
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExcited-state locked amino analogues of the green fluorescent protein chromophore with a giant Stokes shift†
Snizhana O. Zaitsevaa,
Dilara A. Farkhutdinovab,
Nadezhda S. Baleevaa,
Alexander Yu. Smirnov a,
Marina B. Zagudaylovaa,
Aleksander M. Shakhovc,
Artyom A. Astafievcb,
Mikhail S. Baranov*ad and
Anastasia V. Bochenkova*b
a,
Marina B. Zagudaylovaa,
Aleksander M. Shakhovc,
Artyom A. Astafievcb,
Mikhail S. Baranov*ad and
Anastasia V. Bochenkova*b
aInstitute of Bioorganic Chemistry, Russian Academy of Sciences, Miklukho-Maklaya 16/10, 117997 Moscow, Russia. E-mail: baranovmikes@gmail.com
bDepartment of Chemistry, Lomonosov Moscow State University, Leninskie Gory 1/3, 119991 Moscow, Russia. E-mail: bochenkova@phys.chem.msu.ru
cSemenov Institute of Chemical Physics of RAS, Moscow, Russia
dPirogov Russian National Research Medical University, Ostrovitianov 1, Moscow 117997, Russia
First published on 26th November 2019
Abstract
We design a novel class of excited-state locked GFP chromophores by introducing an amine group at the double exo-bond and a difluoroboryl bridge. We show that these chromophores intrinsically exhibit a very large Stokes shift of 1 eV. Further tuning through chemical modifications of their aryl substituents makes them environmentally sensitive.
Introduction
Organic fluorophores have attracted much attention in recent years.1 These compounds are widely used in fluorescence microscopy where they help to visualize various biological processes.2 Among the diversity of these fluorophores, various methine dyes are of particular interest.3 Derivatives of the Green Fluorescent Protein (GFP) chromophore are the most interesting examples of methine dyes.4 Like many other methine dyes, the GFP chromophore and its analogues do not exhibit significant emission in solution as well as in the gas phase at room temperature.5 The fluorescence is quenched by a faster nonradiative decay channel, which is associated with the intramolecular rotation about the double bond of the methine bridge coupled to photoisomerization.The fluorescence can be restored in various environments or by cooling, when the rotation of the arylidene fragment is arrested.6 The chromophore is conformationally rigid inside the native protein environment, which explains the fluorescent properties of GFP at room temperature. The internal rotation can also be blocked through chemical modifications, for example, by introducing an internal bridge (Scheme 1).7 Such locked GFP chromophores can successfully be used to create various fluorescent labels8 and sensors;9 however, their synthesis is not always an easy task.
 | ||
| Scheme 1 Locked derivatives of the GFP chromophore. | ||
Chromophores with large Stokes shifts (LSS), i.e. large gaps between excitation and emission maxima, have many advantages in various bioimaging applications. The LSS fluorophores are characterized by much higher signal-to-noise ratio, since spectral overlap between excitation and emission bands is minimized, thus avoiding reabsorption of emitted photons and significantly diminishing a background noise from scattered light used for excitation. In multicolor bioimaging applications, several fluorophores with similar excitation and different emission energies can simultaneously be used and excited by a single wavelength.10 The use of an LSS donor in Förster resonance energy transfer (FRET) pairs guarantees a small contamination from direct excitation of an acceptor.11
Up until now, the LSS fluorescent protein chromophores have been designed based on their photoacid properties.10 In this case, light absorbing and emitting molecular forms are different and represent protonated (neutral) and deprotonated (anionic) chromophores, which are characterized by shifted absorption and emission bands. Here, we propose a new approach for creating conformationally locked derivatives of the GFP chromophore based on the use of amino-methylene-imidazolones (Scheme 1). In combination with high-level quantum chemistry calculations, we show that this new class of the locked GFP chromophores exhibit remarkable photophysical properties that can be traced to a single molecular form. These chromophores only become planar in the first excited state due to the formation of an extended π-conjugated system, resembling the effect of excited-state aromaticity.12 This results in a very large Stokes shift of about 1 eV, thus making the developed locked amino GFP chromophores appealing for the use in bioimaging applications.
Results and discussion
Among the existing approaches to the synthesis of the GFP chromophore derivatives, those based on condensation of imidazolones with aromatic aldehydes are worth to be mentioned.13 It is well known, that various imidates are also suitable substrates for similar condensations (e.g., with malonic esters14). Therefore, we have decided to combine these two approaches to create derivatives, which contain an additional amino group at the double exo-bond of the GFP chromophore (compounds 4‡ and 7,§ Scheme 2). | ||
| Scheme 2 Synthesis of the GFP chromophore derivatives with an additional amino group at the double exo-bond. | ||
Using imidates 3 and two in situ generated imidazolones 2 and 6, we have obtained a set of compounds containing various residues in the arylidene moiety. To introduce an internal difluoroboryl bridge, we have used the action of the boron fluoride in the presence of a base (Scheme 3).¶ As opposed to the previously reported approach based on C-borylation with boron tribromide, this reaction is characterised by relatively high yields and requires milder conditions. This makes it possible to obtain compounds with labile functional groups, e.g. ether or ester, which cannot be synthesized using BBr3.
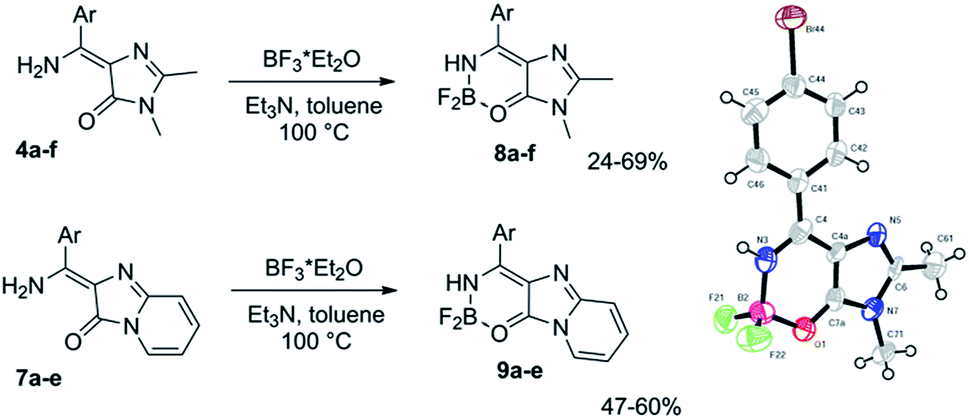 | ||
| Scheme 3 Synthesis of the new locked GFP chromophore derivatives and a general view of X-ray structure of compound 8d with atoms represented as thermal ellipsoids at 50% probability level. | ||
The introduction of the difluoroboryl bridge dramatically increases the quantum yield of fluorescence by 2–3 orders of magnitude. No appreciable emission from non-locked compounds 3 and 7 is observed in some solvents, whereas the fluorescence quantum yields (FQY) sometimes reach 60% and higher for compounds 8 and 9 (Tables 1, S2 and S4†).
The newly synthesized locked amino GFP chromophores exhibit a blue shift of their absorption maxima as compared to their non-locked analogues (Tables S1 and S3†). This distinguishes the lock proposed in this paper from those reported previously (Scheme 1). The latter exhibit a red shift due to the nitrogen-boron coordination of the atom at position N(3) of the imidazolone ring.7 At the same time, the emission maxima of compounds 8 are red shifted with respect to their non-locked predecessors (Tables S1 and S3†).
Compounds 8 and 9 are characterized by relatively short-wavelength positions of the absorption maxima, which is not typical for the GFP chromophore derivatives containing similar aryl groups. Furthermore, all compounds exhibit very close positions of their absorption bands regardless of the introduced aryl substituent (Fig. 1, Table 1). No significant red shift is observed when stronger donor groups are introduced, as it is opposed to the behavior of other derivatives of the GFP chromophore.4a
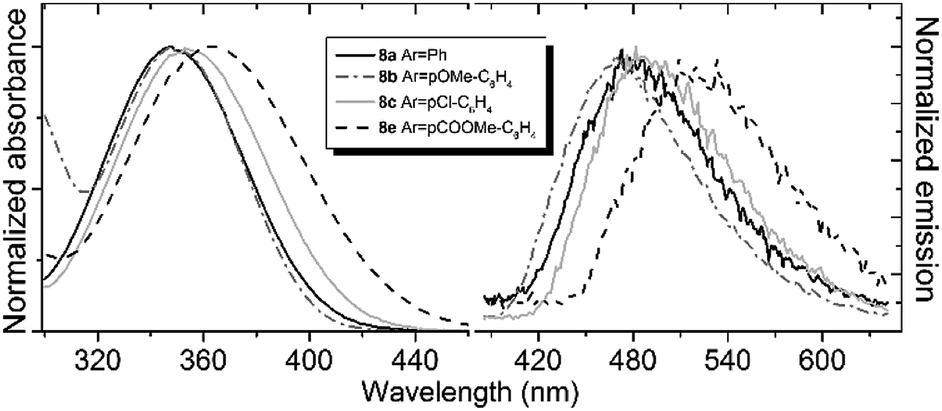 | ||
| Fig. 1 Absorption (left) and emission (right) spectra of compound 8 in acetonitrile. Emission spectra are recorded at an excitation wavelength of 340 nm. | ||
| 8a (Ph) | 8b (pOMe-C6H4) | 8e (pCO2Me-C6H4) | 8f (Pyridin-4-yl) | 9a (Ph) | 9b (pOMe-C6H4) | 9e (pCO2Me-C6H4) | ||
|---|---|---|---|---|---|---|---|---|
| a Peak maximum in nm (extinction coefficient in (mM cm)−1).b Peak maximum in nm (fluorescence quantum yield in %).c Non-fluorescent. | ||||||||
| Water | Absa | 338 (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) 000) |
343 (14000) |
349 (8500) | 348 (6500) | 402 (7500) | 398 (9500) | 423 (6000) |
| Emb | 470 (0.4) | 468 (0.5) | —c | —c | 498 (30) | 495 (13) | —c | |
| MeOH | Absa | 341 (9500) | 343 (12000) |
359 (7000) | 361 (6500) | 409 (7000) | 416 (7500) | 434 (5500) |
| Emb | 472 (1.9) | 460 (3.8) | 517 (0.5) | —c | 498 (30) | 528 (23) | ∼562 (2.1) | |
| CH3CN | Absa | 348 (10500) |
352 (13500) |
365 (7500) | 362 (6500) | 419 (7000) | 410 (9000) | 440 (6000) |
| Emb | 480 (2.8) | 468 (5.1) | 521 (1.7) | 505 (1.2) | 501 (25) | 533 (23) | 545 (5.6) | |
| EtOAc | Absa | 352 (10000) |
353 (12000) |
372 (7000) | 370 (6500) | 426 (8000) | 411 (8500) | 442 (6000) |
| Emb | 470 (4.5) | 461 (9.7) | 466 (6.5) | 465 (5.8) | 502 (30) | 532 (34) | 538 (6.0) | |
| Dioxane | Absa | 351 (10500) |
351 (13000) |
368 (7000) | 370 (6000) | 426 (7000) | 402 (9000) | 445 (6000) |
| Emb | 460 (9.3) | 456 (18) | 430 (22) | 430 (20) | 510 (62) | 530 (43) | 542 (6.0) | |
At the same time, the fluorescence of the new locked compounds is found to be sensitive to variations in the aryl substituents as well as to solvents (Fig. 1 and Table 1). The emission maximum can significantly be altered in different environments, particularly, in the case of the locked compounds with electron-accepting aryl substituents. The bathochromic shift of the emission maximum can be as large as 0.5 eV, as it is observed for compounds 8e upon increasing the solvent polarity.
The substituted locked GFP chromophores also show a significant variation in FQY in various solvents (Table 1). The smallest values of FQY are observed in polar and proton environments. In some cases, the fluorescence is completely lost in water. The bathochromic shifts and fluorescence quenching in polar and protic solvents suggest that these new locked chromophores have large dipole moments in their first excited state, which are larger than those in the ground state. Indeed, the absorption maxima are only weakly dependent on the nature of the solvent, while the emission maxima can significantly be altered. Furthermore, the locked compounds with electron-accepting aryl substituents exhibit a higher degree of the charge transfer character upon excitation, hence the excited-state properties become more sensitive to environment.
Importantly, the new locked GFP chromophore derivatives exhibit very large Stokes shifts of about 1 eV for compounds 8 and 0.6 eV for compounds 9. High-level quantum chemistry calculations based on the extended multiconfiguration quasi-degenerate perturbation theory XMCQPDT2 (ref. 15) are used to explain the origin of the large Stokes shift observed in the new class of the locked GFP chromophore derivatives. The equilibrium structures of compound 8a in the ground (S0) and first excited singlet (S1) states are shown in Fig. 2. Remarkably, the ground-state geometry is non-planar regardless the lock introduced to arrest the internal rotation. Here, the double bond is locked; however, the aryl fragment can be rotated about the single bond (C4–C5) in the methine bridge. The lack of conjugation between the two parts of the molecule in the ground state explains insensitivity of the absorption maxima of the new locked compounds to aryl substituents.
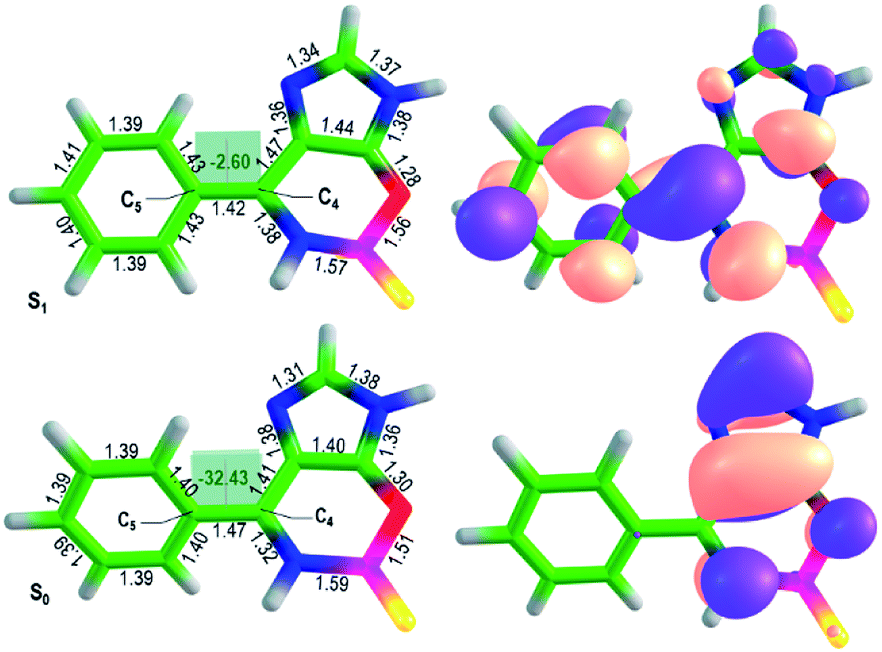 | ||
| Fig. 2 Optimized ground-state and excited-state structures of compound 8a (left) and XMCQDPT2 natural orbitals involved in the S0–S1 transition (right). The color code stands for the carbon, hydrogen, oxygen, boron, nitrogen, and fluorine atoms depicted in green, white, red, magenta, blue, and yellow, respectively. The bond lengths are shown in Å, the dihedral angles are in degrees. | ||
Upon excitation, the equilibrium geometry changes significantly and adopts an almost planar conformation. The excited-state molecule also exhibits a shorter length of the C4–C5 bond compared to that in the ground state. These structural changes suggest that the molecule becomes more conjugated upon excitation. The formation of an extended π-conjugated system leads to a smaller S1–S0 energy gap and hence to a large Stokes shift.
The calculated vertical excitation and emission energies are 3.4 and 2.6 eV (Table S5†), respectively, and the calculated Stokes shift is 0.8 eV, which agrees well with the experimental value of LSS in dioxane (0.8 eV, Table 1). The calculated average dipole moments in the ground (5.6 Debye) and first excited (7.2 Debye) states are relatively high and reveal a partial charge-transfer character in S1 (Δμ = 5.2 Debye). The molecule becomes more conjugated and more polar upon excitation, which is in accord with the observed sensitivity of the emission maxima to the aryl substituents and variations in polarity and proticity of the solvents. The natural orbitals involved in the S0–S1 transition support the redistribution of the electron density over the entire conjugated system upon excitation (Fig. 2). These results show that LSS is an intrinsic photophysical property of the newly developed class of the locked GFP chromophores.
In summary, we have designed and synthesized a new class of the locked amino derivatives of the GFP chromophore. The new lock is introduced at the double exo-bond of the chromophore. Remarkably, these chromophores appear to be fully locked only in the excited state, rather than in the ground state – as one might expect for the native GFP chromophore. These derivatives exhibit a large Stokes shift, which is found to be their intrinsic photophysical property. The LSS origin can be traced to the formation of an extended π-conjugated system in the excited state, ensuring the planarity of the entire molecule. The LSS property makes the new locked GFP chromophores appealing for the use in multicolor bioimaging applications. However, since many of these chromophores absorb in the UV, two-photon excitation should be employed. Indeed, the calculated two-photon absorption cross-sections are similar to those of the neutral GFP chromophores (Table S6†).
Furthermore, the locked amino GFP derivatives become more polar upon excitation, indicating a charge-transfer character of their first excited sate. This can be used to further tune the photophysical properties of the newly developed LSS GFP-based fluorophores by introducing various electron-accepting groups to the aryl moiety, thus enhancing the charge-transfer character of the first excited state. This makes their emission properties sensitive to environment, and hence they can be used as sensors to medium polarity and other fluorogenes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support from the Russian Foundation for Basic Research Grants No. 17-00-00404 (17-00-00401, 17-00-00402, 17-00-00403) KOMFI. DAF and AVB acknowledge support from the Lomonosov Moscow State University Program of Development for providing HPC computing resources. We thank Victor B. Rybakov and Konstantin A. Lyssenko for their help with X-ray analysis.Notes and references
- (a) T. Terai and C. P. Nagano, Pflügers Arch., 2013, 465, 347 CrossRef CAS PubMed; (b) V. I. Martynov, A. A. Pakhomov, N. V. Popova, I. E. Deyev and A. G. Petrenko, Acta Naturae, 2016, 8, 33 CrossRef CAS PubMed.
- (a) H. Sahoo, RSC Adv., 2012, 2, 7017 RSC; (b) C. P. Toseland, Journal of Chemical Biology, 2013, 6, 85 CrossRef PubMed; (c) E. A. Specht, E. Braselmann and A. E. Palmer, Annu. Rev. Physiol., 2017, 79, 93 CrossRef CAS PubMed; (d) A. Ettinger and T. Wittmann, Methods Cell Biol., 2014, 123, 77 Search PubMed.
- H. Berneth, Ullmann's Encyclopedia of Industrial Chemistry, 2002, vol. 23, pp. 49–109 Search PubMed.
- (a) C. L. Walker, K. A. Lukyanov, I. V. Yampolsky, A. S. Mishin, A. M. Duraj-Thatte, A. Bahareh, L. M. Tolbert and K. M. Solntsev, Curr. Opin. Chem. Biol., 2015, 27, 64 CrossRef CAS PubMed; (b) N. S. Baleeva and M. S. Baranov, Chem. Heterocycl. Compd., 2016, 52, 444 CrossRef CAS.
- (a) K. Addison, I. A. Heisler, J. Conyard, T. Dixon, P. C. Bulman Page and S. R. Meech, Faraday Discuss., 2013, 163, 277 RSC; (b) A. Svendsen, H. V. Kiefer, H. B. Pedersen, A. V. Bochenkova and L. H. Andersen, J. Am. Chem. Soc., 2017, 139, 8766 CrossRef CAS PubMed; (c) A. V. Bochenkova and L. H. Andersen, Faraday Discuss., 2013, 163, 297 RSC.
- (a) A. Baldridge, A. Samanta, N. Jayaraj, V. Ramamurthy and L. M. Tolbert, J. Am. Chem. Soc., 2011, 133, 712 CrossRef CAS PubMed; (b) D. E. Williams, E. A. Dolgopolova, P. J. Pellechia, A. Palukoshka, T. J. Wilson, R. Tan, J. M. Maier, A. B. Greytak, M. D. Smith, J. A. Krause and N. B. Shustova, J. Am. Chem. Soc., 2015, 137, 2223 CrossRef CAS PubMed; (c) J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642 CrossRef CAS PubMed; (d) G. S. Filonov, J. D. Moon, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 16299 CrossRef CAS PubMed; (e) N. G. Bozhanova, M. S. Baranov, N. V. Klementieva, K. S. Sarkisyan, A. S. Gavrikov, I. V. Yampolsky, E. V. Zagaynova, S. A. Lukyanov, K. A. Lukyanov and A. S. Mishin, Chem. Sci., 2017, 8, 7138 RSC.
- (a) M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Slipchenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, J. Am. Chem. Soc., 2012, 134, 6025 CrossRef CAS PubMed; (b) L. Wu and K. Burgess, J. Am. Chem. Soc., 2008, 130, 4089 CrossRef CAS PubMed; (c) A. Baldridge, K. M. Solntsev, C. Song, N. Tanioka, J. Kowalik, K. Hardcastle and L. M. Tolbert, Chem. Commun., 2010, 46, 5686 RSC.
- (a) A. A. Stakheev, D. Y. Ryazantsev, Y. K. Zvezdina, M. S. Baranov and S. K. Zavriev, Biochemistry (Moscow), 2018, 83, 855 CrossRef CAS PubMed; (b) M. Frizler, I. V. Yampolsky, M. S. Baranov, M. Stirnberg and M. Gütschow, Org. Biomol. Chem., 2013, 11, 5913 RSC.
- (a) S. C. Olsen, M. S. Baranov, N. S. Baleeva, M. M. Antonova, K. Johnson and K. M. Solntsev, Phys. Chem. Chem. Phys., 2016, 18, 26703 RSC; (b) N. S. Baleeva, I. V. Yampolsky and M. S. Baranov, Russ. J. Bioorg. Chem., 2016, 42, 453 CrossRef CAS.
- D. M. Shcherbakova, M. A. Hink, L. Joosen, T. W. Gadella and V. V. Verkhusha, J. Am. Chem. Soc., 2012, 134, 7913 CrossRef CAS PubMed.
- (a) H. Ai, K. L. Hazelwood, M. W. Davidson and R. E. Campbell, Nat. Methods, 2008, 5, 401 CrossRef CAS PubMed; (b) J. Yang, L. Wang, F. Yang, H. Luo, L. Xu, J. Lu, S. Zeng and Z. Zhang, PLoS One, 2013, 8, e64849 CrossRef CAS PubMed.
- M. Rosenberg, C. Dahlstrand, K. Kilså and H. Ottosson, Chem. Rev., 2014, 114, 5379 CrossRef CAS PubMed.
- (a) Y. Hsu, Y. Chen, H. Tseng, Z. Zhang, J. Shen, W. Chuang and P.-T. Chou, J. Am. Chem. Soc., 2014, 136, 11805 CrossRef CAS PubMed; (b) S. O. Zaitseva, S. V. Golodukhina, N. S. Baleeva, E. A. Levina, A. Yu. Smirnov, M. B. Zagudaylova and M. S. Baranov, ChemistrySelect, 2018, 3, 8593 CrossRef CAS.
- (a) R. L. Shriner and F. W. Neumann, Chem. Rev., 1944, 35, 351 CrossRef CAS; (b) M. Ikejiri, H. Kojima, Y. Fugono, A. Fujisaka, Y. Chihara and K. Miyashita, Org. Biomol. Chem., 2018, 16, 2397 RSC.
- (a) A. A. Granovsky, J. Chem. Phys., 2011, 134, 214113 CrossRef PubMed; (b) A. A. Granovsky, Firefly, version 8.2.0, http://classic.chem.msu.su/gran/firefly/index.html Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Optical spectra, NMR spectra, extended description of synthetic procedures, and computational results. CCDC 1919301. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra08808c |
| ‡ Synthesis of compounds 4. Amide 1 (3.50 g, 10.0 mmol) was dissolved in dry toluene (50 mL) under argon. Acetic anhydride (1.0 g, 10 mmol) were added and the mixture was stirred at room temperature until the complete dissolution (approx. 40 min). Corresponding imidate hydrochloride 3 (7.0 mmol) was suspended in CHCl3 (50 mL) and treated with sodium hydroxide solution (1 g in 10 mL of H2O). The organic layer was separated, dried over Na2SO4 and the solvent was removed under vacuum. Afterwards molecular sieves (4 Å, 5 g) and the solution of 2,3-dimethyl-3,5-dihydro-4H-imidazol-4-one in toluene 2 (see above) were added to the crude imidate. The mixture was stirred at 100 °C for 15–30 minutes, cooled to the room temperature; molecular sieves were removed by filtration and washed with dichloromethane (2 × 50 mL). The solvent was removed under vacuum and the product was purified by column chromatography (EtOAc–Hexane 1:9). |
| § Synthesis of compounds 7. Pyridin-2-yl-glycine hydrochloride 5 (1 g, 5 mmol) was dissolved in PCl3 (10 mL) under argon. The mixture was refluxed for 3 h and phosphorus trichloride was removed under vacuum. Afterwards pyridine (8 mL), triethylamine (1 mL) and corresponding imidate hydrochloride 3 (4 mmol) were added and the mixture was stirred for 18 h at room temperature. The solvent was removed under vacuum, the residue was dissolved in 200 mL of chloroform and washed with saturated NaHCO3 solution (1 × 100 mL) and brine (1 × 50 mL). The solution was dried over Na2SO4 and the solvent was removed under vacuum. The product was purified by column chromatography (CHCl3). |
| ¶ Synthesis of compounds 8 and 9. Corresponding chromophore 4 or 7 (0.3 mmol) and triethylamine (0.42 mL, 3 mmol) were dissolved in toluene (5 mL). Boron trifluoride diethyl etherate (0.22 mL, 1.8 mmol) was added and the reaction mixture was heated at 100 °C for 2–2.5 hours. After that 20 mL of ethylacetate was added. The solution was washed with water (3 × 10 mL), brine (1 × 10 mL) and dried over Na2SO4. The solvent was removed under vacuum and the product was purified by column chromatography (CHCl3). |
| This journal is © The Royal Society of Chemistry 2019 |