
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient catalytic oxidation of methyl aromatic hydrocarbon with N-alkyl pyridinium salts†
Qiaohong Zhanga,
Honghao Hea,
Huibin Wanga,
Zhan Zhang*b and
Chen Chen *a
*a
aSchool of Material Science and Chemical Engineering, Ningbo University, 818 Fenghua Road, Ningbo, 315211, PR China. E-mail: chenchen@nbu.edu.cn; Fax: +86 574 87609836; Tel: +86 574 87609836
bChina Tobacco Henan Industrial Co. Ltd, No.8 The 3rd Avenue, Zhengzhou, 450001, PR China. E-mail: zhangzhan2059729@126.com
First published on 26th November 2019
Abstract
A series of N-alkyl pyridinium salts were synthesized and employed as metal-free catalyst for the selective oxidation of methyl aromatic hydrocarbon with molecular oxygen. The electronic effect of the substitutes was found to be an important factor for the catalytic performance. With the introduction of electron-donating substitute –N(CH3)2, the conversion of p-xylene and selectivity of p-toluic acid could be simultaneously increased. 1-Benzyl-4-N,N-dimethylaminopyridinium salt showed the highest catalytic activity, and 95% conversion with 84% of selectivity to p-toluic acid could be obtained for the selective oxidation of p-xylene. Several methyl aromatic hydrocarbons could all be efficiently oxidized with the reported catalyst at the absence of any metal species.
Introduction
Selective oxidation of methyl aromatic hydrocarbon is one of the key technologies in transforming petrochemical raw materials into useful oxygenates such as alcohol, aldehyde, ketone, and carboxylic acid, which are valuable products in agricultural, pharmaceutical, polymer industries, etc.1–5 For example, oxidation of toluene to benzoic acid and oxidation of p-xylene to p-toluic acid or terephthalic acid are all important commercial processes in the chemical industry.6,7 For these processes, owing to the inert reactivity of the C–H bond, metal-containing catalytic systems are often utilized as catalysts to facilitate the conversion of aromatic hydrocarbons at elevated temperature or pressure.8–10 One typical example is the Co–Mn–Br homogeneous catalytic system which was firstly developed in the middle of last century and is still being utilized in the present.7,11,12 Metal-containing catalytic systems such as Co, Cu, or Mn-based oxides, etc. showed good catalytic activity at higher reaction temperatures.3,4,13–17 For their surface hydrophilicity, however, oxygenated products such as organic acid are easily adsorbed on their surface, and over-oxidation or coking were likely to happen at higher temperatures. The surface organic modification strategy was illustrated for inorganic heterogeneous catalysts in our previous work to modulate the surface hydrophilicity/hydrophobicity; the hydrophobic or superhydrophobic catalyst showed better lipophilicity and good catalytic activity was obtained at lower temperatures.18–21Compared with those heterogeneous catalysts, homogeneous organic catalysts own better solubility and could be used under mild conditions. For example, metal-complex or the metal-complexation promoted organic N-hydroxyl catalytic systems were reported for the conversion of hydrocarbon.3,4,22–24 For the above mentioned catalytic system, metal species were all involved. Development of the metal-free catalytic system for the selective oxidation of hydrocarbon was a challenge for the chemists. Pure organic catalyst could show better interaction with the organic substrates. Under mild reaction conditions, higher catalytic activity is expected to be obtained and coking or over-oxidation caused by the metal-species might also be avoided. Encouraging progresses have been achieved on the metal-free organocatalysis such as the oxidation of alcohol to aldehyde/ketone catalyzed by 2,2,6,6-tetramethyl-piperidyl-1-oxy (TEMPO), and selective oxidation of hydrocarbon by N-hydroxyphthalimide (NHPI) system, or the other organic molecules.25–29 Besides these systems, it was found that simple nitrogen cation compounds were useful in facilitating the oxidation of hydrocarbon. For example, quaternary ammonium salts could catalyze the oxidation of different hydrocarbon such as cyclohexene, tetralin, and ethylbenzene.30–32 Nitrogen-cation compounds such as methyl violet could be used as the electron-promoter to combine with NHPI in promoting the metal-free oxidation of aromatic hydrocarbon.33,34
In nature, it was found that pyridinium cation analogues (e.g. NAD+) played an important role in the redox processes.35 Compared with the conventional quaternary ammonium salts, quaternary pyridinium salts posses some advantages including better thermal stability and simpler recycling from the reaction mixture.36,37 Seldom studies, however, have focused on their utilization in the selective oxidation of methyl aromatic hydrocarbon. In this study, a series of N-alkyl pyridinium salts with different type of substitutes were designed and synthesized (as shown in Scheme 1). These compounds are 1-benzylpyridin-1-ium bromide (a), 1-benzyl-4-cyanopyridin-1-ium bromide (b), 1-benzyl-4-(dimethylamino)pyridin-1-ium bromide (c), 1-decyl-4-(dimethylamino)pyridin-1-ium bromide (d), 1-dodecyl-4-(dimethylamino)pyridin-1-ium bromide (e), respectively, and their catalytic performance in the oxidation of methyl aromatic hydrocarbon was investigated.
 | ||
| Scheme 1 Structure of different N-alkyl pyridinium salts. | ||
Experimental
General
4-Dimethylamino pyridine (DMAP, 99%), 4-cyano pyridine (99%), 4-tert-butyl toluene (99%), 4-chloro toluene (>98%), p-tolualdehyde (TALD, 99%), p-toulic acid (TA, 99%), 4-carboxylic benzaldehyde (4-CBA, 99%), terephthalic acid (TPA, 99%) were commercial products from J&K Chemical Co. Ltd. n-Decyl bromide (99%) and n-dodecyl bromide (98%) were purchased from Aladdin Chemical Reagents Co. Ltd., China. Other materials and reagents were purchased from Tianjin Kermel Chemical Reagent Co. Ltd., China. Different methyl aromatics were used after removing water by adding 5 wt% of 4 A molecular sieve and being treated for 24 hours. The gas chromatography (GC) was Agilent-7890 equipped with a flame ionization detector. High-performance liquid chromatography (HPLC) measurements were performed on Waters e2695 with UV/Vis 2489 spectrometer. FT-IR spectrums were recorded on Bruker TENSOR 27 spectrometer using KBr pellet (Fig. S1†). 1H NMR and 13C NMR spectrums were recorded on a Bruker AVANCE III NMR spectrometer in DMSO-d6 with tetramethylsilane (TMS) as the internal standard. GC-MS were recorded by Agilent 6890N GC/5973 mass spectrometer (Fig. S2–S6†).General procedure for the synthesis of N-alkyl pyridinium salts
Different N-alkyl pyridinium salts were synthesized by refluxing corresponding pyridines with alkyl bromide in a procedure refereeing to the previously published report.36,38 The synthesis was carried out under the following procedure: 1 mol of pyridine and 80 ml of toluene were placed in the flask, respectively. 1.1 mol of alkyl bromide were added dropwise into the flask at 70 °C and refluxed for 24 hours. Then, the pyridinium salts formed as solid and they were washed by acetone for three times to remove residual materials. Finally they were evaporated and dried at high vacuum under 70 °C overnight. The structure was confirmed by FT-IR measurement, 1H NMR and 13C NMR spectroscopy.General procedure for the catalytic oxidation of aromatic hydrocarbon
Catalytic oxidation reactions were carried out in a 20 ml scale Teflon-lined autoclave equipped with magnetic stirrer and automatic temperature controller. In a typical experiment, 10 mmol of p-xylene and 5 ml of acetonitrile were added into the autoclave followed by 0.5 mmol of c as catalyst. And 0.2 mmol of p-tolualdehyde (TALD) was also introduced as initiator to reduce the induction period. After the autoclave was sealed, the air inside was replaced with O2 for three times, then the autoclave was heated to 160 °C. The pressure was kept at 1.5 MPa during the reaction by continuously recharging of oxygen. After reaction, the autoclave was cooled to room temperature and carefully depressurized. 5 ml of dimethyl formamide (DMF) was used to dissolve the products for analysis. Different products were identified by GC-MS spectrometer and comparing retention time of authentic samples. 4-Carboxylic benzaldehyde (4-CBA) and terephthalic acid (TPA) were quantified by HPLC using prevail organic acid column and oxalic acid as the internal standard. The mobile phase consisted of acetonitrile and water (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70, V/V) by adjusting the pH = 2–3 with phosphoric acid. Other products were quantified by GC using HP-5 column and 1,3-dichlorobenzene as the internal standard.
:70, V/V) by adjusting the pH = 2–3 with phosphoric acid. Other products were quantified by GC using HP-5 column and 1,3-dichlorobenzene as the internal standard.
General procedure for the decomposition of tert-butyl hydroperoxide
In a typical reaction procedure, 7 mmol of TBHP and 1 mmol of N-alkyl pyridinium salt were placed in a 10 ml flask and stirred under 80 °C for 1 h in the dark. Then the reaction mixture was rapidly cooled to room temperature and the conversion of TBHP was determined by iodometric titration.Results & discussion
Initially, the selective oxidation of p-xylene was chosen as a model reaction to test the catalytic performance of these N-alkyl pyridinium salts in the absence of solvent (Scheme 2). And the reaction results were listed in Table 1. It can be observed that the investigated N-alkyl pyridinium salts could all catalyze the oxidation of p-xylene. | ||
| Scheme 2 Oxidation of p-xylene. | ||
| Entry | Catalyst | Conv. (%) | Selectivity (%) | |||
|---|---|---|---|---|---|---|
| TA | TALD | TAOL | Othersb | |||
| a Reaction conditions: 10 mmol of p-xylene, 0.25 mmol of c, 0.2 mmol of p-tolualdehyde as initiator, 1.0 MPa O2, 160 °C, 2 h.b Mainly (4-methylbenzyl)-p-toluates, 4-CBA, and TPA.c 190 °C, 2 h (ref. 41).d 180 °C, 3 h, air pressure, 2.0 mp, with acetic acid as solvent (ref. 42).e 150 °C, 3.5 h, (ref. 43). | ||||||
| 1 | a | 39 | 72 | 13 | 10 | 5 |
| 2 | b | 27 | 70 | 15 | 13 | 2 |
| 3 | c | 52 | 86 | 6 | 2 | 6 |
| 4 | d | 48 | 75 | 14 | 5 | 6 |
| 5 | e | 50 | 74 | 12 | 4 | 10 |
| 6 | TBAB | 4 | 19 | 43 | 31 | 7 |
| 7c | MgCO3 | 28 | 54 | 20 | 19 | 7 |
| 8d | T (p-Cl)PPMnCl | 12 | 73 | 24 | — | 3 |
| 9e | Co(II) (DPDME) | 15 | 14 | 49 | 33 | 4 |
The main product is p-toluic acid (TA). Small amount of p-tolualcohol (TOAL) and p-tolualdehyde (TALD) were also obtained as by-product. The distribution of the product is different with those results obtained with metal-containing catalyst being used, in which deep oxidation happened and then considerable amount of 4-carboxylicbenzaldehyde (4-CBA) and terephthalic acid (TPA) produced.39 For organic catalysts, introduction of substitute was an efficient method to modulate their catalytic activity.28,40 Effect of different substituted groups was studied in the present work. For the oxidation of p-xylene, with catalyst a being employed, the conversion reached 39% (Table 1, Entry 1). Replacing of hydrogen with cyano group, the obtained catalyst b gave a p-xylene conversion of 27% (Table 1, Entry 2). While for catalyst c the p-xylene conversion increased to 52% (Table 1, Entry 3). The conversion of p-xylene increased in the order of –CN < –H < –N(CH3)2 with the modulation of the substituted group on the pyridine ring. And the same order was followed for the changing of the selectivity of TA. The highest selectivity of 86% for TA could be obtained with catalyst c being used. In literature, MnCO3, metalloporphyrin T (p-Cl)PPMnCl, or metallo-deuteroporphyrin Co(II) (DPDME) was reported as good catalyst for the selective oxidation of p-xylene, and the conversion was 28%, 12%, and 15%, respectively, which was obtained even at a much higher reaction temperature of 190 °C (Table 1, Entry 7–9).41–43 By comparison, the reported catalyst in the present work showed better catalytic activity and higher selectivity to TA. In addition, it could also be observed that the introduction of electron-donating group was positive while the introduction of electron-withdrawing group was negative for the obtaining of high catalytic activity and selectivity. Owning to a resonance stabilization of the positive charge delocalization, the introduction of electron-donating group could increase the stability of the pyridinium salts, while the introduction of electron-withdrawing cyano-group acted the contrary effect.44–46 A free-radical mechanism usually involved during the catalytic oxidation of hydrocarbon with the organic catalyst, in which organic hydroperoxide is the primary product which could be decomposed to the free-radical intermediate and the final oxygenated products.31,32 Then, the decomposition of tert-butyl hydroperoxide (TBHP) was used to primarily evaluate the catalytic performance of different N-alkyl pyridinium salts in the present work (Fig. 1). It could be found that dimethylamino group substituted N-alkyl pyridinium salts c exhibited higher activity than a and b, which exhibited similar trends with that in the catalytic oxidation of p-xylene. Similar results were obtained in the process of the decomposition of CHHP.32 The catalytic ability of decomposing organic hydroperoxide is an important factor for the catalytic performance. The above two factors probably led to the difference on the catalytic performance in the present work. Furthermore, with the variation of N-alkyl substituted group, no remarkable differences were obtained. When catalyst d and e were used, comparable p-xylene conversion of 48% and 50% were obtained, respectively (Table 1, Entry 4 and 5). Control experiment using conventional quaternary ammonium salts of tetrabutyl ammonium bromide (TBAB) was also carried out. Under the same reaction conditions, only 4% of p-xylene was converted (Table 1, Entry 6).
 | ||
| Fig. 1 Decomposition of TBHP with different N-alkyl pyridinium salts. | ||
Though high selectivity for TA could be obtained for the solvent-free oxidation of p-xylene, the conversion of p-xylene was not satisfied. It was acknowledged that the solubility of TA in the weak polar aromatic media is low. With the increasing of TA solid products during the reaction, further reaction was difficult to carry on. Then different organic solvents were introduced to improve the reaction, and the results were shown in Fig. 2. To our delight, higher catalytic activity was obtained with the polar solvent such as acetonitrile, acitic acid, or dimethylformamide being used, respectively. The oxidation efficiency was greatly enhanced in acetonitrile, and 88% of p-xylene conversion was obtained with a TA selectivity of 83%, which were higher than those obtained in acetic acid or dimethylformamide. Then, acetonitrile was selected as the solvent for the further study.
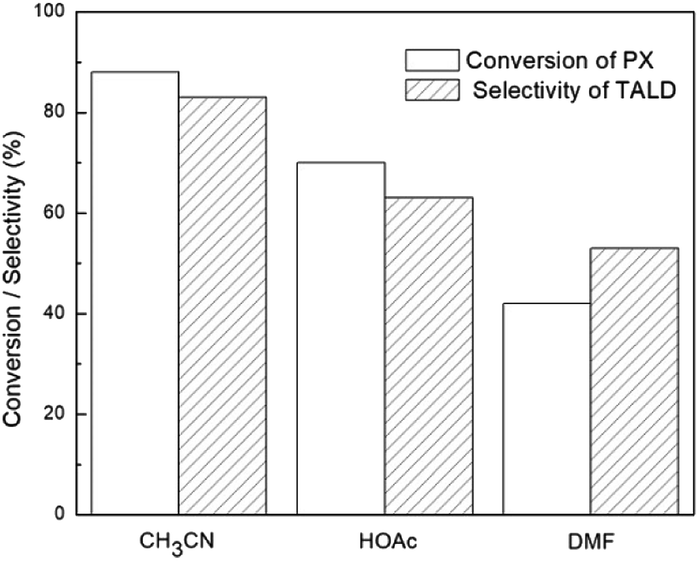 | ||
| Fig. 2 Influence of solvents on p-xylene oxidation. Reaction conditions: 10 mmol of p-xylene, 0.5 mmol of c as catalyst, 0.2 mmol of p-tolualdehyde as initiator, 5 ml of organic solvent, 1.5 MPa O2, 160 °C, 3 h. | ||
In order to better understand the reaction process, detailed study with different reaction time was carried out in acetonitrile using catalyst c (Fig. 3). p-Xylene conversion rapidly increased during the first 3 h, then it became steady after 4 h. For the distribution of oxygenated products, TA was still the dominant product after 4 h. Different from the data under solvent-free conditions, the selectivity for TOAL was negligible in acetonitrile even at the initial time. Selectivity for TALD rapidly decreased with the time manifesting that TALD was converted to TA with the time. At the same time, the selectivity of deep oxygenated products such as 4-CBA or TPA also increased slowly. Under the optimized reaction time, the maximum yield for TA was obtained after 4 h with a p-xylene conversion of 95% and TA selectivity of 84%. In addition, the effects of catalyst concentration and reaction temperature on the catalytic performance were also studied, respectively (Fig. S7 and S8†).
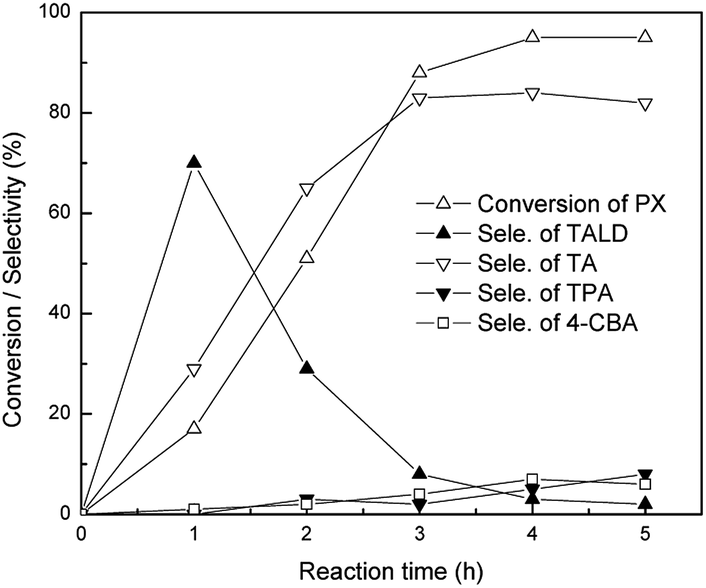 | ||
| Fig. 3 Influence of reaction time on p-xylene oxidation. Reaction conditions: 10 mmol of p-xylene, 0.5 mmol of c, 0.2 mmol of p-tolualdehyde as initiator, 5 ml of acetonitrile, 1.5 MPa O2, 160 °C. | ||
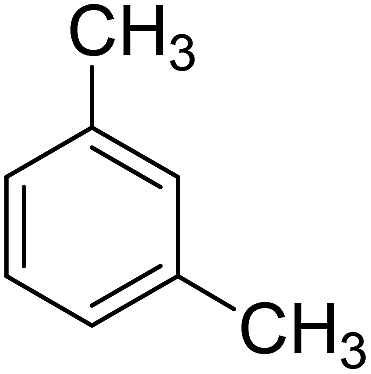
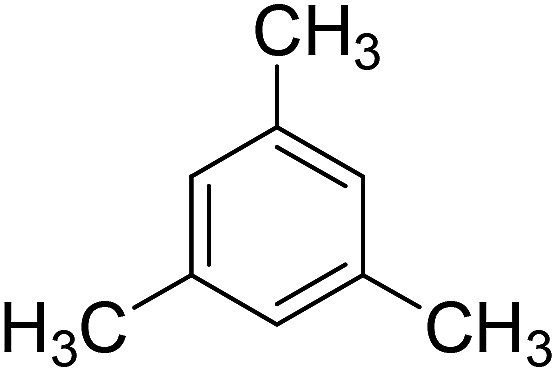
To extend the scope of this catalytic system, the oxidation of different methyl aromatic hydrocarbon were investigated using catalyst c under the optimized conditions (Table 2). o-Xylene and p-tert-butyl toluene could also be oxidized with the conversion of 96% and 93%, respectively (Table 2, Entry 1 and 2). While for the case of m-xylene and mesitylene, the conversion was 63% and 56%, respectively (Table 2, Entry 3 and 4). In the oxidation of methyl aromatics, electro-donating substitute was favorable for the activation and conversion of methyl group. This kind of promotion effect was weaker for the electro-donating substitute when it emerges at the meta position, which caused a lower conversion. Though toluene was even more difficult to be oxidized, 15% of conversion could also be obtained under the same reaction conditions (Table 2, Entry 5). These results indicated that the reported catalyst in the present work could facilitate the efficient catalytic oxidation of different methyl aromatic hydrocarbon.











A possible reaction mechanism was proposed for the oxidation of aromatics, and p-xylene was taken as an example (Scheme 3). Firstly, the p-xylene hydroperoxide was formed as primary product under the condition of heating and with the p-tolualdehyde as the initiator (1). The spontaneous homolytic decomposition of the formed hydroperoxide is very slow at the absence of catalyst. At the presence of N-alkyl pyridinium salts, however, the cation could attack at the more nucleophilic O atom (the inside one, near the organic radical) of the hydroperoxide, which would enhance the homolysis and then accelerate the formation of the free radicals (2).47 The free radicals thus formed were involved in the following oxidation chain, and terminated with the oxidation process finished (3–6). During the above process, the homolytic decomposition efficiency of the formed hydroperoxide is closely with the reaction efficiency, and N-alkyl pyridinium salts facilitated this decomposition step and then realized the higher catalytic activity.
 | ||
| Scheme 3 Proposed reaction mechanism of p-xylene oxidation. | ||
Conclusions
In summary, N-alkyl pyridinium salts were found to be effective for the catalytic oxidation of methyl aromatic hydrocarbon with molecular oxygen in the absence of metal species. Through the comparison of N-alkyl pyridinium salts with different substituted groups, electron-donating dimethylamino group substituted N-alkyl pyridinium salts exhibited a better catalytic activity. Especially with 1-benzyl-4-N,N-dimethylaminopyridinium bromide being used as the catalyst, different methyl aromatics were successfully oxidized in acetonitrile. The reported results offered a simple organocatalytic system for the metal-free oxidation of methyl aromatics. Intensive study on the role of N-alkyl pyridinium salts and its application for the catalytic oxidation of other substrate are underway.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (21872075 and 21873052) and by K. C. Wang Magna Fund in Ningbo University.References
- J. Piera and J.-E. Backvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef CAS PubMed.
- L. Kesavan, R. Tiruvalam, M. H. Ab Rahim, M. I. bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195–199 CrossRef CAS PubMed.
- A. C. Lindhorst, S. Haslinger and F. E. Kuehn, Chem. Commun., 2015, 51, 17193–17212 RSC.
- Y. Zhang, H. Zhang and Y. Yang, J. Chem. Technol. Biotechnol., 2019, 94, 1585–1592 CrossRef CAS.
- X. Yang, Y. Wang, L. Zhou, C. Chen, W. Zhang and J. Xu, J. Chem. Technol. Biotechnol., 2010, 85, 564–568 CrossRef CAS.
- P. Raghavendrachar and S. Ramachandran, Ind. Eng. Chem. Res., 1992, 31, 453–462 CrossRef CAS.
- W. Partenheimer, Catal. Today, 1995, 23, 69–158 CrossRef CAS.
- R. S. Drago, Coord. Chem. Rev., 1992, 117, 185–213 CrossRef CAS.
- Y. Jiang, C. Zhang, Y. Li, P. Jiang, J. Jiang and Y. Leng, New J. Chem., 2018, 42, 16829–16835 RSC.
- S. Nicolae, F. Neatu and M. Florea, C. R. Chim., 2018, 21, 354–361 CrossRef CAS.
- A. Hu, C. Lu, B. Li and T. Huo, Ind. Eng. Chem. Res., 2006, 45, 5688–5692 CrossRef CAS.
- H. Yuan, X. Fang, Q. Ma, J. Mao, K. Chen, Z. Chen and H. Li, J. Catal., 2016, 339, 284–291 CrossRef CAS.
- F. Wang, J. Xu, X. Li, J. Gao, L. Zhou and R. Ohnishi, Adv. Synth. Catal., 2005, 347, 1987–1992 CrossRef CAS.
- X. Li, J. Xu, F. Wang, J. Gao, L. P. Zhou and G. Yang, Catal. Lett., 2006, 108, 137–140 CrossRef CAS.
- P. Eversfield, W. Liu and E. Klemm, Catal. Lett., 2017, 147, 785–791 CrossRef CAS.
- S. Farahmand and M. Ghiaci, Chem. Eng. Sci., 2018, 190, 5–13 CrossRef CAS.
- Y. Deng, T. Zhang, C. Au and S. Yin, Appl. Catal., A, 2013, 467, 117–123 CrossRef CAS.
- C. Chen, S. Shi, M. Wang, H. Ma, L. Zhou and J. Xu, J. Mater. Chem. A, 2014, 2, 8126–8134 RSC.
- C. Chen, J. Xu, Q. Zhang, H. Ma, H. Miao and L. Zhou, J. Phys. Chem. C, 2009, 113, 2855–2860 CrossRef CAS.
- C. Chen, J. Xu, Q. Zhang, Y. Ma, L. Zhou and M. Wang, Chem. Commun., 2011, 47, 1336–1338 RSC.
- M. Wang, C. Chen, Q. Zhang, Z. Du, Z. Zhang, J. Gao and J. Xu, J. Chem. Technol. Biotechnol., 2010, 85, 283–287 CrossRef CAS.
- C. Guo, Q. Liu, X. Wang and H. Hu, Appl. Catal., A, 2005, 282, 55–59 CrossRef CAS.
- L. Chen, B. Li and D. Liu, Catal. Lett., 2014, 144, 1053–1061 CrossRef CAS.
- Q. Zhang, C. Chen, J. Xu, F. Wang, J. Gao and C. Xia, Adv. Synth. Catal., 2011, 353, 226–230 CrossRef CAS.
- G. Dobras and B. Orlinska, Appl. Catal., A, 2018, 561, 59–67 CrossRef CAS.
- A. A. Fokin and P. R. Schreiner, Adv. Synth. Catal., 2003, 345, 1035–1052 CrossRef CAS.
- R. Liu, X. Liang, C. Dong and X. Hu, J. Am. Chem. Soc., 2004, 126, 4112–4113 CrossRef CAS PubMed.
- Z. Zhang, J. Gao, F. Wang and J. Xu, Molecules, 2012, 17, 3957–3968 CrossRef CAS PubMed.
- G. Yang, Y. F. Ma and J. Xu, J. Am. Chem. Soc., 2004, 126, 10542–10543 CrossRef CAS PubMed.
- P. P Toribio, J. M. Campos-Martin and J. L. G. Fierro, Appl. Catal., A, 2005, 294, 290–297 CrossRef.
- L. J. Csanyi and K. Jaky, J. Mol. Catal. A: Chem., 1997, 120, 125–138 CrossRef CAS.
- L. J. Csanyi and K. Jaky, Phys. Chem. Chem. Phys., 2001, 3, 2018–2024 RSC.
- X. Tong, J. Xu, H. Miao and J. Gao, Tetrahedron Lett., 2006, 47, 1763–1766 CrossRef CAS.
- X. Tong, J. Xu, H. Miao, J. Gao, Z. Sun and W. Zhang, J. Chem. Technol. Biotechnol., 2009, 84, 1762–1766 CrossRef CAS.
- J. M. Berg, J. L. Tymoczko, L. Stryer and W. H. Freeman, Biochemistry, New York, 2002 Search PubMed.
- B. Tong, Q. S. Liu, Z. C. Tan and U. Welz-Biermann, J. Phys. Chem. A, 2010, 114, 3782–3787 CrossRef CAS PubMed.
- D. Vuluga, J. Legros, B. Crousse and D. Bonnet-Delpon, Chem. - Eur. J., 2010, 16, 1776–1779 CrossRef CAS PubMed.
- B. A. D. Neto and J. Spencerb, J. Braz. Chem. Soc., 2012, 23, 987–1007 CrossRef CAS.
- Q. Jiang, H. Hu, C. Guo, Q. Liu, J. Song and Q. Li, J. Porphyrins Phthalocyanines, 2007, 11, 524–530 CrossRef CAS.
- G. Yang, Q. Zhang, H. Miao, X. Tong and J. Xu, Org. Lett., 2005, 7, 263–266 CrossRef CAS PubMed.
- J. Gao, X. Tong, X. Li, H. Miao and J. Xu, J. Chem. Technol. Biotechnol., 2007, 82, 62–625 CrossRef.
- Q. Jiang, Y. Xiao, Z. Tan, Q. Li and C. Guo, J. Mol. Catal. A: Chem., 2008, 285, 162–168 CrossRef CAS.
- W. Zhou a, C. Sun, S. Xu and B. Hu, Inorg. Chim. Acta, 2012, 382, 167–170 CrossRef.
- E. F. V. Scriven, Chem. Soc. Rev., 1983, 12, 129–161 RSC.
- N. De Rycke, F. Couty and O. R. P. David, Chem. - Eur. J., 2011, 17, 12852–12871 CrossRef CAS PubMed.
- I. Reetz, V. Bacak and Y. Yagci, Macromol. Chem. Phys., 1997, 198, 19–28 CrossRef CAS.
- L. J. Csányi, K. Jáky, Z. Kóta and T. Páli, J. Mol. Catal. A: Chem., 2004, 209, 59–68 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Influence of temperature on the catalytic oxidation of p-xylene, optimization of reaction temperature, optimization of catalyst concentration, and correlative FT-IR and NMR spectrums. See DOI: 10.1039/c9ra08185b |
| This journal is © The Royal Society of Chemistry 2019 |