Å3/2897.0 Å3 (9.1%)) was considered approximately with two methanol molecules. (#2), a = 7.2510(3) Å, b = 18.4847(8) Å, c = 22.9606(10) Å, α = 66.844(4)°, β = 85.123(3)°, γ = 87.644(3)°, V = 2897.0(2) Å3, Z = 2, Pcalcd. = 1.588 g cm−1, R1(F) = 0.0747 (I > 2(l)), wR2(F2) = 0.2040 (all), GoF = 0.949. Squeeze method was used to subtract the electron densities for disordered solvent molecules. The area volume for potential solvent molecules (263.9 Å3/2897.0 Å3 (9.1%)) was considered approximately with two methanol molecules. |
| This journal is © The Royal Society of Chemistry 2019 |