
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced catalytic activity of inhomogeneous Rh-based solid-solution alloy nanoparticles†
Md Samiul Islam Sarker *a,
Takahiro Nakamura*b,
Satoshi Kameokab,
Yuichiro Hayasakac,
Shu Yinb and
Shunichi Satob
*a,
Takahiro Nakamura*b,
Satoshi Kameokab,
Yuichiro Hayasakac,
Shu Yinb and
Shunichi Satob
aDepartment of Physics, University of Rajshahi, Rajshahi 6205, Bangladesh. E-mail: samiul-phy@ru.ac.bd
bInstitute of Multidisciplinary Research for Advanced Materials (IMRAM), Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan. E-mail: nakamu@tagen.tohoku.ac.jp; Tel: +81-22-217-5146
cInstitute for Materials Research (IMR), Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan
First published on 27th November 2019
Abstract
Catalytic Rh-based alloy nanoparticles (NPs) with inhomogeneous solid-solution structures were prepared from homogeneous solid-solution alloy NPs. Compared with homogeneous alloy NPs, these inhomogeneous alloy NPs exhibited enhanced catalytic activity and superior catalytic durability. Homogeneous solid-solution alloy NPs consisting of Rh and other immiscible noble metals were synthesized by laser-induced nucleation method in metallic ion solutions. STEM elemental mapping and EDS composition analysis of the particles clearly demonstrated that all the constituents were uniformly dispersed within the NPs. Moreover, the compositions of the alloys were nearly identical to the initial feeding ratios of metallic ions in the mixed solutions, strongly indicating the formation of equimolar solid-solution alloy NPs over the entire composition range. Although the catalytic stability of these Rh-based homogeneous alloy NPs during CO oxidation was improved, their catalytic activity was comparable to that of pure metal catalysts, owing to the uniform local structure at the atomic level. However, the catalytic activity of the alloy NPs was enhanced by heat treatment, which introduced inhomogeneity in the atomic distribution within the NPs. The enhanced activity was due to dissimilar interfaces in the inhomogeneous solid-solution alloy NPs.
1. Introduction
Nanoparticles (NPs) of noble metals such as Rh, Pd, and Pt are widely used as catalysts for the oxidation of CO in exhaust gas streams1 and for the preferential oxidation of hydrogen in fuel cells,2 owing to the advanced specific properties of these materials. Rh NPs are well-known high-efficiency catalysts for CO oxidation and NOx reduction.3–7 Despite the high catalytic performance, Rh NPs are easily oxidized under oxidizing conditions and re-clustered under reducing conditions. At high temperatures, though their catalytic activities towards CO/NO redox reactions simultaneously increase, Rh NPs tend to aggregate.8 On the other hand, Pd and Pt NPs are used in the preparation of electrodes in fuel cells and for automobile exhaust gas purification. However, these metal NPs suffer from CO poisoning, resulting in deactivation of their catalytic properties.9–11 Therefore, the effective removal of CO from the surface of these metal catalysts is essential for ensuring catalytic durability. The undesirable properties of pure metal NP catalysts may be improved by alloying with other metals. For example, the oxidation of Rh NPs can be effectively suppressed by alloying with small amounts of Pd.12 Alloy NPs of noble metals have also drawn much attention, not only because they enhance the activity of redox reactions by synergetic effects, but also because the cost of catalysts can be lowered by reducing the amount of expensive noble metals.13,14 While some kinds of binary alloy NPs show systematic changes in catalytic activity with changes in elemental composition, others exhibit an abrupt enhancement in catalytic activity at the optimum elemental composition. For example, the catalytic activity of RhxPt1−x alloy NPs for CO oxidation is between those of pure Rh and Pt NPs.15 By contrast, Rh-rich binary Rh1−xPdx NPs, synthesized by the polyol reduction method with a diameter of 15 nm, are found to exhibit enhanced catalytic activity for CO oxidation compared with that of pure Rh or Pd NPs.16 Moreover, the Rh–Pt core–shell-structured alloy NPs, also synthesized through the polyol reduction method in ethylene glycol (EG), show better CO oxidizing ability than pure Rh or Pt NPs.17 Further, the preferential oxidation of CO in hydrogen in the presence of core–shell-structured RuxPt1−x NPs is much superior to that with pure Ru or Pt NPs.18 Recently, an enhancement of CO oxidizing ability was reported in the presence of Ru–Pd solid-solution alloy NPs, which are immiscible in bulk form, compared with that of pure Ru or Pd NPs, owing to the presence of defect sites in the mixed fcc/hcp structures of Ru and Pd.19To date, chemically synthesized metal and alloy particles have been widely used as catalysts. However, it is well known that NPs with well-tailored shape and composition are often fabricated using toxic reagents. In addition, multi-step procedures are usually required and the use of chemicals, including reducing agents and stabilizers, is essential. For example, relatively strong reducing agents, such as hydrazine, are required to synthesize alloy NPs containing several elements with different reduction potentials. On the other hand, sustaining the high activity of catalytic particles with a large reactive surface area is also a critical problem during industrial use. Normally, metal particles, whether supported or unsupported, are sintered and inactivated at high temperatures.20,21 Many industrial applications, including CO oxidation, steam reforming, and automobile exhaust control, are operated above 300 °C. Therefore, catalytic stability of particles at high reaction temperatures is highly desired for practical applications.22–24
Rh, Pt, and Pd are metals in the 4d transition metal group with fcc structures. It is generally difficult to form homogeneous solid-solution alloys in the bulk form with these metals at room temperature because of the immiscible gap in their binary phase diagrams (Rh–Pd, Rh–Pt, and Pt–Pd), as a result of different electronic structures. Although it has been reported that NPs composed of immiscible metals tend to form solid solutions,25 a special technique is still required for ensuring that the solid-solution alloys are homogeneous, even with the chemical reduction method.26 Recently, we successfully demonstrated the formation of homogeneous solid-solutions of binary27–30 and ternary alloy NPs31,32 with fully tunable compositions using tightly focused femtosecond laser irradiation of mixed solutions containing metallic ions in an atmospheric pressure at room temperature, without using any reducing agents or complicated procedures. In a laser-induced highly intense optical field, solvated electrons and hydrogen radicals are created via the optical decomposition of water molecules. These transient species act as strong reducing agents for the reduction of metallic ions to zero-valent atoms in very short periods of time (∼hundreds of picoseconds),33 resulting in NP formation. We named this novel technique for fabrication of NPs “Laser-induced Nucleation Method (LINM)”. This method is quite simple, eco-friendly, and highly reproducible technique for the formation of noble metal and solid-solution alloy NPs with immiscible elements as long as metal ions can be reduced by transient reactive species. Moreover, the NPs produced are completely free from impurities. At present, there are no reports on the catalytic activity of composition-controlled equimolar solid-solution Rh-based alloy NPs because it is difficult to synthesize such solid-solution alloys with immiscible elements. Moreover, the effect of the local structure of the alloy NPs on their catalytic activity is still unclear.
In the present study, we investigated the catalytic activity of Rh-based alloy NPs fabricated by laser-induced nucleation method in mixed ion solutions. We also discussed origin of the high catalytic activity of alloy NP catalysts from the viewpoint of local structure within NPs.
2. Experimental
2.1. Preparation of metallic ion and mixed ion solutions
Rhodium(III) chloride trihydrate (RhCl3·3H2O, 99.5%, Wako), palladium(II) chloride (PdCl2, 99.9%, Sigma Aldrich), and hexachloroplatinic(IV) acid hexahydrate (H2PtCl6·6H2O, 99.9%, Sigma Aldrich) were purchased and used without further purification. Pure Rh, Pd, and Pt solutions were separately prepared by dissolving salts of these metals in ultrapure water (18.2 MΩ cm, arium® pro UV, Sartorius AG). The concentrations of these metals in the solutions were set at 2.54 × 10−4 mol L−1. In this study, 7.0 wt% of trisodium citrate dihydrate (Na3C6H5O7·2H2O, Wako Pure Chemical Industry Ltd.) was added to all the sample solutions to achieve NPs with uniform diameters. The citrate was removed from the NPs by hydrogen treatment before catalyst testing. Mixed solutions were prepared by simply mixing certain ratios of the Rh, Pd, and Pt ion solutions. The mixing ratios of the Rh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd, Rh:Pt, and Rh:Pd:Pt solutions were 1:1, 1:1, and 1:1:1, respectively. In this paper, the pure metal ion and mixed metal ion sample solutions of Rh, Pd, and Pt are denoted as Rh, Pd, Pt, RhPd, RhPt, and RhPdPt.
:Pd, Rh:Pt, and Rh:Pd:Pt solutions were 1:1, 1:1, and 1:1:1, respectively. In this paper, the pure metal ion and mixed metal ion sample solutions of Rh, Pd, and Pt are denoted as Rh, Pd, Pt, RhPd, RhPt, and RhPdPt.
2.2. Laser irradiation conditions
Pure Rh, Pd, and Pt NPs, as well as Rh-based mixed metal alloy NPs, were prepared by laser-induced nucleation method in the metal ion solutions. A quartz glass cuvette with 3 mL of pure or mixed metal ion solution was used. A Ti:sapphire laser (Spitfire Pro, Spectra Physics Co.), which generates femtosecond laser pulses by the chirped pulse amplifier (CPA) system (wavelength: 800 nm; pulse width: 100 fs; average pulse energy: 5.5 mJ; repetition rate: 100 Hz), was utilized. The beam was tightly focused with an aspheric lens with a focal length of 8 mm and numerical aperture of 0.5. Using the value of 5 mm for the laser beam radius before the focusing lens and 1.33 for the refractive index of water (nwater), the spot size at the focal point was calculated to be 175 μm. The spherical aberration resulting from refractive index mismatch between the cuvette and the solution was considered in this calculation. Assuming no loss in the sample solution and the optics employed in the experimental setup, the beam intensity was estimated to be 2.2 × 1014 W cm−2 at the focal point.2.3 Characterization of nanoparticles
The structure of the fabricated NPs was characterized by electron microscopy and X-ray diffraction. Bright-field images and selected-area electron diffraction (SAED) patterns of the NPs were obtained using a transmission electron microscope (TEM; JEOL, JEM2000EXII, JEOL Ltd.) operated at 200 kV. High-resolution TEM images were acquired with a TITAN80-300 (FEI) in the STEM mode operated at 300 kV. High-angle annular dark field (HAADF) STEM-EDS mappings were recorded using the same instrument to measure the elemental composition and distribution in the NPs. The crystalline structure of the NPs was determined by powder X-ray diffraction (XRD, RINT-V, Rigaku Co.). The elemental compositions of as-prepared alloy nanoparticles were confirmed by inductivity coupled plasma optical emission spectrometry (ICP-OES, Optima 3300XL, PerkinElmer). X-ray Photoelectron Spectroscopy (XPS, ULVAC-PHI) analysis was conducted to evaluate the electronic states of metal elements for alloy NPs.2.4. Preparation of the supported catalyst
To investigate the catalytic activity, the fabricated NPs were uniformly supported on γ-Al2O3 particles, which are typical supports for catalysts, by following a two-step procedure. First, a slurry of γ-Al2O3 was prepared by dispersing 1.0 g of γ-Al2O3 powder (99.97% metal basis, Alfa Aesar) in 33.0 mL of ultrapure water (18.2 MΩ cm). The slurry was then sonicated in an ultrasonic bath for 1 h. Subsequently, 3.0 mL of the γ-Al2O3 slurry was mixed with 9.0 mL of a colloidal solution of NPs prepared by laser irradiation. The remaining γ-Al2O3 solution was then added. This procedure yielded a colloidal solution with 0.059 wt% metal-supported catalyst. The colloidal solution of supported NPs was then dried for 1 day using a freeze dryer to obtain the catalyst in the powder form. The as-prepared catalyst powder was pressed into a pellet using a uniaxial press with a pressure of 16 MPa (Riken Seiki Co. Ltd.) for 7 min. The formed pellet was crushed and sieved to obtain granules with sizes in the range of 180–210 μm. As a result, the effective surface area of the catalyst samples was almost uniform.2.5. Catalysis testing
The activity of the CO oxidation catalytic reaction was measured as follows. The granule catalyst (70 mg) obtained via the procedure described above was loaded into a tubular glass reactor (inner diameter: 3.35 mm) and set on a quartz wool base. The reactor was then connected to a gas chromatograph (GC-8, Shimadzu Co.) for measuring the extent of CO to CO2 conversion. Prior to the catalytic activity measurements, the catalyst sample was treated at 300 °C for 2 h in H2 gas with a flow rate of 30 cm3 min−1 to remove impurities, including citrate, from the surface of the sample. Following hydrogen treatment, a gas mixture containing CO/O2 (1 and 0.5%, respectively) with He as a carrier gas was passed through the reactor at a flow rate of 30 cm3 min−1. After 15 min, the gas at the outlet was collected and analysed by gas chromatography with a thermal conductivity detector. The catalyst was heated at a rate of 5 °C min−1 until CO was completely oxidized to CO2, and then cooled to 150 °C at the same rate. The CO to CO2 conversion rate was recorded as a function of temperature during the heat cycle. CO conversion was then calculated by the simple formula, XCO (%) = (1 − [CO]out/[CO]in) × 100, where [CO]in and [CO]out are the input and output amounts of CO, respectively.2.6. Heat treatment of alloy nanoparticle catalysts
The sample was initially heated to 300 °C in H2 gas with a flow rate of 30 cm3 min−1 to remove adsorbed water molecules and decompose contaminants consisting of citrate present on the surface. Thereafter, 28.8 mg of the sample was placed in a reaction chamber and a mixed gas composed of Ar and O2 was introduced at flow rates of 80 and 320 cm3 min−1, respectively. The temperature of the system was then increased up to 1000 °C at a rate of 10 °C min−1.3. Results and discussion
When the femtosecond laser pulses were incident on the solution through an aspheric lens, a tiny flash of plasma was observed and fine bubbles of oxygen and hydrogen gases were generated around the focal point, indicating the optical breakdown of water molecules. After a certain duration of highly intense femtosecond laser irradiation, alloy NPs were formed in the mixed ion solutions. During laser irradiation, metallic ions were reduced to zero valence atoms (nuclei) by the transient reactive species generated through the photodecomposition of water molecules. Then, NPs were formed through nucleus growth in the solutions. When laser irradiation was stopped before complete reduction of ions in the solution, relatively large particles with a wide size distribution would be formed through autocatalytic action of the nuclei with unreacted ions. At the same time, formed NPs were broken into small pieces by successive laser pulses. Therefore, irradiation time was important to obtain NPs with small particle size and narrow size distribution. The details of the fabrication mechanism have been reported previously.27–32TEM images of the as-prepared NPs of Rh, Pd, Pt, RhPd, RhPt, and RhPdPt are shown in Fig. 1. The mean diameters of the Rh, Pd, Pt, RhPd, RhPt, and RhPdPt NPs calculated by measuring over 100 particles in the TEM images were 16.5 ± 6.5, 8.5 ± 2.1, 9.5 ± 2.3, 10.2 ± 2.3, 11.8 ± 3.3, and 11.5 ± 3.2 nm, respectively. The average determined by EDS analyses were Rh44.2Pd55.8, Rh53.1Pt46.9, and stoichiometries of the RhPd, RhPt, and RhPdPt NPs Rh29.3Pd34.9Pt35.8, respectively. The stoichiometric ratios suggest that the compositions of the NPs are comparable to the initial feed ratios of the metallic ions in the mixed solutions.
 | ||
| Fig. 1 TEM images of the as-prepared NPs, synthesized by laser-induced nucleation method in (a) Rh, (b) Pd, (c) Pt, (d) RhPd, (e) RhPt, and (f) RhPdPt solutions. | ||
STEM-EDS elemental mapping was performed to evaluate the distribution of elements in the alloy NPs fabricated in the mixed solutions. Fig. 2 shows a typical STEM-EDS mapping for the NPs of RhPt. The particles were found to be spherical in the HAADF-STEM image (Fig. 2a). The elemental mappings and reconstructed images of the RhPt NPs (Fig. 2b–d) clearly indicate the homogeneous distribution of each element in the NPs. Moreover, the line scanning profile (Fig. 2f) also shows a uniform distribution of Rh and Pt in the particles. These results clearly demonstrate that homogeneous solid-solution RhPt alloy NPs were formed, despite the immiscible nature of Rh and Pt.
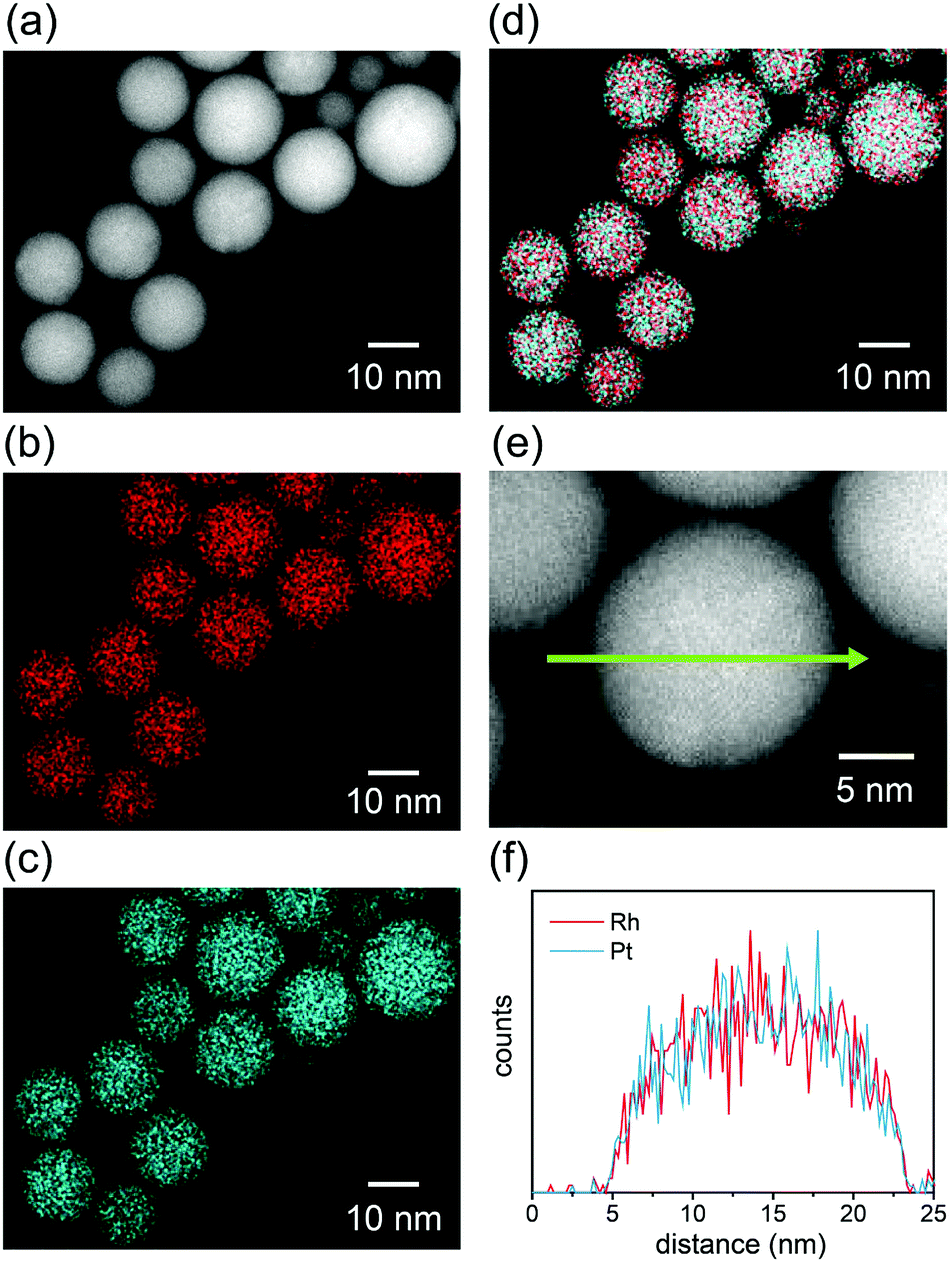 | ||
| Fig. 2 STEM-EDS mapping for the NPs fabricated in RhPt solution. (a) HAADF-STEM image, (b) Rh-L and (c) Pt-L STEM-EDS mappings, (d) reconstructed image of the maps in (b and c), (e) magnified HAADF-STEM image of a particle in (a), and (f) EDS line profile of Rh and Pt along the green arrow shown in (e). | ||
In the same manner, elemental mapping was also conducted for the NPs fabricated in RhPd and RhPdPt mixed solutions, and the results confirm the formation of solid-solution alloys, as illustrated in Fig. S1.† The crystallinity of the NPs was investigated by XRD analysis. Fig. 3 shows the XRD patterns of the NPs fabricated in Rh, Pd, Pt, RhPd, RhPt, and RhPdPt solutions. All the XRD patterns are characteristic of the fcc crystal structure. The interplanar spacings of the Rh, Pd, and Pt NPs calculated using the diffraction peaks originating from the (111) plane of the fcc crystal were 2.20, 2.25, and 2.26 Å, respectively, which agree with the known values for bulk Rh, Pd, and Pt (JCPDS card no. 05-0685, 46-1043, and 04-0802). Further, the interplanar spacings of the NPs formed in the RhPd, RhPt, and RhPdPt solutions were calculated to be 2.22, 2.23, and 2.24 Å, respectively, and these values are proportional to the theoretical values deduced using Vegard's law. Elemental compositions of as-prepared RhPd, RhPt and PhPdPt alloy NPs measured by ICP were Rh49.9Pd50.1, Rh49.0Pt51.0 and Rh32.3Pd34.3Pt33.1, respectively. This also suggested that the elemental compositions of prepared alloy NPs were fairly reflected the feeding ratios of metallic ions in mixed solutions.
 | ||
| Fig. 3 XRD patterns of the NPs fabricated in Rh, Pd, Pt, RhPd, RhPt, and RhPdPt solutions. The positions of the (111) and (200) peaks for Rh (solid), Pd (dotted), and Pt (broken) are indicated. | ||
The detailed structures of the particles were examined by HR-TEM. Fig. S2† shows an HR-TEM image of the particles fabricated in the RhPd solution as a representative example. It is clear that the fabricated particles were polycrystalline with spherical morphologies. In addition, the electric states of Rh, Pd and Pt were identified as metallic states by XPS analysis (Fig. S3†). Peak shift into lower-binding energy side in the spectra could be originated from solid-solution alloy structure of NPs.
Compositional and structural analyses of the fabricated NPs by spectroscopic and diffraction techniques clearly demonstrated the formation of solid-solution binary and ternary alloy NPs, despite the immiscible nature of Rh–Pd, Rh–Pt, and Pd–Pt binary systems. The formation of alloy NPs might be attributed to the high reduction potential (∼−2.84 eV) and very short lifetime (∼hundreds of nanoseconds) of the solvated electrons and hydrogen radicals formed by the photodecomposition of water molecules in the strong optical field. The detailed reaction scheme has been discussed previously.27–30 In the present study, based on in situ UV-visible spectral observations, the metallic ions appeared to be completely reduced in solution within 30 min under irradiation at a repetition rate of 100 Hz.
After the successful formation of the fully tunable homogeneous solid-solution alloys, the as-synthesized metal and alloy particles were supported on γ-Al2O3 particles using the two-step procedure described in the experimental section.
The catalytic CO oxidation activities of the supported NPs were characterized. Fig. S4† shows the TEM images of the γ-Al2O3 supported NPs. The average diameters of the Rh, Pd, Pt, RhPd, RhPt, and RhPdPt alloy NPs estimated from the TEM images were 16.8 ± 4.8, 9.3 ± 2.1, 7.3 ± 2.5, 10.4 ± 2.1, 14.6 ± 5.0, and 10.3 ± 2.7 nm, respectively, which were the same within experimental error to the diameters of the as-prepared NPs shown in Fig. 1. It is obvious that the fabricated NPs were uniformly dispersed on the surface of the γ-Al2O3 support.
Fig. 4 shows the catalytic CO oxidation activities of the Rh, Pd, Pt, RhPd, RhPt, and RhPdPt NP catalysts. The temperatures corresponding to 50% conversion of CO to CO2 (T50) for each catalyst are summarized in Table 1. The T50 values indicate that Rh exhibits the best catalytic performance for CO oxidation among the pure metal catalysts, whereas Pt exhibits the lowest activity. The catalytic activities of the binary and ternary Rh-based alloy NP catalysts are between those of pure Rh and Pt. This order of catalytic activity may be explained as follows. Generally, Rh is known to be an active catalyst for CO oxidation, whereas Pd and Pt suffer from CO poisoning, which was also demonstrated previously by Kalinkin et al.34,35 Moreover, Park et al. found that the CO oxidizing ability of Rh–Pt binary alloy particles could be controlled by changing their composition.15 In both cases, the activity of pure Rh was found to be much higher than that of pure Pt, whereas the activities of the alloy catalysts were between those of the individual pure metals. In addition, the surface reaction between adsorbed CO (COads) and O (Oads) on both noble metals was very efficient. However, it was reported that differences in the rates of O2 dissociative adsorption on polycrystalline surfaces of Pt and Rh metals existed. The dissociative sticking probabilities of Pt and Rh were about 0.2 and 1.0, respectively.36,37 Therefore, at low temperatures, the competitive adsorption of CO and O2 led to more efficient formation of Oads at the vacant sites on the Rh surface than those on the Pt surface, resulting in the faster formation of vacant sites through the reaction between neighboring COads and Oads. As shown in Fig. 4, the activity of the Pt catalyst is much improved in our experiments by alloying with Rh or Rh and Pd in the binary and ternary alloy catalysts, and the T50 values significantly decrease from 313 °C for the Pt catalyst to 275 and 266 °C for the RhPt and RhPdPt alloy catalysts, respectively. This result indicates that CO adsorbed on the surface of the Pt atoms was also effectively oxidized on the surface of other noble metal atoms through the Langmuir–Hinshelwood mechanism.
 | ||
| Fig. 4 Catalytic CO conversion on (a) Rh, (b) Pd, (c) Pt, (c) RhPd, (d) RhPt, (e) RhPt, and (f) RhPdPt NP catalysts at different temperatures. | ||
| T50±0.5(CO) (°C) | |
|---|---|
| (a) Rh | 262 |
| (b) Pd | 266 |
| (c) Pt | 314 |
| (d) RhPd | 260 |
| (e) RhPt | 276 |
| (f) RhPdPt | 267 |
The catalytic stability of the NP catalysts was also investigated by hysteresis analysis for CO conversion during a heating–cooling process. Fig. S5† shows the catalytic performance of the Rh, Pd, and Pt pure metal NP catalysts and the RhPd, RhPt, and RhPdPt alloy catalysts. The catalytic activities decreased during the catalysis testing for pure Rh and Pt, owing to oxidation and clustering of Rh and CO poisoning of Pt. In contrast, the catalytic performance of the alloy NP catalysts remained unchanged. Although cycle test must be required for evaluation of durability of catalyst, the tentative catalytic durability was improved in the alloy catalysts. The stability of the alloy NP catalysts might be caused by differences in the binding properties of the constituents in the alloy NP, with strong metal–metal interactions influencing the bonding between catalyst surfaces and reactants.
The morphologies of the NPs after catalysis were examined by TEM (Fig. S6†). In the case of the Rh NP catalyst, large aggregated particles were observed even after one cycle catalysis. This morphology was completely different from that of the as-prepared NPs (Fig. S4†). This change is reported to be due to the oxidation and/or re-clustering of Rh at high temperatures.26 However, no significant morphological changes or re-clustering effects were observed for the other NP catalysts after catalysis. In addition, the elemental mappings of the alloy NP catalysts after catalysis provided visual evidence of the homogeneous structures of the solid-solution alloy NPs, as shown in Fig. S7.† The compositional line profiles of the alloy NPs also confirmed that each element was homogeneously distributed within the particles. These results indicate that the binary and ternary Rh-based alloy NPs were stable at the atomic level throughout the catalysis process.
Although the catalytic stability was improved by alloying, the actual catalytic activity of the alloy NPs was not remarkably improved compared to those of pure Rh and Pd in the present study. This result could be explained by related previous studies. Alayoglu and Eichorn found that the catalytic activity of RhPt core–shell alloy particles was much higher than that of homogeneous alloy particles.17 They argued that the enhanced activities might be attributed to the architectural configuration of the particles, which influences the electronic structure and facilitates alternate reactions on the surface of the particles. Similarly, the synergistic effects of core–shell RhPd particles was also demonstrated by Araya and Díaz, who concluded that the synergetic effects heavily depended on the distribution of Rh and Pd on the particle surface.38 On the other hand, Kusada et al. reported that RuPd solid-solution alloy NPs showed outstanding catalytic activity compared with those of pure Ru and Pd.19 The authors claim that the improved catalytic activity was caused by the presence of defect sites in the alloy particles composed of fcc and hcp crystal structures. In fact, an inhomogeneous distribution of Ru and Pd was confirmed by EDS elemental mapping of the Ru–Pd alloy NPs. In other words, the unusual synergetic effect was attributed to the inhomogeneous local structure of the alloy NPs.
To validate this viewpoint, we attempted to synthesize inhomogeneous alloy NPs with defect sites and/or structural inhomogeneity by heat treatment of the homogeneous solid-solution alloy NPs. First, as a representative example, the thermal stability of the RhPd NP catalyst during heat treatment was measured by thermogravimetric-differential thermal analysis (TG-DTA). The results of the TG-DTA analysis for the RhPd alloy NP catalyst are presented in Fig. 5. It is difficult to observe clear weight changes in the TG curve (dashed line), except for an endothermic peak around 100 °C, which was attributed to the evaporation of water adsorbed on the sample. On the other hand, a large endothermic peak at 624.2 °C is observed in the DTA curve (solid line), in addition to a small peak around 100 °C. It is known that Rh and Pd are oxidized in an oxidizing atmosphere via an exothermic reaction at high temperatures and the oxidized metals return to the metallic state at much higher temperatures via an endothermic reaction. Therefore, although further investigations are needed, it is predicted that local structural changes in the homogeneous solid-solution alloy NPs occur around 600 °C. Based on the TG-DTA analysis, the homogeneous alloy NP catalysts were heat treated at 600 °C. The homogeneous alloy NP catalysts (70 mg) were placed in a reaction chamber and subjected to H2 treatment using the same procedure described earlier. O2 gas was subsequently passed through the reactor and the samples were heated to 600 °C at a rate of 5 °C min−1. Following the O2 treatment, the samples were subjected to H2 treatment once again at 300 °C to facilitate the reduction of the oxidized portion of the catalyst. Since the oxidation–reduction properties depended on element, the elemental distributions tend to be disordered due to preferential oxidation and reduction processes during above-mentioned heat treatment.
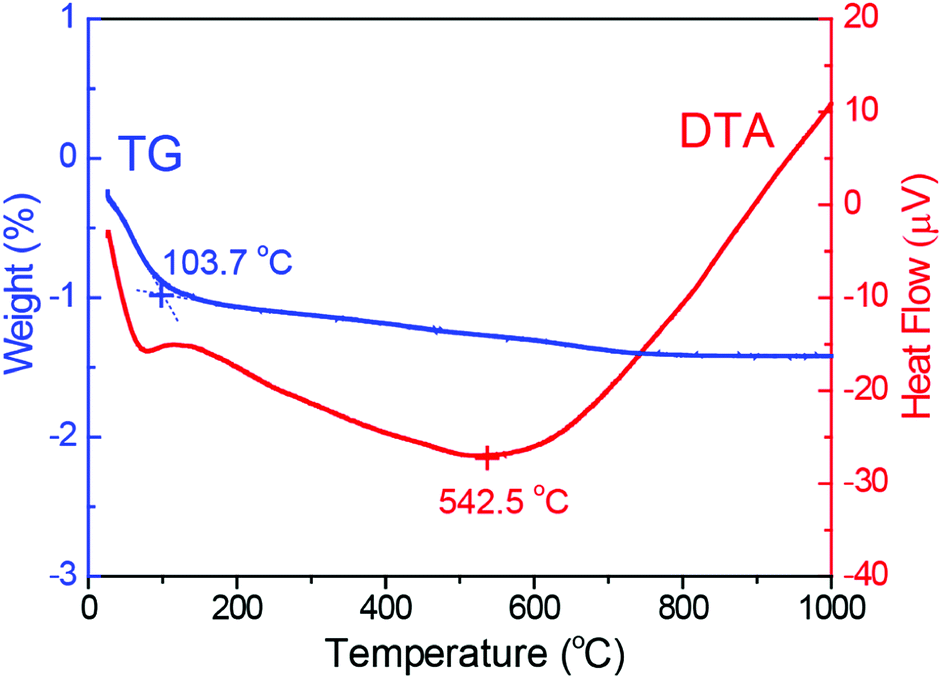 | ||
| Fig. 5 Thermogravimetric analysis and differential thermal analysis of RhPd alloy NPs supported on γ-Al2O3. | ||
The local structure of alloy NPs after heat treatment was evaluated by EDS elemental mapping. Fig. 6 shows the STEM-EDS mapping for the RhPt NPs, as a representative case. An inhomogeneous distribution of all the elements is clearly observed in the NPs. This result strongly demonstrates that inhomogeneous alloy NPs were successfully prepared by the heat treatment of the homogeneous solid-solution alloy NPs. In the same manner, elemental mappings were also obtained for the RhPd and RhPdPt NPs, and the results confirmed the formation of inhomogeneous solid-solution alloys, as shown in Fig. S8.†
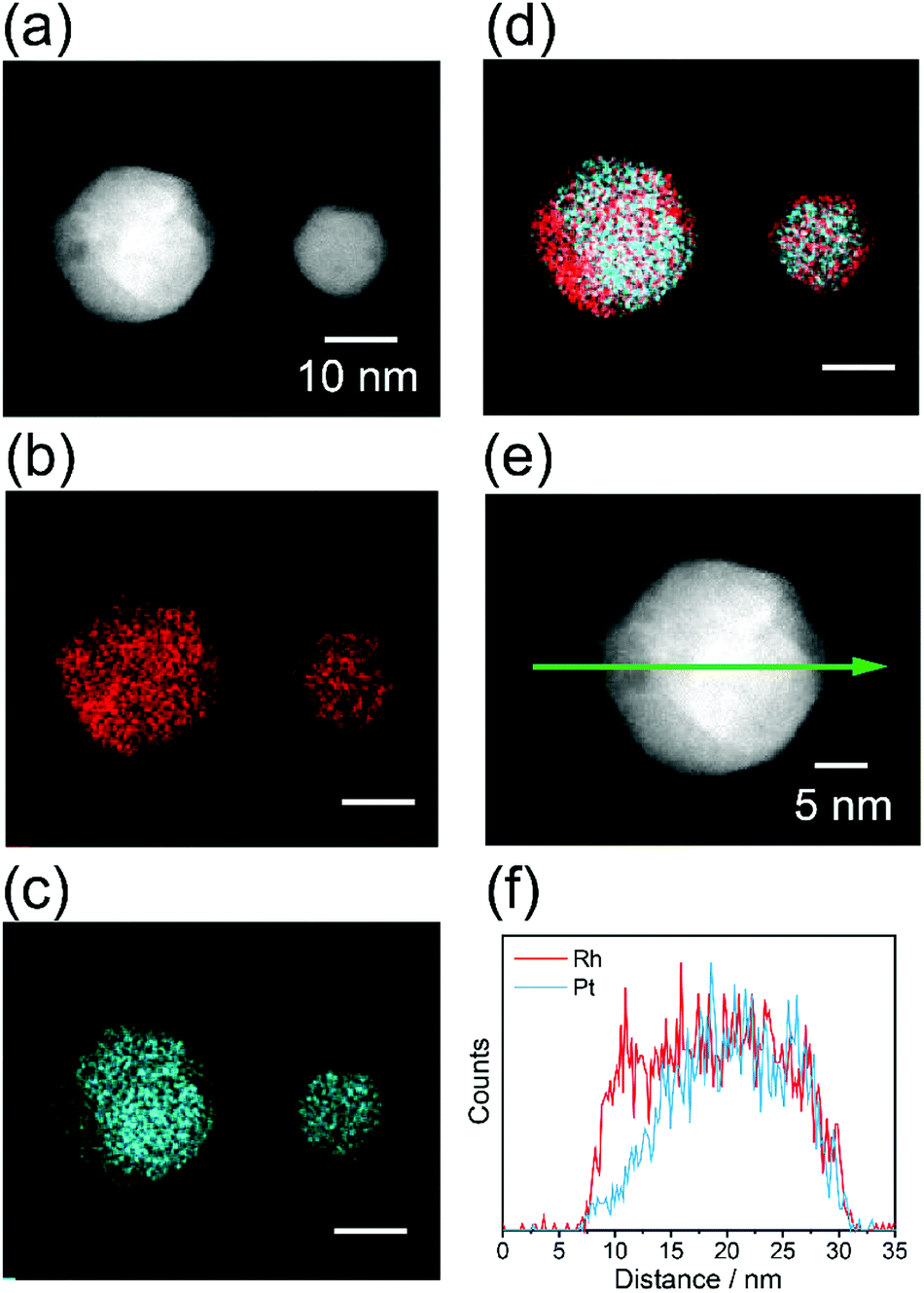 | ||
| Fig. 6 STEM-EDS mapping of the RhPt alloy NPs after heat treatment of the homogeneous RhPt alloy NPs. (a) HAADF-STEM image, (b) Rh-L and (c) Pt-L STEM-EDS mapping, (d) reconstructed image of the maps in (b and c), (e) magnified HAADF-STEM image of a particle in (a), and (f) EDS line profiles of Rh and Pt NP along the green arrow in (e). | ||
Fig. 7 shows the catalytic CO conversion performance of the inhomogeneous alloy NP catalysts. The T50 values for these catalysts are summarized in Table 2. It should be noted that the catalytic activities of the inhomogeneous alloy NP catalysts show improvement in comparison with the corresponding homogeneous NP catalysts (Fig. 4). For example, the active catalytic conversion of CO occurred at a lower temperature with the inhomogeneous RhPd alloy NP catalyst than with the homogeneous RhPd catalyst. In addition, the T50 value for the inhomogeneous RhPd alloy was 249 °C, whereas that for the homogeneous RhPd catalyst was 260 °C. This catalytic enhancement could be attributed to a dissimilar interface in the inhomogeneous solid-solution alloy NPs, which alters the electronic structures of the surface. As a result, CO oxidation is more facile on the inhomogeneous alloy catalysts than on the pure metal catalysts. This result is analogous to the behaviour of other inhomogeneously structured alloy NP catalysts, such as core–shell17,18 and partly phase-segregated catalysts.19
 | ||
| Fig. 7 Catalytic CO conversion on the alloy NPs before and after heat treatment. Catalytic CO conversion on the as-prepared (a) RhPd, (b) RhPt, and (c) RhPdPt alloy NPs, and on the corresponding heat-treated (d) RhPd, (e) RhPt, and (f) RhPdPt alloy NPs at various temperatures. | ||
| T50±0.5(CO) (°C) | |
|---|---|
| (a) RhPd H.T. | 249 |
| (b) RhPt H.T. | 262 |
| (c) RhPdPt H.T. | 251 |
The catalytic stability of the inhomogeneous alloy NPs was also investigated by hysteresis analysis of the catalytic CO conversion reaction during heating and cooling processes, as shown in Fig. S9.† The catalytic activities of all the inhomogeneous alloy NP catalysts remained unchanged during the heating and cooling processes, indicating their improved catalytic stability. Therefore, it could be concluded that the high catalytic performance of the alloy NP catalysts is due to the inhomogeneous local structure of the alloy NPs, whereas the catalytic stability is attributed to the solid-solution.
4. Conclusions
In this study, inhomogeneous solid-solution alloy NP catalysts with high catalytic activity and improved stability were successfully fabricated by femtosecond laser irradiation of mixed ion solutions, followed by application of an appropriate heat treatment. The local structures of the alloy NPs formed through high-intensity laser irradiation of the solutions were evaluated by STEM-EDS elemental mapping and EDS compositional analyses. The results of these analyses allowed us to confirm the equimolar solid-solution structures with uniform distribution of elements and controllable compositions. Catalytic CO oxidation was carried out for the pure metals as well as the homogeneous alloy NPs. Although the catalytic stability of the pure metal NPs was significantly improved by alloying with other noble metal elements, the catalytic activities of the alloy NPs were not very different from that of pure Rh, owing to the uniform local structure. On the other hand, the catalytic activity of inhomogeneous alloy NP catalysts prepared by heat treatment of the homogeneous catalysts was remarkably enhanced. These inhomogeneous catalysts also exhibited improved stability. These results are attributed to the preferential catalytic oxidation reaction of CO at the interface between different phases. To the best of our knowledge, this study is the first to demonstrate the origin of the high catalytic activity observed for alloy NP catalysts from the viewpoint of the local structure within NPs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Young Scientists (A) (No. 23686102) from the Japan Society for the Promotion of Science. We thank Mr Masaya Hino, Ms Erika Furukawa and Mr Takumi Akiyama for their help with ICP measurements. We thank Ms Sayaka Ogawa for her help with XPS measurements.Notes and references
- J. T. Kummer, Use of noble metals in automobile exhaust catalysts, J. Phys. Chem., 1986, 90, 4747–4752 CrossRef CAS.
- L. Carrette, K. A. Friedrich and U. Stimming, Fuel cells: Principles, types, fuels, and applications, ChemPhysChem, 2000, 1, 162–193 CrossRef CAS PubMed.
- M. Bowker, Automotive catalysis studied by surface science, Chem. Soc. Rev., 2008, 37, 2204–2211 RSC.
- M. E. Grass, Y. Zhang, D. R. Butcher, J. Y. Park, Y. Li, H. Bluhm, K. M. Bratlie, T. Zhang and G. A. Somorjai, A reactive oxide overlayer on rhodium nanoparticles during CO oxidation and its size dependence studied by in situ ambient-pressure X-ray photoelectron spectroscopy, Angew. Chem., Int. Ed., 2008, 47, 8893–8896 CrossRef CAS PubMed.
- Y. Zhang, M. E. Grass, W. Huang and G. A. Somorjai, Seedless polyol synthesis and CO oxidation activity of monodisperse (111)- and (100)-oriented rhodium nanocrystals in sub-10 nm sizes, Langmuir, 2010, 26, 16463–16468 CrossRef CAS PubMed.
- J. Gustafson, R. Westerström, A. Mikkelsen, X. Torrelles, O. Balmes, N. Bovet, J. N. Andersen, C. J. Baddeley and E. Lundgren, Sensitivity of catalysis to surface structure: The example of CO oxidation on Rh under realistic conditions, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 045423 CrossRef.
- G. Ertl, Reactions at surfaces: From atoms to complexity (Nobel lecture), Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS PubMed.
- A. J. Dent, J. Evans, S. G. Fiddy, B. Jyoti, M. A. Newton and M. Tromp, Rhodium dispersion during NO/CO conversions, Angew. Chem., Int. Ed., 2007, 46, 5356–5358 CrossRef CAS PubMed.
- U. A. Paulus, U. Endruschat, G. J. Feldmeyer, T. J. Schmidt, H. Bönnemann and R. J. Behm, New PtRu alloy colloids as precursors for fuel cell catalysts, J. Catal., 2000, 195, 383–393 CrossRef CAS.
- E. Antolini, Formation of carbon-supported PtM alloys for low temperature fuel cells: A review, Mater. Chem. Phys., 2003, 78, 563–573 CrossRef CAS.
- G.-Q. Lu and A. Wieckowski, Heterogeneous electrocatalysis: A core field of interfacial science, Curr. Opin. Colloid Interface Sci., 2000, 5, 95–100 CrossRef CAS.
- M. A. Newton, B. Jyoti, A. J. Dent, S. Diaz-Moreno, S. G. Fiddy and J. Evans, The impact of phase changes, alloying and segregation in supported RhPd catalysts during selective NO reduction by H2, ChemPhysChem, 2004, 5, 1056–1058 CrossRef CAS PubMed.
- N. Toshima and T. Yonezawa, Bimetallic nanoparticles–Novel materials for chemical and physical applications, New J. Chem., 1998, 22, 1179–1201 RSC.
- R. Ferrando, J. Jellinek and R. L. Johnston, Nanoalloys: From theory to applications of alloy clusters and nanoparticles, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- J. Y. Park, Y. Zhang, M. Grass, T. Zhang and G. A. Somorjai, Tuning of catalytic CO oxidation by changing composition of Rh–Pt bimetallic nanoparticles, Nano Lett., 2008, 8, 673–677 CrossRef CAS PubMed.
- J. R. Renzas, W. Huang, Y. Zhang, M. E. Grass, D. T. Hoang, S. Alayoglu, D. R. Butcher, F. Tao, Z. Liu and G. A. Somorjai, Rh1−xPdx nanoparticle composition dependence in CO oxidation by oxygen: Catalytic activity enhancement in bimetallic systems, Phys. Chem. Chem. Phys., 2011, 13, 2556–2562 RSC.
- S. Alayoglu and B. Eichorn, Rh–Pt bimetallic catalysts: Synthesis, characterization, and catalysis of core–shell, alloy, and monometallic nanoparticles, J. Am. Chem. Soc., 2008, 130, 17479–17486 CrossRef CAS PubMed.
- S. Alayoglu, A. U. Nilekar, M. Mavrikakis and B. Eichhorn, Ru–Pt core–shell nanoparticles for preferential oxidation of carbon monoxide in hydrogen, Nat. Mater., 2008, 7, 333–338 CrossRef CAS PubMed.
- K. Kusada, H. Kobayashi, R. Ikeda, Y. Kubota, M. Takata, S. Toh, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka and H. Kitagawa, Solid solution alloy nanoparticles of immiscible Pd and Ru elements neighboring on Rh: Changeover of the thermodynamic behaviour for hydrogen storage and enhanced CO-oxidizing ability, J. Am. Chem. Soc., 2014, 136, 1864–1871 CrossRef CAS PubMed.
- C. T. Campbell, S. C. Parker and D. E. Starr, The effect of size-dependent nanoparticle energetics on catalyst sintering, Science, 2002, 298, 811–814 CrossRef CAS PubMed.
- S. H. Joo, J. Y. Park, C.-K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Thermally stable Pt/mesoporous silica core–shell nanocatalysts for high-temperature reactions, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed.
- A. Cao and G. Veser, Exceptional high-temperature stability through distillation-like self-stabilization in bimetallic nanoparticles, Nat. Mater., 2010, 9, 75–81 CrossRef CAS PubMed.
- H. Bönnemann, G. Braun, W. Brijoux, R. Brinkmann, A. S. Tilling, K. Seevogel and K. Siepen, Nanoscale colloidal metals and alloys stabilized by solvents and surfactants: Preparation and use as catalyst precursors, J. Organomet. Chem., 1996, 520, 143–162 CrossRef.
- P. Qiao, S. Xu, D. Zhang, R. Li, S. Zou, J. Liu, W. Yi, J. Li and J. Fan, Sub-10 nm Au–Pt–Pd alloy trimetallic nanoparticles with a high oxidation-resistant property as efficient and durable VOC oxidation catalysts, Chem. Commun., 2014, 50, 11713–11716 RSC.
- G. C. Bond, The electronic structure of platinum–gold alloy particles, Platinum Met. Rev., 2007, 51, 63–68 CrossRef CAS.
- H. Zhang and N. Toshima, Preparation of novel Au/Pt/Ag trimetallic nanoparticles and their high catalytic activity for aerobic glucose oxidation, Appl. Catal., A, 2011, 400, 9–13 CrossRef CAS.
- Y. Herbani, T. Nakamura and S. Sato, Synthesis of near-monodispersed Au–Ag nanoalloys by high intensity laser irradiation of metal ions in hexane, J. Phys. Chem. C, 2011, 115, 21592–21598 CrossRef CAS.
- T. Nakamura, Y. Herbani and S. Sato, Fabrication of solid-solution gold–platinum nanoparticles with controllable compositions by high-intensity laser irradiation of solution, J. Nanopart. Res., 2012, 14, 785 CrossRef.
- M. S. I. Sarker, T. Nakamura, Y. Herbani and S. Sato, Fabrication of Rh based solid-solution bimetallic alloy nanoparticles with fully-tunable composition through femtosecond laser irradiation in aqueous solution, Appl. Phys. A, 2013, 110, 145–152 CrossRef CAS.
- T. Nakamura and S. Sato, Green and facile synthesis of Pd–Pt alloy nanoparticles by laser irradiation of aqueous solution, J. Nanosci. Nanotechnol., 2015, 15, 426–432 CrossRef CAS PubMed.
- Y. Herbani, T. Nakamura and S. Sato, Synthesis of platinum-based binary and ternary alloy nanoparticles in an intense laser field, J. Colloid Interface Sci., 2012, 375, 78–87 CrossRef CAS PubMed.
- M. S. I. Sarker, T. Nakamura and S. Sato, Composition-controlled ternary Rh–Pd–Pt solid-solution alloy nanoparticles by laser irradiation of mixed solution of metallic ions, J. Mater. Res., 2014, 29, 856–864 CrossRef.
- S. Pommeret, F. Gobert, M. Mostafavi, I. Lampre and J.-C. Mialocq, Femtochemistry of the hydrated electron at decimolar concentration, J. Phys. Chem. A, 2001, 105, 11400–11406 CrossRef CAS.
- A. V. Kalinkin, A. V. Pashis and V. I. Bukhtiyarov, CO oxidation over the Pt–Rh system. 2. Reaction on an alloy, React. Kinet. Catal. Lett., 2003, 78, 107–112 CrossRef CAS.
- A. V. Kalinkin, A. V. Pashis and V. I. Bukhtiyarov, CO oxidation over the Pt–Rh system. 3. Reaction on a heterophase surface, React. Kinet. Catal. Lett., 2003, 78, 121–127 CrossRef CAS.
- W. M. Daniel, Y. Kim, H. C. Peebles and J. M. White, Adsorption of Ag, O2 and N2O on Ag/Rh(100), Surf. Sci., 1981, 111, 189–204 CrossRef CAS.
- C. R. Helms, H. P. Bonzel and S. Kelemen, The effect of the surface structure of Pt on its electronic properties and the adsorption of CO, O2, and H2: A comparison of Pt(100) – (5×20) and Pt(100) – (1×1), J. Chem. Phys., 1976, 65, 1773–1782 CrossRef CAS.
- P. Araya and V. Díaz, Synergism in the reaction of CO with O2 on bimetallic Rh–Pd catalysts supported on silica, J. Chem. Soc., Faraday Trans., 1997, 93, 3887–3891 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: STEM-EDS elemental mappings of the nanoparticles fabricated in RhPd or RhPdPt solution as-fabricated, after heat treatment, and after catalytic reaction; HR-TEM images of the nanoparticles fabricated in RhPd solution; XPS spectra for RhPdPt alloy NPs; TEM images of the γ-Al2O3 supported catalysts before and after measurement of catalytic activity; catalytic stability of the as-prepared and heat-treated nanoparticle catalysts during CO conversion measured by hysteresis analyses during the heating and cooling process. See DOI: 10.1039/c9ra06167c |
| This journal is © The Royal Society of Chemistry 2019 |